

Abstract
Epidermal growth factor receptor (EGFR) is overexpressed in head and neck squamous cell carcinoma (HNSCC) where it has been shown to promote tumor cell invasion upon phosphorylation. One mechanism by which EGFR promotes tumor progression is by activating signal cascades that lead to loss of E-cadherin, a transmembrane glycoprotein of the cell-cell adherence junctions; however mediators of these signaling cascades are not fully understood. One such mediator, RhoC, is activated upon a number of external stimuli, such as epidermal growth factor (EGF), but its role as a mediator of EGF-stimulated migration and invasion has not been elucidated in HNSCC. In the present study, we investigate the role of RhoC as a mediator of EGF-stimulated migration and invasion in HNSCC. We show that upon EGF stimulation, EGFR and RhoC were strongly activated in HNSCC. This resulted in activation of the phosphatidylinositol 3-Kinase Akt pathway (PI3K-Akt), phosphorylation of GSK-3β at the Ser9 residue, and subsequent down regulation of E-cadherin cell surface expression resulting in increased tumor cell invasion. Knockdown of RhoC restored E-cadherin expression and inhibited EGF-stimulated migration and invasion. This is the first report in HNSCC demonstrating the role RhoC plays in mediating EGF-stimulated migration and invasion by down-regulating the PI3K-Akt pathway and E-cadherin expression. RhoC may serve as a treatment target for HNSCC.
Abbreviations: EGFR, epidermal growth factor receptor; EGF, epidermal growth factor; HNSCC, head a neck squamous cell carcinoma; PI3K, phosphatidylinositol 3-kinase; STAT3, signal transducer and activator of transcription 3; EMT, epithelial mesenchymal transition; GSK3β, glycogen synthase kinase 3 beta; epithelial cadherin, E-cadherin; HOK, human oral keratinocytes; MAPK, mitogen-activated protein kinase; ERK, extracellular-signal-regulated kinase
Introduction
According to the World Health Organization (WHO), head and neck squamous cell carcinoma (HNSCC) is amongst the most prevalent cancers worldwide, consistently ranking in the top twelve of most common cancers. It is estimated that in 2014 there will be over 42,000 new cases of oropharyneal cancer in the United States alone, which will account for more than 8000 deaths [1]. Unfortunately, despite advancements in chemotherapy, radiation therapy and surgical approaches, the survival rates have not improved for decades, most likely due to identification of most of these cancers at a very late stage when tumors have already undergone locoregional and distant spread. This is further complicated by the fact that aggressive treatments are selected on the basis of tumor size and spread rather than on the biology of individual lesions. However, two tumors that receive the same treatment may vary dramatically in biologic behavior. Therefore, characterization of signaling cascades that are responsible for tumor development, invasion and metastasis in HNSCC will help to identify aggressive tumors earlier in the disease process and will aid in the development of new treatment approaches.
Studies have shown that aberrant activation of Rho family of GTPases, members of the Ras homology protein family, promotes uncontrolled cellular proliferation as well as increases the invasive and metastatic potential of tumor cells [2], [3], [4]. Specifically, Rho GTPases have been shown to cause tumorigenic transformation of rodent fibroblasts [5], as well as promote T lymphoma cell invasion [6], [7], breast cancer cell invasion [8] and melanoma cell metastatic growth [9]. Rho accomplishes this by shuttling between inactive guanine diphosphate (GDP) and active GTP-bound forms. It is the active GTP-bound form that triggers a cell response.
Of the Rho family members (RhoA, RhoB, RhoC, Rac1, Rac2, Rac3, and CDC42), RhoC is increasingly being implicated in a variety of malignant tumor types, including HNSCC. Studies have shown that RhoC overexpression is linked to metastatic behavior in HNSCC where selective RhoC inhibition markedly decreases cell motility and invasion in vitro and in vivo [10]. Additionally, Islam et al., 2014, recently demonstrated that RhoC plays an important role in signal transduction and activation of transcription 3 (STAT3) phosphorylation and the activation of core cancer stem cell transcription factors [11]. Despite the emerging evidence establishing RhoC as a key player in HNSCC invasion and metastasis, little is known about the intracellular signaling cascades that lead to RhoC activation and the subsequent effects on downstream signaling molecules.
In order for a tumor cell to invade surrounding tissues leading to metastatic disease, it must acquire motile and invasive characteristics—namely, these cells undergo transition to a more mesenchymal, fibroblast-like morphology where cell-to-cell contacts are disrupted. This process is collectively referred to as epithelial-to-mesenchymal transition (EMT). Epithelial cadherin (E-cadherin) is a transmembrane glycoprotein of the cell-cell adhesion transmembrane molecule and its down-regulation leading to disruption of adherence junctions has been shown to contribute to the EMT process. Furthermore, E-cadherin plays important roles in cellular signal transduction in collaboration with receptor tyrosine kinases such as the Epidermal Growth Factor Receptor (EGFR), which is highly expressed in HNSCC and is inversely correlated with patient survival [12], [13]. Studies have shown that EGFR phosphorylation, subsequent to Epidermal Growth Factor (EGF) binding, can lead to transcriptional regulation of E-cadherin via the phosphatidylinositol 3-kinase/Akt/GSK-3β (PI3K/Akt/GSK3β) pathway. The PI3K/Akt/GSK3β signaling cascade is involved in the development and progression of HNSCC and plays a role in tumor resistance to radiotherapy and chemotherapy [14], [15]. E-cadherin transcription is regulated by multiple processes, including hypermethylation and repression of promoter activity of the Snail superfamily of zinc-finger transcription factors, Snail and Slug. Growing evidence suggests that the epidermal growth factor receptor family and its downstream signaling pathways, such as PI3K/Akt/GSK3β, up-regulate Snail leading to underexpression of E-cadherin [15].
In this study, we explored the role RhoC plays in EGFR/PI3K/Akt/GSK3β signaling and the cell–cell adhesion function of E-cadherin. We demonstrate that RhoC mediates EGFR/PI3K/Akt/GSK3β signaling in HNSCC to reduce E-cadherin expression, thus promoting a more invasive phenotype. To the best of our knowledge, the role of RhoC in EGFR/PI3K/Akt/GSK3β signaling pathways has not yet been reported in HNSCC. Our findings add to the growing evidence supporting the promise of RhoC as a prognostic marker and potential therapeutic target for HNSCC.
Materials and Methods
Cell Culture
Human oral SCC cell lines were grown in Dulbecco’s modified Eagle’s medium/F-12 (DMEM/F-12, Thermo scientific) containing 2.5 mM l-Glutamine, 15 mM HEPES buffer, 10% fetal bovine serum, 100 μg/ml penicillin, 100 μg/ml streptomysin. The primary human oral keritinocyte cell line (HOK, Science cell research Laboratories) was maintained in Oral Keratinocyte Medium (OKM) containing oral keratinocyte growth supplementation (OKGS) and penicillin/streptomysin (Science cell research Laboratories).
Western Blot Analysis
Cells with or without treatment with Epidermal Growth Factor (EGF) (Sigma Aldrich) were washed with ice-cold phosphate- buffered saline and lysed in RIPA buffer (25mM Tris.HCl pH 7.6, 150mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing Halt protease inhibitor cocktail (Thermo scientific). For phospho – antibodies the lysis buffer was supplemented with Halt phosphotase inhibitor cocktail (Thermo scientific). Cell lysates were scraped into Microfuge tubes and left for 20 min with occasional vortexing, and was pelleted by centrifugation at 12,000 rpm for 20 min at 4 °C. The supernatant was collected, and protein content was measured by the Coomassie (Bradford) Protein Assay Kit (Thermo scientific). Equal amount of protein (25 μg) were electrophoresed on 8 or 12% SDS-PAGE gels and transferred to PVDF membranes (BioRad). The membranes were blocked with 5% nonfat milk or with 3% BSA in Tris - buffered saline containing 0.1% Tween-20 (TBS-T) (Fisher) for 1h at room temperature (RT) to block nonspecific binding. Membranes were incubated with the primary antibody for 2h at RT or overnight at 4 °C. Primary antibody concentrations were as follows: EGF Receptor (D38B1) XP Rabbit monoclonal antibody (1:1000), Phospho - EGF Receptor (Tyr992) antibody (1:500), Phospho - EGF Receptor (Tyr1068) (D7A5) XP Rabbit monoclonal antibody (1:1000), RhoC (D40E4) Rabbit monoclonal antibody (1:1000), RhoA (67B9) Rabbit monoclonal antibody (1:1000), E-Cadherin (24E10) Rabbit monoclonal antibody (1:5000), Phospho-GSK-3β (ser9) (5B3) Rabbit monoclonal antibody (1:2000), GSK-3β (27C10) Rabbit monoclonal antibody (1:2000), phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (D13.14.4E) XP Rabbit monoclonal antibody (1:2000), p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (137F5) Rabbit monoclonal antibody (1:2000), Phospho-Akt (Ser473) (D9E) XP Rabbit monoclonal antibody (1:2000), Akt (pan) (C67E7) Rabbit monoclonal antibody (1:2000), Snail (C15D3) Rabbit monoclonal antibody (1:500). Membranes were washed in TBS-T. Anti-rabbit IgG, HRP-linked antibody used as a secondary antibody to detect primary antibodies. Visualization of immunoreactive proteins was accomplished by superSignal West pico Chemiluminescent substrate (Thermo Scientific) and exposure to film or VersaDoc (Biorad). For Akt inhibition experiments, UM-SCC-6 cells were pre-treated with the AKT inhibitor, Ly294002, for 30 minutes followed by stimulation with EGF for 5 minutes. Densitometric analysis was performed and densitometric units (DU) are presented for each immunoblot.
Rho Pull-Down Assay
Cells were treated with EGF for 5 minutes to 2 hours. Then GTP bound Rho proteins were pulled down using a rhotekin binding and detection kit (Thermo Scientific) according to manufacturer’s instruction.
Small Interfering RNA (siRNA) Transfection
To knock down RhoC, cells were transfected with ON-Targetplus SMARTpool RhoC (50 nM) (Dharmacon Reseach, Inc) using Lipofectamine2000 (Invitrogen) in complete medium without antibiotics. The siCONTROL Non-Targetingpool siRNA (Dhamacon) was used as a transfection control. Western Blot samples were prepared 72 h after siRNA transfection.
Wound Healing Assay
Cell migration was assessed by the ability of cells to migrate into a cell-free area. Once the cells reached confluence, the medium was changed to FBS free medium for overnight starvation. The monolayers were then wounded by using a 200ul pipet tip and washed with PBS twice. After washing, the cells were incubated in the medium with or without EGF for the desired time period. The migration was observed under the microscope (NiKON Eclipse TS100, Japan). The images were taken by using cyberlink PowerDirector 10 softwear at 4 × magnification. The width of the scratches was observed and measured using Image J software. The relative distance was calculated as a mean width of the cell scratch.
Invasion Assay
Cell invasion was measured by using QCM 24-well Collagen-Based Cell Invasion Assay (Millipore Corporation). siRNA transfected cells were treated according to manufacturer’s instruction. Cells were stimulated with EGF for 48h. The results were read by using a microplate reader at the 562nM wavelength.
Immunofluorescent Staining
Transfected cells were seeded into 35mm glass bottom culture dishes (MatTek corporation). When the cells reached 70% confluence, they were starved and cultured in the medium with or without EGF for the desired time. Cells were then washed once with phosphate-buffered saline (PBS) and fixed with HistoChoiceMB (Molecular Biology) tissue fixative (Amresco) for 20 minutes at RT. Cells were washed with PBS three times and permeabilized with 0.5% Triton X-100 in PBS for 10 minutes at RT. After washing cells twice with PBS, the cells were blocked with 5% BSA in PBS for 1h at RT and incubated with purified mouse anti-E-Cadherin mAb (BD Transduction laboratories) diluted in 1% BSA in PBS (1:50). Alexa Fluor546-labeled donkey anti-mouse IgG (Invitrogen) was used as a secondary antibody (1:500). Finally, the cells were mounted with Vectashield mounting medium for fluorescence with DAPI (Vector Laboratories, Inc.) and were examined using a confocal microscope (Nikon TE2000-U).
Statistical Analysis
Student’s t test or paired t test was used to evaluate the significance of differences between two groups. The differences were considered statistically significant if P < .05.
Results
EGFR is Highly Expressed in HNSCC and is Robustly Phosphorylated upon EGF Binding
The importance of EGFR expression, phosphorylation, and activation of downstream effectors has been well characterized in a variety of tumor types [16], [17], [18], [19], but the role of small GTPases in mediating these important signaling cascades is not as clear. In this study, initial experiments sought to confirm EGFR expression and signaling in non-malignant human oral keratinocytes (HOK) and in a panel of well-characterized HNSCC cell lines: UM-SCC-1, UM-SCC-5, UM-SCC-6, UM-SCC-10B, UM-SCC-17B, SCC-0, and SCC-1483. As expected, EGFR protein levels were variably expressed across all cell lines, with the highest level of expression seen in UM-SCC-6 and UM-SCC-10B cells (Figure 1A). Of the cell lines tested, UM-SCC-6 cells were chosen for subsequent experiments based on the observation that 1) total EGFR was highly expressed in these cells; 2) endogenous EGFR phosphorylation was relatively low in the absence of exogenous epidermal growth factor (EGF) stimulation (Figure 1B); and 3) RhoC protein was highly expressed in these cells (Figure 3A).
Figure 1.
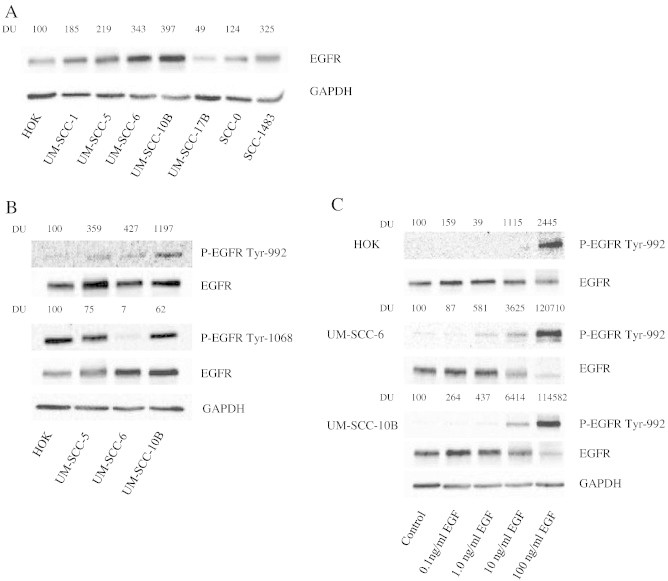
EGFR is expressed and phosphorylated upon EGF binding in HNSCC cell lines. (A) Whole cell lysate of HOK, UM-SCC-(1,5,6,10B, 17B), and SCC-(0, 1483) were separated by PAGE and blotted with EGFR antibodies. The same membrane was subsequently blotted with GAPDH antibody to verify equivalency of loading. DU = densitometic units. (B) HOK and UM-SCC-5, -6, and -10B cells were analyzed for endogenous expression of phospho-EGFR at Tyr992 and phospho-EGFR at Tyr1068. Total EGFR and GAPDH were used as loading controls. (C) Dose response of EGF-mediated phosphorylation of EGFR. HOK, UM-SCC-6, and -10B cells were stimulated with 1 to 100 ng/ml of EGF for 5 min and then were analyzed for EGFR-Tyr 992 phosphorylation.
Figure 3.
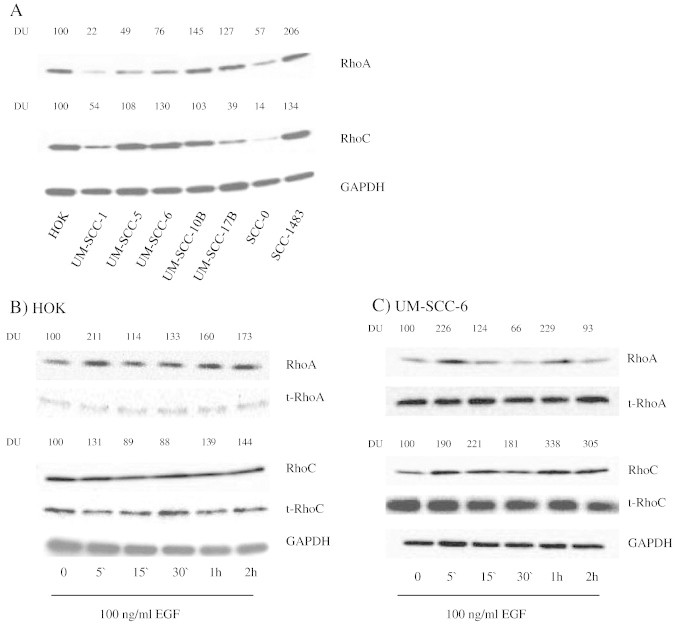
RhoC is expressed in HNSCC and is activated by EGFR. (A) Whole cell lysate of HOK, UM-SCC-(1, 5, 6, 10B, 17B), and SCC-(0, 1483) were separated by PAGE and blotted with RhoA and RhoC antibodies. GAPDH was used as loading control. (B) HOK and UM-SCC-6 cells were stimulated with 100 ng/ml of EGF for varying durations from 5 minutes to 2 hours and RhoA and RhoC activation was evaluated by a pull down assay. Total RhoA, RhoC and GAPDH were used as loading controls. Active, GTP-bound Rho was quantified by densitometry and normalized to total normalized Rho with respect to GAPDH.
We next determined whether we could activate EGFR upon treatment with increasing concentrations of EGF (0.1–100 ng/ml). Upon activation by its growth factor ligands, EGFR undergoes a transition from an inactive monomeric form to active homodimer and heterodimers. EGFR dimerization stimulates its intrinsic intracellular protein-tyrosine kinase activity and autophosphorylation of several tyrosine residues in the C-terminal domain of EGFR occurs [20]. EGFR phosphotyrosine residues Tyr992 and Tyr1068 were selected for investigation based on their known association with tumor progression. EGFR autophosphorylation elicits downstream activation and signaling by several other proteins that associate with the phosphorylated tyrosines through their own phosphotyrosine-binding SH2 domains [20]. Of the possible tyrosine residues phosphorylated upon EGF binding, we chose Tyr992 for further investigation due to its known effects on increasing the duration of interaction with effector molecules, its low endogenous phosphorylation status in non-malignant cells (Figure 1B, top panel), its described role in linking EGFR to a diversity of downstream signaling pathways, including the PI3K pathway, and its effects on cytoskeletal remodeling, suggesting a potential role in tumor progression [21]. In the presence of 100 ng/ml of EGF, EGFR was strongly phosphorylated at residue Tyr992 in HOK, UM-SCC-6, and UM-SCC-10B cells, however only the HNSCC cells demonstrated Tyr992 phosphorylation at concentrations as low as 1.0 ng/ml (Figure 1C). This finding is consistent with other studies where EGFR phosphorylation was determined to be more sensitive to lower concentrations of EGF depending on differential phosphotyrosine residue activation and cell type [22].
EGFR Phosphorylation at Tyr992 Activates the PI3K Pathway and Up-regulates Snail Protein Expression
E-cadherin is frequently lost during advanced stages of epithelial cancer progression due to up-regulation of the transcription factor, Snail, following EGFR activation. Increased Snail expression has been correlated with HNSCC invasion and local recurrence following therapy, which is consistent with its role in diminishing cell-to-cell contact by binding to the E-cadherin promoter and repressing transcription [23]. In agreement with these findings, we observed expression of Snail in UM-SCC-6 cells whereas none was seen in HOK cells (Figure 2A). Furthermore, Snail expression was greatly stabilized in as little as three hours following EGF stimulation in UM-SCC-6 cells (Figure 2B). Previous studies in other tumor types have shown that Snail up-regulation and subsequent E-cadherin loss is mediated by glycogen synthase kinase-3β phosphorylation at Ser9 (GSK-3βser9), which is a well-known downstream effector of the PI3K/Akt pathway and several other pathways (a notable example being the mitogen-activated protein kinase (MAPK) pathway), and has been shown to stabilize Snail leading to its localization into the nucleus [23]. Indeed, we observed phosphorylation of Akt and GSK-3βser9 in UM-SCC-6 cells at 5 minutes following treatment with EGF (Figure 2C). Given that a number of other pathways can also phosphorylate GSK-3βser9, we next set out to confirm that GSK-3βser9 is indeed phosphorylated by an EGFR/PI3K/Akt mechanism in the UM-SCC-6 cells. To do so, we measured EGF-stimulated GSK-3βser9 phosphorylation in the presence or absence of a well-established PI3K pathway inhibitor, Ly294002. As expected, GSK-3βser9 phosphorylation was enhanced following stimulation with 100ng/ml concentration of EGF in the absence of Ly294002 but was reduced by over 40% upon inhibition of the PI3-kinase pathway with Ly294002 (Figure 2D), confirming the important role the PI3K/Akt pathway plays in GSK-3βser9 signaling in these cells.
Figure 2.
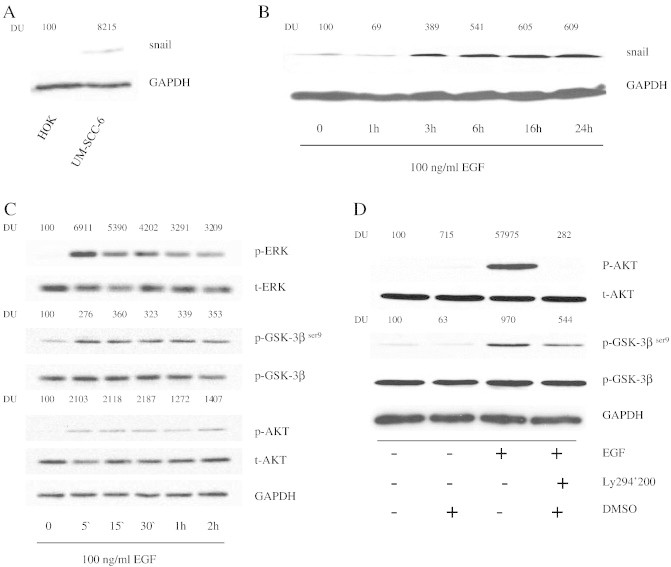
EGFR phosphorylation at Tyr992 activates the PI3-kinase pathway and up-regulates Snail protein expression. (A) Snail expression in HOK and UM-SCC-6 was examined via immunoblot analysis. (B) Snail expression is up-regulated following EGF stimulation. UM-SCC-6 cells were stimulated with 100 ng/ml of EGF for varying durations from 1 to 24 hours. Whole cell lysates were blotted to detect Snail expression. GAPDH was used as loading control. (C) ERK, GSK-3βser9 and AKT are phosphorylated following EGF stimulation. UM-SCC-6 cells were stimulated with 100 ng/ml EGF for 5 min to 2 h. Whole cell lysates were blotted with phospho-ERK, phospho-GSK-3βser9 and phospho-AKT antibodies followed by total ERK, AKT and GSK-3βser9 antibodies, respectively. (D) EGFR phosphorylates GSK-3βser9 via the PI3-kinase pathway. UM-SCC-6 cells were pre- incubated for 1 hour with 25 μM of LY294002 or DMSO as vehicle control followed by 100ng/ml EGF stimulation. Akt and GSK-3βser9 activation were evaluated by immunoblot analysis of whole cell lysates with phospho-Akt and phosphor-GSK-3βser9, respectively. The membrane was subsequently blotted with total Akt, GSK-3βser9, and GAPDH antibodies as loading controls.
RhoC is Expressed in HNSCC and is Activated by EGFR Signaling
RhoC has been linked to increased metastatic behavior in a variety of tumor types, but its specific role in cell signaling pathways that lead to tumor progression is poorly understood. Building on the fact that Rho GTPases have been previously shown to activate PI3K/Akt in other cell systems, we hypothesized that perhaps RhoC plays an important role in linking EGFR activation to PI3K activation, leading to E-cadherin loss in HNSCC. To test this hypothesis, we first determined the level of RhoC expression (as well as RhoA expression for comparison) in the same panel of HNSCC cell lines used in our EGFR expression studies. We then assessed RhoC activation status following EGF treatment. Protein expression was confirmed using isoform-specific Rho antibodies. As seen in Figure 3A, primary and malignant kertinocytes express RhoC and RhoA at variable levels, however when compared with the corresponding GAPDH signals that served as loading controls, it is apparent that UM-SCC-6 is one of the strongest expressers of RhoC and one of the weakest expressers of RhoA. The effect of EGF on Rho-GTP was analyzed using a previously described Rho pull-down assay, which only binds the active form of Rho proteins. Interestingly, EGF stimulation increased the level of active RhoA in both primary keratinocytes and UM-SCC-6 cells; however, the active form of RhoC was only increased in UM-SCC-6 cells following EGF stimulation (Figure 3, B and C). These results suggest different roles for RhoA and RhoC in normal and cancer cells.
RhoC Knock-Down Decreases EGF-stimulated Akt and GSK-3βser9 Phosphorylation
Since RhoC was significantly up-regulated by EGF in UM-SCC-6 cells, we next knocked down RhoC expression in the same cells using siRNA gene silencing techniques and then looked at the resultant downstream effects on the PI3K pathway (and the MAPK pathway for comparison). First, we confirmed the specificity of RhoC knockdown by comparing immunoblot expression of RhoC versus RhoA following siRNA transfection. As seen in Figure 4A, following siRNA transfection RhoC expression was significantly reduced to almost undetectable levels whereas RhoA protein expression remained unaffected, confirming the specificity of the siRNAs for RhoC. Next, we determined the effects of RhoC knock-down on EGF-stimulated Akt and GSK-3βser9phosphorylation. Knocking down RhoC decreased EGF-induced phosphorylation of Akt and GSK-3β ser9 by 31% and 34%, respectively (Figure 4B), but had no effect on the phosphorylation of MAPK family member, extracellular-signal-regulated kinase (ERK; Figure 4B), suggesting a greater role for RhoC as a mediator in the EGFR-PI3K signaling cascade as compared to the EGFR-MAPK pathway.
Figure 4.
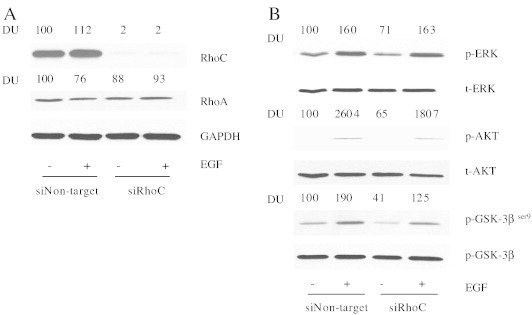
RhoC knock-down decreases EGF-stimulated Akt and GSK-3βser9 phosphorylation. (A) Expression of RhoC in HNSCC cells transfected with siRhoC. Plasmid expressing either control siRNA (siNon-target) or RhoC-specific siRNA (siRhoC) was transfected into UM-SCC-6 cells as described in the Materials and Methods. The cells were then simulated with 100 ng/ml EGF. RhoA and RhoC expression were confirmed by immunoblot analysis with GAPDH serving as a loading control. (B) At 72 hours following transfection with either siNon-target or siRhoC, UM-SCC-6 cells were stimulated with 100 ng/ml EGF or vehicle control for 5 minutes. Whole cell lysates were then blotted with phospho-ERK, phospho-GSK-3βser9 and phospho-AKT antibodies followed by total ERK, AKT and GSK-3βser9 antibodies, respectively.
RhoC knock-down rescues E-cadherin loss and inhibits cell migration and invasion
As described above, RhoC serves as a mediator of the EGFR/PI3k/Akt/GSK-3β ser9 signaling cascade, but what is the biological significance of this newly described role for RhoC in HNSCC? To answer this question we evaluated E-cadherin expression and determined the effects on migration and invasion in RhoC knock-down cells. As expected, E-cadherin expression was reduced in the UM-SCC-6 parent cell line (Figure 5A) and in cells expressing control siRNA (siNon-target) (Figure 5B) six days following EGF stimulation, whereas cells expressing RhoC-specific siRNA (siRhoC) showed no such E-cadherin loss (Figure 5B). Similarly, confocal microscopic analysis revealed that RhoC knock-down cells maintained a greater degree of cell-to-cell contact and E-cadherin membrane localization (Figure 5C) and were significantly less invasive (Figure 6A, P ≤ .05) and motile (Figure 6B, P ≤ .0001) in comparison to their siNon-target counterparts.
Figure 5.
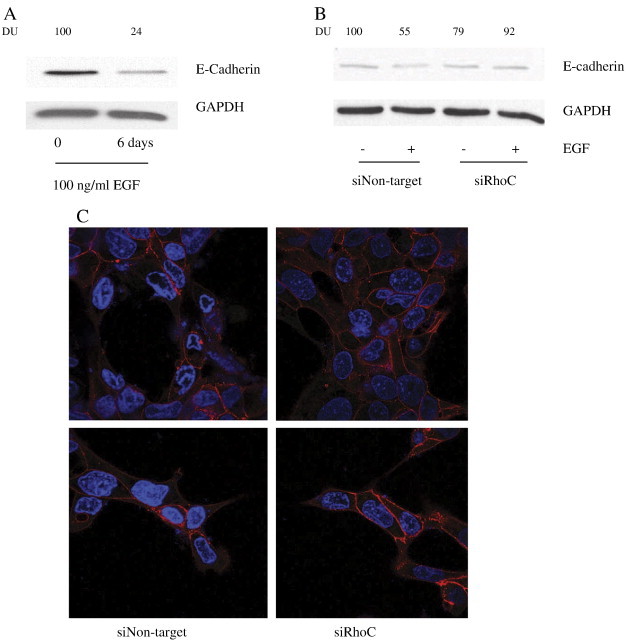
Effects of RhoC knock-down on E-cadherin expression. (A and B) UM-SCC-6 cells were stimulated for 6 days with 100 ng/ ml EGF followed by immunoblot analysis of E-cadherin expression in non-transfected and siRhoC or control-transfected siNon-target UM-SCC-6 cells. GAPDH served as a loading control. (C) Subcellular localizations E-cadherin was examined using immunofluorescence analysis. Of note, knocking down RhoC expression caused reduced E-cadherin membrane localization. Images were obtained at 40 × magnification.
Figure 6.

RhoC knock-down inhibits EGF-stimulated cell migration and invasion. Scratch assay and transwell chamber assay were used to compare the migratory and invasive capabilities, respectively, of siRhoC or control-transfected siNon-target UM-SCC-6 cells. (A) The effect of RhoC knockdown on cell invasion was investigated by comparing the invasion index of siRhoC-transfected cells against siNon-target controls, where siNon-target invasion was set at 100% (⁎P < .05). (B) Knocking down RhoC with siRNA prevented migration of cells to the scratched area as compared to siNon-target control. The width of the scratches was observed and measured using Image J software. The relative distance was calculated as a mean width of the cell scratch. Images were obtained at 4 × magnification.
Discussion
The three Rho GTPases of higher vertebrates, RhoA, B, and C, share 85% amino acid sequence identity [24]. Different Rho proteins are not functionally redundant in the cell but play different roles in cell physiology [25], [26]. Of the three Rho GTPases, RhoC is most convincingly linked with cancer metastasis. Association between RhoC and tumor progression has been described in a variety of tumor types, including breast cancer [27], non-small cell lung carcinoma [28], colon carcinoma [29], malignant melanoma [2], and hepatocellular carcinoma [30]. In HNSCC, RhoC overexpression has been linked to metastatic behavior in HNSCC [10]. The role of RhoA in tumor progression is not so clear, however. Although a number of studies have implicated RhoA in processes leading to transformation and migration in cancer [31], [32], [33], recent reports have shown that although excess RhoA can result in cellular transformation, suppression of RhoA activity is essential for the ability of tumor cells to migrate efficiently [29], [34]. Our findings support this idea in a couple of ways. First, out of the seven SCC cell lines examined, four (UM-SCC-1, 5, and 6) demonstrated higher RhoC expression relative to RhoA expression, whereas HOK cells, despite expressing a somewhat high level of RhoC and RhoA protein overall, expressed both GTPases in relatively equal proportions to one another (Figure 3A). And second, RhoC and RhoA activation status following 100 ng/ml of EGF stimulation differed between primary oral keratinocytes and SCC cells. In UM-SCC-6 cells, both RhoA and RhoC were immediately activated in as little as five minutes following the addition of EGF (Figure 3C), whereas in HOK cells RhoA was activated while RhoC remained unchanged (Figure 3B). These findings suggest that not only do RhoA and RhoC have different expression profiles and serve potentially different purposes in a given cell, but that the two proteins might play different roles in malignant versus non-malignant oral keratinocytes. Further studies will be necessary to more comprehensively characterize these differences.
To our knowledge, this is the first report describing a specific role for RhoC in HNSCC cell signaling that governs transcription of E-cadherin and loss of cell-to-cell contact leading to tumor cell migration and invasion. Despite an increasing number of reports describing the relationship between RhoC overexpression and prognostic outcomes across multiple tumor types, few studies have explored the consequences and regulation of RhoC activation in cancer and even fewer have explored the specific role it plays in signaling pathways known to be important in HNSCC, like those associated with EGFR and PI3K activation. For example, Bellovin et al., 2006, identified RhoC as a prognostic marker of colon carcinoma where they showed up-regulation of RhoC expression following epithelial to mesenchymal transition, a process by which epithelial cells acquire mesenchymal features that promote tumor progression. They went on to show that RhoC expression and activation correlated with a loss of E-cadherin expression and enhanced cell migration. Despite these compelling findings, RhoC expression and activation were not shown to be directly linked to E-cadherin loss, making it difficult to rule out the involvement of parallel pathways. In the current study, we demonstrate that knock-down of RhoC results in a return of E-cadherin expression (Figure 5B) and a reduction of in vitro invasion and migration (Figure 6, A and B, respectively), supporting a direct upstream role for RhoC in regulating E-cadherin expression.
Down-regulation of E-cadherin promotes tumor cell migration and invasion by dismantling the cell-to-cell adhesion complex, resulting in cell polarity loss and enhanced motility. Loss of E-cadherin as a tumor progresses can occur as a result of both genetic and epigenetic mechanisms; however cell signaling dysregulation serves as the primary mechanism by which E-cadherin trafficking and transcription occur. Specifically, initiation of cell signaling cascades leading to increased gene expression of transcription factors that bind the E-cadherin promoter, like the Snail superfamily, serves as one of the most well documented mechanisms driving down-regulation of E-cadherin. Consistent with these observations, we show that as Snail protein levels rise (Figure 2B) following EGF stimulation, E-cadherin protein levels and membrane localization are substantially reduced (Figure 5, A and C, respectively).
Although our data suggests transcriptional regulation of E-cadherin in HNSCC, it is important to consider that control of E-cadherin activity could also stem from posttranslational modifications, including phosphorylation and ubiquitination of E-cadherin leading to degradation [35], [36]. For example, receptor tyrosine kinases, like EGFR and fibroblast growth-factor receptor (FGFR), can directly induce phosphorylation of E-cadherin, leading to disassembly of the adhesion complex and disruption of the cadherin-mediated cell-to-cell junctions [37]. Additionally, β-catenin plays a critical structural role in cadherin-based adhesions and is involved in direct transcriptional activation of the Slug gene, which encodes a transcriptional repressor of E-cadherin [38]. Recent studies in colorectal cancer have demonstrated that Slug can be regulated by PI3K/Akt/GSK-3β-mediated activation of β-catenin, leading to its nuclear localization and subsequent transcriptional up-regulation of Slug [39]. Moreover, studies have shown that EGFR interacts with the cadherin–catenin complex, and upon ligand binding induces tyrosine phosphorylation of β-catenin and disruption of cadherin-catenin binding [40]. Next steps in our laboratory will begin to decipher the impact these important competing pathways have on E-cadherin loss in HNSCC, and the potential role RhoC plays in mediating these signaling cascades.
Accumulating evidence indicates that the EGFR family and its downstream signaling pathways, such as PI3K-Akt and MAPK pathways, up-regulate Snail leading to underexpression of E-cadherin [41], [42]. Similarly, our results in HNSCC show clear activation of PI3K/Akt/GSK-3β and ERK following EGFR phosphorylation (Figure 2C). Activation of PI3K-Akt and subsequent phosphorylation of GSK-3β is significant here as a wealth of literature exists, across multiple tumor types, confirming the importance of GSK-3β phosphorylation in stabilizing Snail so that it can translocate to nucleus and participate in transcriptional regulation. However, a variety of signaling cascades, in addition to the PI3K-Akt pathway, have been shown to phosphorylate GSK-3β [43]. To rule out the possibility of a parallel pathway bypassing the EGFR/PI3K/AKT cascade, we employed a well-known PI3K inhibitor, Ly294002, and looked at the effects it had on GSK-3βser9 phosphorylation. As shown in Figure 2D, GSK-3βser9 phosphorylation was reduced upon PI3K-Akt inhibition, supporting our hypothesis that PI3K-Akt plays a significant role in GSK-3βser9 regulation in these cells. Furthermore, in RhoC knock-down experiments, Akt and GSK-3β phosphorylation was abrogated whereas ERK phosphorylation was not (Figure 4B), suggesting a mediating role for RhoC in EGFR-PI3K signaling but not in the EGFR-MAPK pathway.
In our study, we observed a dose-dependent effect of EGF on EGFR phosphorylation at Tyr992. In HNSCC cells, EGFRTyr992 phosphorylation was seen at EGF concentrations as low as 1.0 ng/ml. In contrast, in HOK cells EGFRTyr992 phosphorylation was not seen until EGF concentrations exceeded 10 mg/ml. These results are in agreement with reports that EGF is a multifunctional cytokine that elicits different and even opposite cellular responses between cell types, and can even have different effects within the same cell type, depending on concentration [26], [44], [45]. Based on these findings, one might speculate that perhaps these differences in EGFR responsiveness according to EGF concentration might help us to understand how EGF availability in the oral cavity contributes to tumorigenesis and metastasis in HNSCC. For example, the physiologic concentration of EGF in whole saliva has been shown to be around 1 ng/ml and this concentration has an ability to promote oral epithelial cell chemotaxis [46], [47]. Given that we observed significantly different responses to lower concentrations of EGF in HNSCC versus HOK, perhaps acquisition of EGF responsiveness might be an important step in HNSCC tumor development.
Conclusions
In this study we provide important insights into the molecular mechanisms mediating the EGF-induced down-regulation of E-cadherin leading to increased cell invasion in HNSCC. Ours is the first report demonstrating that activation of RhoC signaling, following EGFR phosphorylation, likely plays a significant role in down-regulation of cell surface E-cadherin in HNSCC. Specifically, activated RhoC, acting via PI3K signaling, stabilizes Snail and down-regulates E-cadherin, which contributes to EGF-induced cell invasion (Figure 7). Our data, taken together with an emergence of literature elucidating the role of Rho family members in tumor metastasis, argue that RhoC is a promising target in the development of novel strategies aimed at halting head and neck cancer progression.
Figure 7.
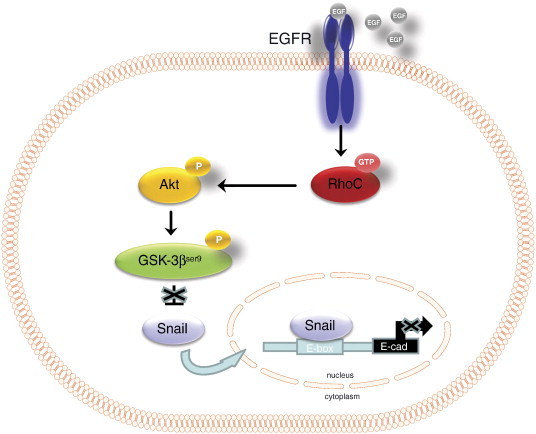
Proposed model for RhoC-mediated EGFR signaling in the regulation of invasion in HNSCC.
Acknowledgements
We would like to thank Dr. Thomas Carey for the provision of UM-SCC cell lines and the Western University of Health Sciences College of Osteopathic Medicine of the Pacific and College Dental Medicine for providing financial support for the project.
References
- 1.Howlader N., N. A., Krapcho M., Garshell J., Miller D., Altekruse S.F., Kosary C.L., Yu M., Ruhl J., Tatalovich Z. National Cancer Institute; 2014. SEER Cancer Statistics Review, 1975–2011. [Google Scholar]
- 2.Clark E.A., Golub T.R., Lander E.S., Hynes R.O. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 3.van Golen K.L., Wu Z.F., Qiao X.T., Bao L.W., Merajver S.D. RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res. 2000;60:5832–5838. [PubMed] [Google Scholar]
- 4.Suwa H., Ohshio G., Imamura T., Watanabe G., Arii S., Imamura M., Narumiya S., Hiai H., Fukumoto M. Overexpression of the rhoC gene correlates with progression of ductal adenocarcinoma of the pancreas. Br J Cancer. 1998;77:147–152. doi: 10.1038/bjc.1998.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khosravi-Far R., Solski P.A., Clark G.J., Kinch M.S., Der C.J. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol Cell Biol. 1995;15:6443–6453. doi: 10.1128/mcb.15.11.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michiels F., Habets G.G., Stam J.C., van der Kammen R.A., Collard J.G. A role for Rac in Tiam1-induced membrane ruffling and invasion. Nature. 1995;375:338–340. doi: 10.1038/375338a0. [DOI] [PubMed] [Google Scholar]
- 7.Habets G.G., Scholtes E.H., Zuydgeest D., van der Kammen R.A., Stam J.C., Berns A., Collard J.G. Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell. 1994;77:537–549. doi: 10.1016/0092-8674(94)90216-x. [DOI] [PubMed] [Google Scholar]
- 8.Keely P.J., Westwick J.K., Whitehead I.P., Der C.J., Parise L.V. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- 9.Hynes R.O., Zhao Q. The evolution of cell adhesion. J Cell Biol. 2000;150:F89–F96. doi: 10.1083/jcb.150.2.f89. [DOI] [PubMed] [Google Scholar]
- 10.Islam M., Lin G., Brenner J.C., Pan Q., Merajver S.D., Hou Y., Kumar P., Teknos T.N. RhoC expression and head and neck cancer metastasis. Mol Cancer Res. 2009;7:1771–1780. doi: 10.1158/1541-7786.MCR-08-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Islam M., Sharma S., Teknos T.N. RhoC regulates cancer stem cells in head and neck squamous cell carcinoma by overexpressing IL-6 and phosphorylation of STAT3. PLoS One. 2014;9:e88527. doi: 10.1371/journal.pone.0088527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicholson R.I., Gee J.M., Harper M.E. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl. 4):S9–S15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 13.Ang K.K., Berkey B.A., Tu X., Zhang H.Z., Katz R., Hammond E.H., Fu K.K., Milas L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62:7350–7356. [PubMed] [Google Scholar]
- 14.Bussink J., van der Kogel A.J., Kaanders J.H. Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer. Lancet Oncol. 2008;9:288–296. doi: 10.1016/S1470-2045(08)70073-1. [DOI] [PubMed] [Google Scholar]
- 15.Matta A., Ralhan R. Overview of current and future biologically based targeted therapies in head and neck squamous cell carcinoma. Head Neck Oncol. 2009;1:6. doi: 10.1186/1758-3284-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng J.C., Chang H.M., Leung P.C. Epidermal growth factor induces human oviductal epithelial cell invasion by down-regulating E-cadherin expression. J Clin Endocrinol Metab. 2012;97:E1380–E1389. doi: 10.1210/jc.2011-2751. [DOI] [PubMed] [Google Scholar]
- 17.Forastiere A.A., Burtness B.A. Epidermal growth factor receptor inhibition in head and neck cancer–more insights, but more questions. J Clin Oncol. 2007;25:2152–2155. doi: 10.1200/JCO.2007.10.9017. [DOI] [PubMed] [Google Scholar]
- 18.Lo H.W., Xia W., Wei Y., Ali-Seyed M., Huang S.F., Hung M.C. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res. 2005;65:338–348. [PubMed] [Google Scholar]
- 19.Sharafinski M.E., Ferris R.L., Ferrone S., Grandis J.R. Epidermal growth factor receptor targeted therapy of squamous cell carcinoma of the head and neck. Head neck. 2010;32:1412–1421. doi: 10.1002/hed.21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emlet D.R., Moscatello D.K., Ludlow L.B., Wong A.J. Subsets of epidermal growth factor receptors during activation and endocytosis. J Biol Chem. 1997;272:4079–4086. doi: 10.1074/jbc.272.7.4079. [DOI] [PubMed] [Google Scholar]
- 21.Holbrook M.R., O'Donnell J.B., Jr., Slakey L.L., Gross D.J. Epidermal growth factor receptor internalization rate is regulated by negative charges near the SH2 binding site Tyr992. Biochemistry. 1999;38:9348–9356. doi: 10.1021/bi990195r. [DOI] [PubMed] [Google Scholar]
- 22.Guo L., Kozlosky C.J., Ericsson L.H., Daniel T.O., Cerretti D.P., Johnson R.S. Studies of ligand-induced site-specific phosphorylation of epidermal growth factor receptor. J Am Soc Mass Spectrom. 2003;14:1022–1031. doi: 10.1016/S1044-0305(03)00206-X. [DOI] [PubMed] [Google Scholar]
- 23.Xiao D., He J. Epithelial mesenchymal transition and lung cancer. J Thorac Dis. 2010;2:154–159. doi: 10.3978/j.issn.2072-1439.2010.02.03.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wheeler A.P., Ridley A.J. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res. 2004;301:43–49. doi: 10.1016/j.yexcr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 25.Jaffe A.B., Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 26.Han J., Li L., Hu J., Yu L., Zheng Y., Guo J., Zheng X., Yi P., Zhou Y. Epidermal growth factor stimulates human trophoblast cell migration through Rho A and Rho C activation. Endocrinology. 2010;151:1732–1742. doi: 10.1210/en.2009-0845. [DOI] [PubMed] [Google Scholar]
- 27.van Golen K.L., Davies S., Wu Z.F., Wang Y., Bucana C.D., Root H., Chandrasekharappa S., Strawderman M., Ethier S.P., Merajver S.D. A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin Cancer Res. 1999;5:2511–2519. [PubMed] [Google Scholar]
- 28.Shikada Y., Yoshino I., Okamoto T., Fukuyama S., Kameyama T., Maehara Y. Higher expression of RhoC is related to invasiveness in non-small cell lung carcinoma. Clin Cancer Res. 2003;9:5282–5286. [PubMed] [Google Scholar]
- 29.Bellovin D.I., Simpson K.J., Danilov T., Maynard E., Rimm D.L., Oettgen P., Mercurio A.M. Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene. 2006;25:6959–6967. doi: 10.1038/sj.onc.1209682. [DOI] [PubMed] [Google Scholar]
- 30.Hakem A., Sanchez-Sweatman O., You-Ten A., Duncan G., Wakeham A., Khokha R., Mak T.W. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005;19:1974–1979. doi: 10.1101/gad.1310805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B., Antonyak M.A., Zhang J., Cerione R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene. 2012;31:4740–4749. doi: 10.1038/onc.2011.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giang Ho T.T., Stultiens A., Dubail J., Lapiere C.M., Nusgens B.V., Colige A.C., Deroanne C.F. RhoGDIalpha-dependent balance between RhoA and RhoC is a key regulator of cancer cell tumorigenesis. Mol Biol Cell. 2011;22:3263–3275. doi: 10.1091/mbc.E11-01-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y.S., Huang W.L., Chang S.H., Chang K.W., Kao S.Y., Lo J.F., Su P.F. Enhanced filopodium formation and stem-like phenotypes in a novel metastatic head and neck cancer cell model. Oncol Rep. 2013;30:2829–2837. doi: 10.3892/or.2013.2772. [DOI] [PubMed] [Google Scholar]
- 34.Bellovin D.I., Bates R.C., Muzikansky A., Rimm D.L., Mercurio A.M. Altered localization of p120 catenin during epithelial to mesenchymal transition of colon carcinoma is prognostic for aggressive disease. Cancer Res. 2005;65:10938–10945. doi: 10.1158/0008-5472.CAN-05-1947. [DOI] [PubMed] [Google Scholar]
- 35.Swaminathan G., Cartwright C.A. Rack1 promotes epithelial cell-cell adhesion by regulating E-cadherin endocytosis. Oncogene. 2012;31:376–389. doi: 10.1038/onc.2011.242. [DOI] [PubMed] [Google Scholar]
- 36.Delva E., Kowalczyk A.P. Regulation of cadherin trafficking. Traffic. 2009;10:259–267. doi: 10.1111/j.1600-0854.2008.00862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cavallaro U., Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- 38.Conacci-Sorrell M., Simcha I., Ben-Yedidia T., Blechman J., Savagner P., Ben-Ze'ev A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J Cell Biol. 2003;163:847–857. doi: 10.1083/jcb.200308162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suman S., Kurisetty V., Das T.P., Vadodkar A., Ramos G., Lakshmanaswamy R., Damodaran C. Activation of AKT signaling promotes epithelial-mesenchymal transition and tumor growth in colorectal cancer cells. Mol Carcinog. 2014;53(Suppl. 1):E151–E160. doi: 10.1002/mc.22076. [DOI] [PubMed] [Google Scholar]
- 40.Hoschuetzky H., Aberle H., Kemler R. Beta-catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J Cell Biol. 1994;127:1375–1380. doi: 10.1083/jcb.127.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou B.P., Deng J., Xia W., Xu J., Li Y.M., Gunduz M., Hung M.C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 42.Grille S.J., Bellacosa A., Upson J., Klein-Szanto A.J., van Roy F., Lee-Kwon W., Donowitz M., Tsichlis P.N., Larue L. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 2003;63:2172–2178. [PubMed] [Google Scholar]
- 43.Zheng H., Li W., Wang Y., Liu Z., Cai Y., Xie T., Shi M., Wang Z., Jiang B. Glycogen synthase kinase-3 beta regulates Snail and beta-catenin expression during Fas-induced epithelial-mesenchymal transition in gastrointestinal cancer. Eur J Cancer. 2013;49:2734–2746. doi: 10.1016/j.ejca.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 44.Staun-Ram E., Goldman S., Gabarin D., Shalev E. Expression and importance of matrix metalloproteinase 2 and 9 (MMP-2 and − 9) in human trophoblast invasion. Reprod Biol Endocrinol. 2004;2:59. doi: 10.1186/1477-7827-2-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maheshwari G., Wells A., Griffith L.G., Lauffenburger D.A. Biophysical integration of effects of epidermal growth factor and fibronectin on fibroblast migration. Biophys J. 1999;76:2814–2823. doi: 10.1016/S0006-3495(99)77435-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Royce L.S., Baum B.J. Physiologic levels of salivary epidermal growth factor stimulate migration of an oral epithelial cell line. Biochim Biophys Acta. 1991;1092:401–403. doi: 10.1016/s0167-4889(97)90019-7. [DOI] [PubMed] [Google Scholar]
- 47.Ohshima M., Sato M., Ishikawa M., Maeno M., Otsuka K. Physiologic levels of epidermal growth factor in saliva stimulate cell migration of an oral epithelial cell line, HO-1-N-1. Eur J Oral Sci. 2002;110:130–136. doi: 10.1034/j.1600-0722.2002.11179.x. [DOI] [PubMed] [Google Scholar]