

Abstract
Drug combinations are common in cancer treatment and are rapidly evolving, moving beyond chemotherapy combinations to combinations of signal transduction inhibitors. For the delivery of drug combinations, i.e., multi-drug delivery, major considerations are synergy, dose regimen (concurrent versus sequential), pharmacokinetics, toxicity, and safety. In this contribution, we review recent research on polymeric micelles for multi-drug delivery in cancer. In concurrent drug delivery, polymeric micelles deliver multi-poorly water-soluble anticancer agents, satisfying strict requirements in solubility, stability, and safety. In sequential drug delivery, polymeric micelles participate in pretreatment strategies that “prime” solid tumors and enhance the penetration of secondarily administered anticancer agent or nanocarrier. The improved delivery of multiple poorly water-soluble anticancer agents by polymeric micelles via concurrent or sequential regimens offers novel and interesting strategies for drug combinations in cancer treatment.
KEY WORDS: controlled release, drug combination, drug delivery, drug solubilization, polymeric micelles
INTRODUCTION
Tumor heterogeneity is increasingly becoming a major consideration in drug delivery, particularly the delivery of drug combinations that target hallmarks of cancer. Cancer is one of the most complex diseases, involving extensive phenotypic and genetic variation (1). Solid tumors are architecturally and micro-environmentally diverse due to the fact that distribution of vasculature, proliferation profile/rate, and the expression site/rate of biomarkers are some of the variables (1–5). Host variability (sex, age, hormonal status, and genetics), origin/types of cancer, and stages of cancer development introduce intertumoral heterogeneity. Solid tumors are also intratumorally heterogeneous, potentially caused by genetic alteration and instability. Burrell et al. emphasized the significance of genetic variation and instability as a major cause of intratumor heterogeneity in cancer evolution (1). Within a solid tumor, genetic variation causes subclonal populations of cells, outgrowth of specific clones that have a phenotypic advantage over other clones, and amplification of a population over neighboring tissues and organs. Not surprisingly, consequences of tumor heterogeneity appear to be drug resistance, poor drug delivery, and failure of cancer treatment.
The primary goals of drug combination in anticancer treatment are to overcome tumor heterogeneity, reduce chemoresistance and achieve additive or more desirable synergistic anticancer efficacy without overlapping toxicity. In 1965, Frei et al. proved that a combination therapy consisting of two cytotoxic agents was therapeutically more effective than monotherapy in children with acute leukemia: combination treatments of methotrexate/6-mercaptopurine, prednisone/6-mercaptopurine, and prednisone/vincristine resulted in complete remission for 45%, 82%, and 84% in children with acute leukemia, respectively, whereas monotherapy of methotrexate, 6-mercaptopurine, vincristine, and prednisone, individually, resulted in 21%, 27%, 47%, and 57% of complete remission, respectively (6,7).
Since then, clinically effective drug combinations, that would improve the efficacy of oncology treatment, have been widely researched. One of the most effective chemotherapy regimens for adult Hodgkin’s lymphoma is ABVD: adriamycin, bleomycin, vinblastine, and dacarbazine (8,9). A combination of a platinum-derivative (cisplatin (CDDP) or carboplatin) and a taxane (paclitaxel (PTX) or docetaxel (DTX)) is regularly used to treat non-small cell lung (NSCLC) and ovarian cancers (10–12). A combination of doxorubicin (DOX), taxane, and platinum-derivative is recognized as one of the most effective treatment options in metastatic breast cancer (13).
In 2000, Weinberg et al. simplified and highlighted hallmarks of cancer comprising six biological capabilities: Self-sufficiency in growth signals, insensitivity to antigrowth signals, tissue invasion and metastasis, limitless replicative potential, sustained angiogenesis, and evading apoptosis (3). In 2011, Weinberg et al. added two more hallmarks, deregulating cellular energetics and evading immune destruction, to encompass pathogenesis of cancer (2,4). Thus, continuing interests in complex biology and architecture of cancer, elaborate genetic alterations, and complex molecular mechanisms in cancer evolution increasingly provide compelling hypotheses for drug combination cancer treatment.
Recent drug combinations have distinctive “targeted” mechanisms of actions, intervene in multiple biological pathways, prevent cross-talk between different membrane receptors, overcome multi-drug resistance (MDR), and minimize overlapping toxicity. Addition of a P-glycoprotein (P-gp) inhibitor to conventional chemotherapeutic drugs was a popular strategy to overcome MDR, suppressing drug efflux from cancer cells and increasing anticancer sensitivity. However, it was accompanied by unacceptable toxicity due to a lack of P-gp tumor-specific inhibition. Recently, chemotherapy and signal transduction inhibitors (“targeted drugs”) have been combined, aiming for enhanced anticancer efficacy, lower drug resistance, and reduced side-effects relative to highly dosed chemotherapy (14).
Delivery of drug combinations, i.e., multi-drug delivery, is challenging, and major considerations include synergism, optimum dose regimen (concurrent versus sequential), pharmacokinetics (PK), multi-drug toxicity, and safety, e.g., drug precipitation and vehicle toxicity.
Many of the clinically used cytotoxic and chemotherapeutic agents, such as PTX, DTX, and etoposide (ETO), have bulky polycyclic structures and are poorly water-soluble (15,16). Many signal transduction inhibitors are also poorly water-soluble. Common techniques for solubility enhancement are physical modification (e.g., particle size reduction, complexation, and solid dispersion), chemical modification (pH adjustment, salt formation, and prodrugs), and addition of excipient (co-solvents and/or surfactants and nanoparticle) (16,17). A typical vehicle for poorly water-soluble chemotherapeutic drugs is commonly a mixture of organic solvent and non-ionic surfactant, such as Cremophor EL and Tween 80. For example, Taxol® (Bristol-Myers Squibb) contains PTX dissolved in a 1:1 v/v mixture of Cremophor EL and anhydrous ethanol and is diluted with 5% dextrose prior to use. Taxotere® (Sanofi-Aventis) constitutes DTX mixed with Tween 80 and anhydrous ethanol (1:1 v/v). Although mixtures of organic solvent and non-ionic surfactant have been successfully used for poorly water-soluble chemotherapeutics, several issues have arisen concerning toxicity: Cremophor EL in Taxol® triggers severe hypersensitivity reactions in >30% patients (18). Cremophor EL interacts with the plastic components of polyvinyl chloride bags and infusion lines and causes toxicity (16). Similarly, Tween 80 induces anaphylaxis, dyspnea, and hypotension in patients (19,20).
In summary, drug combination anticancer treatments continue to evolve, raising high hopes for unprecedented antitumor responses and reduced toxicity. Moving forward, it is likely that more and more interesting anticancer drug combinations will be tested in preclinical models and humans. Current drug delivery technology is adequate for many of these poorly water-soluble anticancer agents; caveats being that toxicity associated drug vehicles is problematic, especially in drug combination, and bioavailability is often low and erratic. Current drug delivery technology is also not meant for drug targeting. Alternatively, drug vehicles such as polymeric micelles permit the delivery of multiple water-insoluble anticancer agents and may replace the toxic formulations of multiple drugs with a single, safe vehicle.
There are six major requirements to consider when determining the successful vehicle development for multi-drug delivery of anticancer drugs. These requirements are as follows: (1) Vehicles carrying multiple drugs maintain long-term physical and chemical stability of drugs throughout manufacturing, storage, and administration. (2) After being administered, vehicles must be inert, biocompatible, biodegradable, and non-toxic. (3) Water solubility of highly hydrophobic drugs is easily enhanced by the addition of or incorporation into vehicles in injectable aqueous solution. (4) Incorporated multiple drugs are released from designed vehicles in a rapid, slow, concurrent, or sequential pattern, depending on a balance between toxicity and efficacy. (5) Drug vehicles carry payloads preferentially to solid tumors without off-target effects. (6) Vehicles are simple in design and have a facile manufacturing process.
Liposomes, polymer-drug conjugates, nanoparticles, dendrimers, and polymeric micelles are being studied for multi-drug delivery (21). Liposomes were the first dual-drug-loaded vehicle for combination of chemotherapy (22–24). Based on the concept of “ratiometric drug dosing,” liposomes containing two hydrophilic drugs, cytarabine/daunorubicin or irinotecan/fluoxuridine, have entered phase II/III clinical trials (25,26). Duncan et al. first proposed dual-drug delivery of poly(N-hydroxypropyl methacrylamide) (pHPMA)-drug conjugates, synthesizing a pHPMA conjugate containing DOX and aminoglutethimide, an aromatase inhibitor (27). Sengupta et al.. devised “nanocells” that have a poly(lactide-co-glycolide) (PLGA) core and a lipid coating for distinct dual drug release patterns (28). Tekade et al. co-loaded hydrophobic methotrexate and hydrophilic all-trans retinoic acid in a fourth-generation (G4) polyamidoamine-based dendrimer and achieved concurrent in vitro release of payloads (29). Prior review articles have covered concepts in multi-drug delivery, advantages and disadvantages of drug vehicles, and the enhanced permeability and retention (EPR) effect for solid tumors (21,22,24). In this review article, we focus on polymeric micelles and micelle-forming polymer-drug conjugates for concurrent or sequential multi-drug delivery of poorly water-soluble anticancer agents and discuss opportunities and challenges for the development of successful polymer-based multi-drug vehicles for treatment of cancer.
POLYMERIC MICELLES FOR DRUG DELIVERY
Polymeric micelles are spherically shaped nanoparticles composed of amphiphilic block copolymers (ABCs) consisting of hydrophobic and hydrophilic blocks in aqueous medium (Fig. 1) (30–35). Amphiphilic di- or tri-block copolymers spontaneously self-assemble into a supramolecular core/shell structure above the critical micelle concentration (CMC) driven primarily by hydrophobic interaction. A hydrophobic core acts as a reservoir for solubilization of poorly water-soluble drugs, whereas a hydrophilic shell interfaces the aqueous milieu. The most widely used hydrophilic block is poly(ethylene glycol) (PEG). A dense brush of PEG ensures micelle solubility in an aqueous milieu. There is versatility in selecting hydrophobic blocks, such as poly(ester)s and poly(l-amino acid)s, and this flexibility provides versatile and predictive properties of polymeric micelles for drug delivery. For example, poly(ε-caprolactone) (PCL) is a semicrystalline polymer with a melting temperature (Tm) of 55°C, whereas poly(d,l-lactic acid) (PLA) is amorphous with a glass transition temperature (Tg) of 50°C (36). Thus, poly(ethylene glycol)-block-poly(ε-caprolactone) (PEG-b-PCL) micelles have a “solid-like” and lower core polarity, resulting in drug solubilization for non-polar anticancer agents and slower drug release in comparison to poly(ethylene glycol)-block-poly(d,l-lactic acid) (PEG-b-PLA) micelles.
Fig. 1.
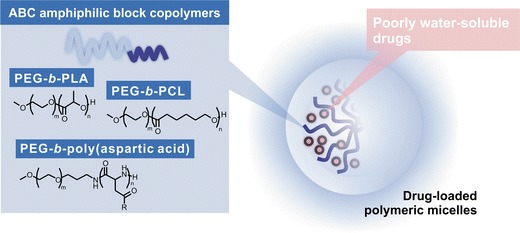
Polymeric micelles for drug delivery
The rationale of polymeric micelles for drug delivery is based on their ability to increase the aqueous solubility of many hydrophobic anticancer agents that possess poor water solubility and safely permit concurrent or sequential intravenous (IV) infusion of anticancer agents. Intermolecular interaction between drug and core-forming block drives drug solubilization, and a sense of drug-polymer compatibility may be gained by the calculation of drug and core-forming block solubility parameters (37). Physical and chemical methods for the loading of drugs in polymeric micelles have been described (38). It is noted that many drug candidates have poor aqueous solubility, and for preclinical studies, drug levels >1.0 mg/mL are required for toxicity evaluation in rodents. Thus, DMSO and surfactant/cosolvent are common in preclinical studies on anticancer agents. In clinical trials, poor aqueous solubility is problematic; bioavailability is often insufficient for therapeutic efficacy (39).
Key physicochemical considerations of polymeric micelles for drug delivery include nanoscopic dimensions, drug-loading capacity, release kinetics, and physical stability against drug precipitation. Physicochemical properties of polymeric micelles may be manipulating by adjusting the mass ratio of hydrophilic and hydrophobic blocks, chemical nature of the hydrophobic block, e.g., side chain modification, and reversible crosslinking (34,40).
Several polymeric micelles are being evaluated in clinical trials for cancer (41). For example, a phase II clinical trial on Genexol-PM, a Cremophor EL-free PEG-b-PLA polymeric micelle for PTX, showed favorable efficacy in patients with metastatic breast cancer (45.3–72.3% response rate), compared with the conventional Cremophor EL-based PTX (21–54% response rate) (42). Genexol-PM is approved in Asia and will enter a phase III bioequivalence clinical trial versus Abraxane® in the USA. A phase I clinical trial on NC-6004, a PEG-b-poly(glutamate) micelle for CDDP, showed that NC-6004 did not induce significant nephrotoxicity at a high dose of 120 mg/m2, noting that nephron-toxicity is commonly caused by a medium-high dose of CDDP (41,43). NC-6004 is under evaluation in phase III clinical trials. A phase II clinical trial on NK-105, a PEG-b-poly(aspartate) micelle for PTX exhibited high antitumor efficacy in patients with advanced stomach cancer and high tolerability, justifying entry into phase III clinical trials. Lastly, NC-6300, a PEG-b-poly(aspartate hydrazide) (PEG-b-p(Asp-Hyd)) with pendant epirubicin, has entered phase I clinical trials in Japan (43). NC-6300 is unique in that epiribicin is covalently bound onto PEG-b-p(Asp-Hyd) by a pH-sensitive hydrazine linkage, enabling stability in blood, but drug release in the interior of acidic endosomes and lysosomes of cancer cells.
CONCURRENT DRUG DELIVERY
Poorly water-soluble anticancer agents can be loaded chemically or physically into polymeric micelles for concurrent multi-drug delivery (Fig. 2). Clinically, many poorly water-soluble anticancer agents are infused sequentially or are orally administered and separately infused. Concurrent delivery by polymeric micelles simplifies the task of multi-drug delivery, improves safety, and may permit anticancer agents to act at solid tumors at the same time, aiming for synergistic drug interaction (44–47).
Fig. 2.
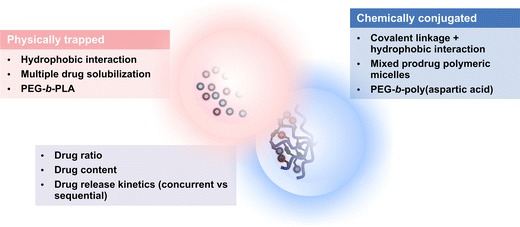
Multi-drug polymeric micelles for drug delivery
For chemical conjugation, a reversible covalent linkage is required between an anticancer agent and a block copolymer, such as PEG-b-p(Asp-Hyd). In this prodrug strategy, anticancer agents are coupled onto a block copolymer and assembled into multi-drug polymeric micelles, or alternatively, anticancer agents are coupled onto separate block copolymers and assembled into mixed polymeric micelles (48,49). The latter approach is favored due to the ease of characterization of a single drug-block copolymer conjugate, e.g., 1H NMR spectroscopy of drug content, versus a two drug-block copolymer conjugate, which may have overlapping peaks in its NMR spectrum.
Drug release from multi-prodrug polymeric micelles may occur by simple hydrolysis or stimuli- /environment-dependent release and may be tuned by choice of chemical linkage, e.g., esters, and spacer groups (50,51). Ideally, multi-drug prodrug polymeric micelles will circulate for prolonged periods in blood to facilitate passive tumor targeting by the EPR effect; enter cancer cells by endocytosis; and release drug. Multi-drug release may be triggered by an acidic pH and/or lysosomal enzymes and be concurrent or sequential, aiming for synergistic activity.
Concurrent multi-drug delivery may also be realized by the physical loading of an anticancer agent in a polymeric micelle in step one and of a second anticancer agent in a second polymeric micelle in step two, followed by mixing and concurrent drug delivery. Alternatively, multi-drug-loaded polymeric micelles are feasible (46). This approach has the advantage of being a one-step drug-loading process for two or three drugs. In both cases, chemical modification of drugs is not a requirement for drug loading.
Multi-drug polymeric micelles may release physically loaded drug(s) by disassembly of polymeric micelles in blood and/or by diffusion. Drug release tends to be rapid in vivo, and in vitro results must be interpreted with caution due to poor in vitro/in vivo correlation. Thus, while EPR targeting of solid tumors is feasible with physically loaded drug, e.g., PTX in NK-105, it is regarded as challenging, and research on multi-drug polymeric micelles for tumor targeting deserves more attention.
Bae et al. synthesized PEG-b-p(Asp-Hyd)s with pendant DOX (anthracycline antibiotic) and wortmannin (WOR, PI3K inhibitor) and formed multi-drug polymeric micelles (Fig. 3, Table I) (48). By adjusting the initial feed ratio, drug ratios, i.e., % WOR on PEG-b-p(Asp-Hyd), were 0, 20, 51, 79, and 100%. Drug ratios of PEG-b-p(Asp-Hyd) micelles were also adjusted by separate attachment of each drug on PEG-b-p(Asp-Hyd), and assembly into mixed prodrug polymeric micelles. At 50% WOR, PEG-b-p(Asp-Hyd) with pendant DOX and WOR formed polymeric micelles with a diameter of ca. 90 nm, whereas mixed polymeric micelles with PEG-b-p(Asp-Hyd) with pendent DOX and PEG-b-p(Asp-Hyd) with pendent WR were larger, ca. 220 nm. While a size difference was observed, equivalent cytotoxicity against MCF-7 breast cancer cells was demonstrated in vitro, suggesting a similar mechanism of action.
Fig. 3.

Synthetic scheme for PEG-b-p(Asp-Hyd) with pendant DOX and WOR (Bae et al., 48). Reprinted from (48) Copyright © 2007, with permission from Elsevier B.V.
Table I.
Multi-Drug Polymeric Micelles for Concurrent Drug Delivery
| Polymer | Drugs | Indication | Status | Reference |
|---|---|---|---|---|
| PEG-b-p(Asp-Hyd)a | Doxorubicin/wortmannin | MCF-7 breast cancer | In vitro | (48) |
| PEG-b-p(Asp-Hyd)a | Doxorubicin/17-hydroxethylamino-17-demethoxygeldanamycin | MCF-7 breast cancer | In vitro | (49) |
| PEG-b-PLA | Paclitaxel/17-AAG/rapamycin | A549 NSCLC and MDA-MB-231 breast cancers | In vivo | (46) |
| PEG-b-p(γ-benzyl l-glutamate) + PEG-b-p(l-lactide) | Doxorubicin/etoposide, Doxorubicin/paclitaxel | CT-26 murine colorectal cancer | In vivo | (52) |
| PEG-b-PLGA | Doxorubicin/paclitaxel | A549 NSCLC, B16 mouse melanoma, and HepG2 liver cancers | In vitro | (53) |
| Poly(2-methyl-2-oxazoline)-b-poly(2-butyl-2-oxazoline)-b-poly(2-methyl-2-oxazoline) | Paclitaxel/17-AAG/etoposide, Paclitaxel/17-AAG/bortezomib | MCF-7 and MDA-MB-231 breast, PC3 prostate, and HepG2 liver cancers | In vitro | (54) |
| PEG-b-poly(carbonate-co-lactic acid) | Cyclopamine/gefitinib | L3.6pl and MIA PaCa-2 pancreatic cancers | In vivo | (55) |
| PEG-b-PCL | Paclitaxel/cyclopamine/gossypol | ES-2-luc and SKOV-3-luc ovarian cancers | In vivo | (44) |
| PEG-DSPE/TPGS | Paclitaxel/17-AAG | SKOV-3 ovarian cancer | In vivo | (56) |
| PEG-b-poly-(glutamic acid)-b-poly(phenylalanine) | Paclitaxel/cisplatin | A2780 ovarian cancer | In vivo | (57) |
| PLGA-b-PEG-b-PLGA | Paclitaxel/17-AAG/rapamycin | ES-2-luc ovarian cancer | In vivo | (58) |
aChemical loading (conjugation) of drugs
Bae et al. also synthesized PEG-b-p(Asp-Hyd) with pendant DOX and PEG-b-p(Asp-Hyd) with pendant 17-hydroethylamino-17-demethoxygeldanamycin (GDM-OH, heat shock protein 90 (Hsp90) inhibitor) and formed mixed prodrug polymeric micelles (49). To attain pH-sensitivity for GDM-OH, a spacer group (levulinic acid) was attached on GDM-OH and its distal ketone reacted with PEG-b-p(Asp-Hyd). PEG-b-p(Asp-Hyd) micelles containing DOX and GDM-OH were ca. 100 nm in diameter and released both drugs in a pH-dependent manner. The IC50 value for PEG-b-p(Asp-Hyd) micelles containing DOX and GDM-OH against MCF-7 breast cancer cells was 600 nM, whereas PEG-b-p(Asp-Hyd) micelles containing DOX and PEG-b-p(Asp-Hyd) micelles GDM-OH had IC50 values of 620 and 1240 nM, respectively.
Shin et al. physically co-incorporated three hydrophobic drugs, PTX (microtubule stabilizer), rapamycin (RAPA, mammalian target of rapamycin (mTOR) inhibitor), and 17-allylamino-17-demethoxygeldanamycin (17-AAG, heat shock protein 90 (Hsp90) inhibitor), in PEG-b-PLA micelles using a simple solvent evaporation method in various drug(s)-in-micelle combinations (46). Three drug-loaded (3-in-1) PEG-b-PLA micelles, termed Triolimus (Fig. 4), had a 10–10,000-fold increase in aqueous solubility of PTX, RAPA, and 17-AAG (up to 7.2, 3.3, and 7.3 mg/mL, respectively). PEG-b-PLA micelles delayed release of three drugs compared to single drug-loaded (1-in-1) micelles in vitro. Concurrent delivery of PTX, RAPA, and 17-AAG via 3-in-1 PEG-b-PLA micelles (z-average particle size of ca. 30–40 nm) at 60, 30, and 60 mg/kg, respectively (intravenous (IV) injection, q4d × 3) caused durable antitumor responses in A549 human non-small cell lung cancer (NSCLC) and MDA-MB-231 human breast cancer xenograft models with acceptable acute toxicity (45). Notably, Taxol® has a maximum tolerated dose of 20 mg/kg in mice (q4d × 3). RAPA and 17-AAG may concurrently inhibit PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways, overcoming negative feedback mechanisms and enhancing cancer cell killing by PTX.
Fig. 4.
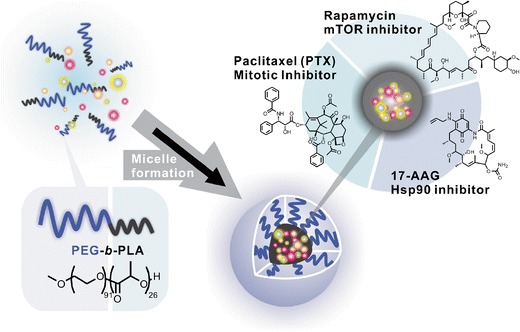
Triolimus: PEG-b-PLA micelles for the concurrent delivery of PTX, RAPA, and 17-AAG (Hasenstein et al., 45). Reprinted from (47), Copyright © 2012, with permission from Elsevier B.V.
At 60, 30, and 60 mg/kg, concurrent delivery of PTX, RAPA, and 17-AAG via 3-in-1 PEG-b-PLA micelles in mice resulted in a 1.7- and 1.6-fold increase in AUC for PTX and RAPA, respectively, over single-drug injected PEG-b-PLA micelles, whereas the AUC for 17-AAG was unchanged (47). 17-AAG may be a better substrate for CYP3A4 than RAPA, leading to increase its AUC after concurrent delivery of 3-in-1 PEG-b-PLA micelles. At more modest clinically relevant dose of PTX, RAPA, and 17-AAG (10, 5, and 10 mg/kg, respectively, via 3-in-1 PEG-b-PLA micelles), PK profiles were similar to single-drug PEG-b-PLA micelles, indicating a lack of drug interaction. However, at both doses, PEG-b-PLA micelles were relatively unstable in blood, disassembled rapidly, and had high clearance values and short half-lives, which are not believed to effectively support delivery of PTX, RAPA, and 17-AAG to solid tumors by the EPR effect.
Na et al. incorporated DOX, ETO (topoisomerase II antagonist), and PTX, individually, in the polymeric micelles composed of PEG-b-poly(γ-benzyl l-glutamate) (GEG, prepared by a dialysis method) or PEG-b-poly(l-lactide) (LE, prepared by a dry-film method) (52). Mixtures of PTX-loaded LE micelles (average diameter of ca. 50 nm) and DOX-loaded GEG micelles (average diameter of ca. 80 nm), and mixtures of DOX-loaded GEG micelles and ETO-loaded GEG micelles (average diameter of ca. 80 nm) induced synergistic cytotoxicity in CT-26 murine colorectal cancer cells in vitro. Drug combination consisting of PTX (2.5 mg/kg) and DOX (2.5 mg/kg) delivered by a mixture of PTX-loaded LE micelles and DOX-loaded GEG micelles resulted in a superior anticancer effect compared free single drug, free DOX or free PTX, at two-fold higher concentrations (5 mg/kg each) in a CT-26-bearing xenograft model.
Wang et al. co-loaded PTX (core) and DOX (shell) in the pH-responsive core/shell nanoparticles composed of PEG-PLGA prepared by an improved double emulsion (W/O/W) method (53). PEG-PLGA nanoparticles carrying PTX and DOX permitted simultaneous release of drugs. The release of PTX and DOX was slow and sustained, resulting in 90% of the total drug release in 300 h, whereas the release of PTX and DOX was much faster at acidic environment, resulting in nearly 100% of the total drug release in 48 h. PEG-PLGA nanoparticles carrying PTX and DOX effectively accumulated in A549 NSCLC and HepG2 liver cancer cells. PTX and DOX at 2:1 (w/w) ratio induced the highest and the most synergistic cytotoxicity in A549, B16 (mouse melanoma cells), and HepG2 cancer cells.
Han et al. developed polymeric micelles using amphiphilic poly(2-oxazoline) (POx) tri-block copolymers, poly(2-methyl-2-oxazoline)-block-poly(2-butyl-2-oxazoline)-block-poly(2-methyl-2-oxazoline) (P(MeOx-b-BuOx-b-MeOx)) in which PBuOx formed the core of polymeric micelles and PMeOx behaved like PEG, providing high hydrophilicity and exhibiting stealth properties in blood stream (54). POx polymeric micelles permitted fine-tuning of the hydrophilic (PMeOx)-hydrophobic (PBuOx) balance by varying length of side alkyl chains and allowed physical incorporation of PTX, DTX (microtubule stabilizer), 17-AAG, ETO, and bortezomib (BTZ, proteasome inhibitor), individually and in combination (PTX/17-AAG, DTX/17-AAG, PTX/ETO, ETO/17-AAG, PTX/BTZ, BTZ/17-AAG, PTX/17-AAG/ETO, PTX/17-AAG/BTZ and DTX/PTX). POx polymeric micelles provided high capacity for drug loading (% drug(s)weight/polymerweight). For example, total drug loading, was >30% for 1-in-1 POx micelles, > 70% for 2-in-1 POx micelles, and > 90% for 3-in-1 POx micelles. POx polymeric micelles surprisingly enabled solubilization of PTX in water, up to 38.7 mg/mL, which resulted in 6.4-fold increased water solubility of PTX compared to PEG-b-PLA micelles. POx micelles containing ETO or BTZ, individually, were relatively unstable over time and aggregated, whereas 2-in-1 and 3-in-1 POx micelles consisting of either ETO or BTZ in combinations with either/both PTX or/and 17-AAG improved their physical stability and displayed more uniform particle sizes over 1-in-1 POx micelles. The authors hypothesized that the major reason for high loading capacity and stability of 3-in-1 POx micelles was favorable drug-drug interaction in the core of micelles and in part, hydrogen bonding between amide bonds of the BuOx block and hydrogen bond donors from drug molecules in micelles.
POx micelles permitted incorporation of multi-drugs in various ratios, allowing for the investigation of ratio-dependent synergism of multiple drugs in vitro. 2-in-1 POx micelles containing ETO/17-AAG and BTZ/17-AAG at certain weight ratios displayed synergistic anticancer effect in MCF-7 breast cancer cells: For example, 1:1 (w/w) ratio of ETO:17-AAG clearly showed synergistic anticancer effect (combination index (CI): 0.45), whereas 1:0.5 induced slight antagonism (CI: 1.03). BTZ/17-AAG at 1:0.6 (CI: 0.099), and 1:1 ratios (CI: 0.40) were strongly synergistic, while 0.3:1 (CI: 6.3) were antagonistic in anticancer effect (CI < 1: synergistic, CI = 1: additive, CI > 1 antagonistic). This study clearly demonstrated that polymeric micelles composed of tunable polymers are versatile, easy to optimize drug ratios/compositions, and capable of many combinations.
Chitkara et al. efficiently co-loaded cyclopamine (CYP, hedgehog inhibitor) and gefitinib (GEF, epidermal growth factor receptor (EGFR) inhibitor) into PEG-b-poly(carbonate-co-lactic acid) (PEG-b-p(CB-co-LA)) micelles, having an average diameter of ca. 54 nm and low polydispersity (55). CYP and GEF exerted synergistic activity against L3.6pl pancreatic cancer cells and additive activity against MIA PaCa-2 pancreatic cancer cells. Further, intratumoral injection of PEG-b-p(CB-co-LA) micelles containing CYP and GEF decreased the rate of tumor growth in a L3.6pl pancreatic tumor model. Co-delivery of two poorly water-soluble signal transduction inhibitors that concurrently target sonic hedgehog and EGFR signaling is an important precedent and another validation of multi-drug delivery via polymeric micelles.
Cho et al. reported another example of a 3-in-1 micelle system, PEG-b-PCL micelles carrying PTX, CYP, and gossypol (GSP, Bcl-2 inhibitor) (Fig. 5) (44). PEG-b-PCL micelles carrying PTX, CYP, and GSP were nanoscopic (z-average diameter of ca. 80–90 nm), able to increase solubility of three payloads in water (up to 6, 6, and 6 mg/mL, respectively); they were fairly stable in aqueous solution; and they were capable of concurrent delivery of PTX, CYP, and GSP via intraperitoneal (IP) administration in xenograft models. PEG-b-PCL micelles simultaneously carrying PTX, CYP, and GSP disaggregated three-dimensional ES-2 ovarian cancer cell spheroids in vitro, presumably by a synergistic anticancer effect, whereas ES-2 cell spheroids stayed intact after treatments of 1-in-1 and 2-in-1 micelles. IP injections of 3-in-1 micelles containing PTX, CYP, and GSP (q7d × 3) at 60, 60, and 60 mg/kg, respectively, were highly effective in IP metastatic ES-2-luc and SKOV-3-luc-bearing ovarian cancer models, eradicating peritoneal tumors, removing ascites, and prolonging survival of treated xenograft models over control and PEG-b-PCL micelles containing PTX alone. These results clearly showed the unique potential of IP locoregional delivery of polymeric micelles carrying multi-drugs for the treatment of aggressive and metastatic ovarian cancers.
Fig. 5.
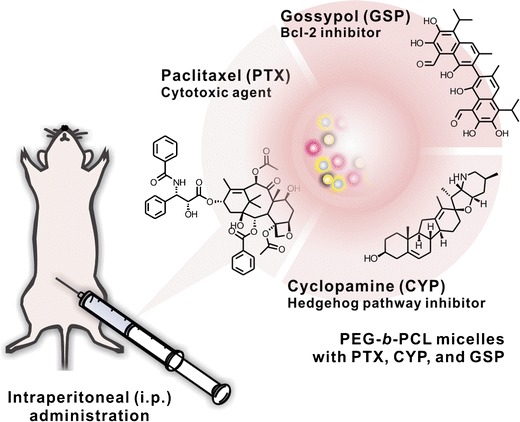
PEG-b-PCL micelles for the concurrent delivery of paclitaxel, cyclopamine, and gossypol (Cho et al., 44). Reprinted from (44), Copyright © 2012, with permission from Elsevier B.V.
Katragadda et al. detailed PEG-distearoylphosphatidylethanolamine/tocopheryl PEG (PEG-DSPE/TPGS) mixed micelles physically carrying both PTX and 17-AAG and emphasized the superior benefits of dual-drug-loading polymeric micelles over free drugs dissolved in solvent (56). Systemically delivered PEG-DSPE/TPGS micelles dual-loaded with PTX (20 mg/kg) and 17-AAG (37.5 mg/kg) resulted in ten-fold increase of PTX level and three-fold increase of 17-AAG level in plasma in comparison with free PTX and 17-AAG. PTX in micelles had one tenth lower total clearance (CLT, mL/kg/min) and one tenth higher area under the curve (AUC, μM/min) compared to free PTX. Levels of PTX and 17-AAG in SKOV-3 tumor tissues were 3.5- and 1.7-fold, respectively, increased without significant alteration of drug levels in normal organs when PTX and 17-AAG were delivered via polymeric micelles in comparison with free drugs dissolved in DMSO solution.
A combination treatment regimen, a weekly IV injection of PTX (20 mg/kg, q7d × 3) and twice-weekly IV injection of 17-AAG (37.5 mg/kg, BIW × 3) in PEG-DSPE/TPGS mixed micelles (dual-loaded micelles followed by 17-AAG-loaded micelles) caused near-complete arrest of tumor growth in SKOV-3 ovarian cancer model at day 42 post initiation of treatment, whereas tumor volumes in groups of control (no treatment) and same regimen of treatment in free forms of PTX and 17-AAG increased approximately eight- and five-fold, respectively, compared to initial tumor volume at day 0. Metabolomic analysis of tumors treated with PTX/17-AAG-loaded micelles showed distinct and significant alterations of metabolomics associated with attenuation of Akt signaling and low Ki-67 proliferation index, which were prominent compared to activities of metabolomics of tumors treated with PTX/17-AAG in free forms.
Desale et al. synthesized PEG-b-poly-(glutamic acid)-b-poly(phenylalanine) (PEG-b-PGlu-b-PPhe) for the dual delivery of PTX and CDDP (alkylating agent) (57). Upon assembly, PEG-b-PGlu-b-PPhe micelles were crosslinked by amide bonds in the intermediate PGlu layer. PEG-b-PGlu-b-PPhe micelles bound PTX in the PPhe core and cisplatin in the intermediate PGlu layer, resulting in 9% and 15% w/w loading, respectively. Interestingly, PEG-b-PGlu-b-PPhe micelles containing both PTX and cisplatin were more potent than PEG-b-PGlu-b-PPhe micelles containing PTX and PEG-b-PGlu-b-PPhe micelles containing cisplatin mixed at the same drug ratio against A2780 ovarian cancer cells. IV injections of PEG-b-PGlu-b-PPhe micelles containing both PTX and cisplatin resulted in a durable antitumor response in an A2780 ovarian cancer xenograft model. Besides multi-drug delivery, crosslinking of PEG-b-PGlu-b-PPhe micelles provides stability against disassembly, as a way of enhancing tumor delivery by the EPR effect.
The concepts of physical drug loading and concurrent delivery of multi-drugs have been extended to sol-gel drug delivery. PLGA-b-PEG-b-PLGA sol-gel for PTX, RAPA, and 17-AAG dissolved 6, 3, and 6 mg/mL of PTX, RAPA, and 17-AAG, respectively, in water as a sol and form a stable gel at 37°C (Fig. 6) (58). In this way, PLGA-b-PEG-b-PLGA hydrogels carrying PTX, RAPA, and 17-AAG (Triogel) permitted locoregional delivery of three drugs in peritoneal cavity of an IP metastatic ES-2-luc ovarian cancer-bearing xenograft model. Importantly, Triogel showed ca. 70- and 80-fold superior potency in tumor regression upon a single IP injection compared to injections (IP or IV) of Triolimus (PEG-b-PLA micelles carrying PTX, RAPA, and 17-AAG, same drug components in solution) and empty vehicles. A single IP injection of Triogel significantly prolonged survival of treated mice bearing ES-2-luc ovarian cancer compared to IP and IV treatments of Triolimus. This promising result led to a conclusion that locally disseminated cancer can be effectively treated with a locoregional drug combination chemotherapy gradually and concurrently released drugs from a gel depot.
Fig. 6.
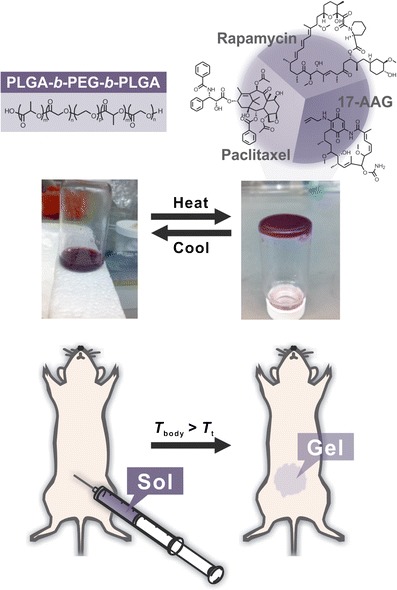
Triogel: PLGA-b-PEG-b-PLGA sol–gel for the sustained intraperitoneal delivery of paclitaxel, rapamycin, and 17-AAG (Cho et al., 58). Adapted with permission of (58), Copyright © 2014, Informa Healthcare
SEQUENTIAL DRUG DELIVERY
Sequential drug delivery, i.e., sequential drug infusion, is commonplace in cancer treatment due to risk of drug incompatibility in IV admixtures, risk of drug precipitation, and overlapping toxicity. In this context, separately formulated polymeric micelles could be used for sequential drug delivery, replacing existing vehicles as safer alternatives for combination anticancer treatment (vide supra).
Perhaps more importantly, strategies involving sequential drug delivery may increase the delivery of anticancer agents into solid tumors. The first injection or drug administration causes a pharmacological response in solid tumors, e.g., decrease in interstitial fluid pressure. As a result, delivery of a second drug or drug vehicle may have enhanced tumor penetration and enhanced antitumor efficacy. The goal is to increase intratumoral drug delivery beyond the scope of the EPR effect for polymeric micelles, other injectable vehicles, and even synergistic drug combinations. Strategies that seek to enhance delivery to solid tumors have been reviewed elsewhere (59,60); we highlight several examples that show potential of polymeric micelles and other nanocarriers in pretreatment strategies for tumor targeting.
Fang et al. have reviewed the EPR effect and discussed the unique features of blood vessels in solid tumors, factors, and strategies for the augmentation of the EPR effect (61). In one strategy, angiotensin II (AT-II) was used to induce hypertension, i.e., constriction of normal blood vessels. Given that tumor blood vessels lack smooth muscle cells, AT-II increased blood flow at solid tumors and enhanced the EPR effect. For example, an 125I-A7 antibody had ca. 14% dose/g tumor uptake in a SW1116 colon carcinoma xenograft model, whereas pretreatment with AT-II lead to a value of ca. 20% dose/g tumor. When AT-II was combined with an angiotensin-converting enzyme (ACE) inhibitor, enalapril, the value increased to ca. 30% dose/g tumor, without increases in normal tissues.
Jain et al. proposed a strategy called “vascular normalization” in 2004 (59). Blocking pro-angiogenic molecules, such as vascular endothelial growth factor (VEGF), decreased vessel density and diameter in tumor tissue, removed immature vessels, and restructured the vasculature into that resembling normal vessels. This normalization of tumor vasculature reduced interstitial fluid pressure and in turn, induced deeper penetration of secondarily delivered anticancer agents into solid tumors. In orthotopic 4T1 and E0771 breast cancer models, a VEGF receptor-2 blocking antibody (DC101), increased the transcellular flux of 12 nm diameter nanoparticles by 3.1-fold in 4T1 tumors, but did not increase the transcellular flux of 60 and 125 nm nanoparticles. Similar results were found in the E0771 breast cancer model. These results strongly suggest that size of nanoparticles is a key consideration in the vascular normalization strategy.
Au et al. proposed a strategy called “tumor priming,” where IV pretreatment by a potent drug inducing apoptosis, such as PTX, could reduce tumor cell density, expand interstitial space, and enhance the intratumoral uptake of a second drug (60,62). For example, in a rat MAT-LyLu prostate cancer model, high dose PTX achieved a higher fraction of apoptotic cells, lower tumor cell density, rapid drug penetration, higher drug accumulation, and more uniform drug distribution in tumor tissues in comparison to continuous infusion-like delivery of the same total dose of PTX over 24 h (60).
In a FaDu human hypopharyngeal carcinoma xenograft, 40 mg/kg of Taxol® increased the tumor-selective delivery of 100 and 200 nm nanoparticles by ca. 1.6-fold, but not 500 nm nanoparticles (62). In addition, tumor priming with 40 mg/kg of Taxol® increased the delivery of DOX subsequently injected 20 mg/kg of Doxil® (DOX liposomes with a diameter of ca. 85 nm) by 1.4-fold, resulting in enhanced therapeutic efficacy. Importantly, tumor priming by PTX appears to be selective for solid tumors, consistent with the fact that tumor cells are prone to apoptosis relative to normal tissues.
Ait-Ouhia et al. set up a quantitative PK/pharmacodynamics (PD) model to analyze tumor priming by PTX and delivery of sterically stabilized liposomes (SSL) containing DOX (63). Simulations predicted that PTX injected 24 h before SSL could increase tumor exposure (AUC) by 2.5-fold, consistent with experimental results. Importantly, this PK/PD approach could be used for evaluating other types of tumor priming strategies and nanocarriers in drug delivery, including polymeric micelles.
Cho et al. showed a powerful tumor priming effect for Triolimus (PTX, RAPA, 17-AAG) in a LS180 colon carcinoma xenograft model (Fig. 7) (64). Using whole-body optical imaging, solid tumors were visualized by 50 nm diameter PEG-b-PCL micelles carrying a near-infrared (NIR) imaging agent (1,1′-dioctadecyl tetramethyl indotricarbocyanine iodide, DiR). Tumor priming by a single IV injection of Triolimus caused apoptosis at solid tumors that peaked over 24 to 48 h and reduced tumor volume by 1.6-fold with <10% body weight change. As a result, PEG-b-PCL micelles carrying DiR had a 2.1-fold higher optical signal at solid tumors versus no tumor priming (i.e., delivery by the EPR effect).
Fig. 7.
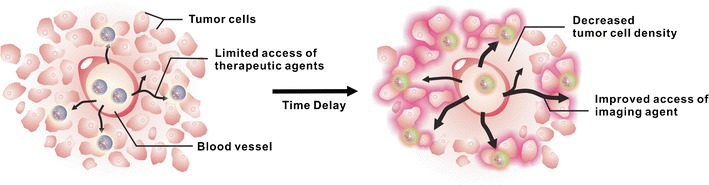
Delivery of an NIR emitting PEG-b-PCL micelle to a solid tumor by the EPR effect (left) and tumor priming (right) (Cho et al., 64). Reprinted with permission from (64), Copyright © 2011, American Chemical Society
Cabral et al. studied a tumor priming effect of a transforming growth factor-β (TGF-β) inhibitor (LY364947) in a hypovascular BxPC3 pancreatic xenograft model (65). LY364947 decreased pericyte coverage of endothelium in neovasculature of pancreatic tumors, decreasing barrier properties associated with tumor stroma. Intratumoral uptake of 70 nm polymeric micelles was low. However, LY364947 increased the intratumoral uptake of 70 nm polymeric micelles, achieving intratumoral uptake found for 30 nm polymeric micelles. In a highly permeable C26 colon carcinoma xenograft model, by contrast, intratumoral uptake of polymeric micelles ranging in size from 30 to 100 nm in diameter was equivalent without the addition of a TGF-β inhibitor.
In summary, strategies, such as vascular normalization or tumor priming, may improve drug delivery into solid tumors beyond the EPR effect and merits consideration in context of sequential drug delivery by polymeric micelles and other nanocarriers. Limitations may include size constraints for nanoparticles in intratumoral penetration. Consideration also has to be given to the potential for toxicity of anticancer agents injected sequentially only 1 or 2 days apart. In this case, injection of slow-release nanocarriers, such as Doxil®, in the second step may blunt toxicity and take advantage of an enhanced EPR effect for tumor targeting.
CONCLUSIONS AND FUTURE DIRECTIONS
Drug combinations have been very successful in the treatment of cancer, and they continue to be widely researched in preclinical and clinical studies with expanding focus on targeting aberrant signaling pathways in solid tumors. Key criteria for the justification of a drug combination are synergy, biologic rationale, selectivity, safety, and favorable PK (66). In this review article, several examples illustrated how polymeric micelles can be used to deliver drug combinations, especially for poorly water-soluble anticancer agents. PEG-b-PLA micelles safely replace existing vehicles, such as co-solvents and surfactants, and they contain physically loaded anticancer agents for first-in-class concurrent multi-drug delivery. In this way, anticancer agents may have overlapping PK profiles and act concurrently at solid tumors, aiming for synergy. Drug interactions for concurrent multi-drug delivery may occur, affecting PK and PD of anticancer agents and thus potentially affecting antitumor efficacy and toxicity (67).
A PEG-b-p(Asp-Hyd) micelle contains pendent anticancer agents in its core, exhibits pH-sensitive release, and also can be used for concurrent multi-drug delivery. Compared to physically loaded drugs, chemically conjugated drugs are less likely to prematurely release drugs after injection or infusion and therefore are more likely to be available for tumor targeting by the EPR effect. In both cases, polymeric micelles disassemble into unimers that undergo renal clearance; however, crosslinking strategies are available that maintain the integrity of polymeric micelles in blood for tumor targeting (68).
Sequential drug delivery of polymeric micelles may be used for “tumor priming” to enhance tumor penetration and uptake of anticancer agents beyond the scope of the EPR effect. These PK strategies that seek to enhance drug delivery into solid tumors are interesting and will likely expand with a greater insight into intratumoral barriers. Polymeric micelles are uniquely suited for multi-drug delivery in the search for synergistic, selective, and safe anticancer drug combinations.
ACKNOWLEDGMENTS
This work was partially supported by Global Innovative Research Center program of the National Research Foundation of Korea and by the Intramural Research Program (Global RNAi Carrier Initiative) of Korean Institute of Science and Technology.
REFERENCES
- 1.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–45. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 2.Hainaut P, Plymoth A. Targeting the hallmarks of cancer: towards a rational approach to next-generation cancer therapy. Curr Opin Oncol. 2013;25:50–1. doi: 10.1097/CCO.0b013e32835b651e. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Heppner GH. Tumor heterogeneity. Cancer Res. 1984;44(6):2259–65. [PubMed] [Google Scholar]
- 6.Frei E, 3rd, Karon M, Levin RH, Freireich EJ, Taylor RJ, Hananian J, et al. The effectiveness of combinations of antileukemic agents in inducing and maintaining remission in children with acute leukemia. Blood. 1965;26:642–56. [PubMed] [Google Scholar]
- 7.Hersh EM, Carbone PP, Wong VG, Freireich EJ. Inhibition of the primary immune response in man by anti-metabolites. Cancer Res. 1965;25:997–1001. [PubMed] [Google Scholar]
- 8.Canellos GP, Abramson JS, Fisher DC, LaCasce AS. Treatment of favorable, limited-stage Hodgkin’s lymphoma with chemotherapy without consolidation by radiation therapy. J Clin Oncol. 2010;28:1611–5. doi: 10.1200/JCO.2009.25.3260. [DOI] [PubMed] [Google Scholar]
- 9.Canellos GP, Gollub J, Neuberg D, Mauch P, Shulman LN. Primary systemic treatment of advanced Hodgkin’s disease with EVA (etoposide, vinblastine, doxorubicin): 10-year follow-up. Ann Oncol. 2003;14:268–72. doi: 10.1093/annonc/mdg076. [DOI] [PubMed] [Google Scholar]
- 10.Martoni A, Cacciari N, Angelelli B, Zamagni C, Pannuti F. Chemotherapy of advanced ovarian cancer. Front Biosci. 1997;2:g20–6. [PubMed] [Google Scholar]
- 11.McGuire WP., 3rd Current status of taxane and platinum-based chemotherapy in ovarian cancer. J Clin Oncol. 2003;21:133s-5s. doi: 10.1200/JCO.2003.01.066. [DOI] [PubMed] [Google Scholar]
- 12.Rigas JR. Taxane-platinum combinations in advanced non-small cell lung cancer: a review. Oncologist. 2004;9:16–23. doi: 10.1634/theoncologist.9-suppl_2-16. [DOI] [PubMed] [Google Scholar]
- 13.Guarneri V, Conte PF. The curability of breast cancer and the treatment of advanced disease. Eur J Nucl Med Mol I. 2004;31:S149–61. doi: 10.1007/s00259-004-1538-5. [DOI] [PubMed] [Google Scholar]
- 14.Melisi D, Troiani T, Damiano V, Tortora G, Ciardiello F. Therapeutic integration of signal transduction targeting agents and conventional anti-cancer treatments. Endo-Relat Cancer. 2004;11:51–68. doi: 10.1677/erc.0.0110051. [DOI] [PubMed] [Google Scholar]
- 15.Ku MS. Use of the biopharmaceutical classification system in early drug development. AAPS J. 2008;10:208–12. doi: 10.1208/s12248-008-9020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narvekar M, Xue HY, Eoh JY, Wong HL. Nanocarrier for poorly water-soluble anticancer drugs-barriers of translation and solutions. AAPS Pharm Sci. 2014;15:822–3. doi: 10.1208/s12249-014-0107-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savjani KT, Gajjar AK, Savjani JK. Drug solubility: importance and enhancement techniques. ISRN Pharm. 2012;2012:195727. doi: 10.5402/2012/195727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590–8. doi: 10.1016/S0959-8049(01)00171-X. [DOI] [PubMed] [Google Scholar]
- 19.Coors EA, Seybold H, Merk HF, Mahler V. Polysorbate 80 in medical products and nonimmunologic anaphylactoid reactions. Ann Allerg Asthma Im. 2005;95:593–9. doi: 10.1016/S1081-1206(10)61024-1. [DOI] [PubMed] [Google Scholar]
- 20.Shelley WB, Talanin N, Shelley ED. Polysorbate 80 hypersensitivity. Lancet. 1995;345:1312–3. doi: 10.1016/S0140-6736(95)90963-X. [DOI] [PubMed] [Google Scholar]
- 21.Peer D, Karp JM, Hong S, FaroKHzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–60. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 22.Hu CM, Aryal S, Zhang L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther Deliv. 2010;1:323–34. doi: 10.4155/tde.10.13. [DOI] [PubMed] [Google Scholar]
- 23.Lee JH, Nan A. Combination drug delivery approaches in metastatic breast cancer. J Drug Deliv. 2012;2012:915375. doi: 10.1155/2012/915375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parhi P, Mohanty C, Sahoo SK. Nanotechnology-based combinational drug delivery: an emerging approach for cancer therapy. Drug Discov Today. 2012;17:1044–52. doi: 10.1016/j.drudis.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 25.Batist G, Gelmon KA, Chi KN, Miller WH, Chia SKL, Mayer LD, et al. Safety, pharmacokinetics, and efficacy of CPX-1 liposome injection in patients with advanced solid tumors. Clin Cancer Res. 2009;15:692–700. doi: 10.1158/1078-0432.CCR-08-0515. [DOI] [PubMed] [Google Scholar]
- 26.Lancet JE, Cortes JE, Hogge DE, Tallman MS, Kovacsovics TJ, Damon LE, et al. Phase II, multicenter, randomized, open label trial of CPX-351 (cytarabine:daunorubicin) liposome injection versus cytarabine and daunorubicin in patients with untreated AML 60–75 years of age. Blood. 2014;123:3239–46. doi: 10.1182/blood-2013-12-540971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greco F, Vicent MJ, Gee S, Jones AT, Gee J, Nicholson RI, et al. Investigating the mechanism of enhanced cytotoxicity of HPMA copolymer-Dox-AGM in breast cancer cells. J Control Release. 2007;117:28–39. doi: 10.1016/j.jconrel.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 28.Sengupta S, Eavarone D, Capila I, Zhao GL, Watson N, Kiziltepe T, et al. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436:568–72. doi: 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- 29.Tekade RK, Dutta T, Gajbhiye V, Jain NK. Exploring dendrimer towards dual drug delivery: pH responsive simultaneous drug-release kinetics. J Microencapsul. 2009;26:287–96. doi: 10.1080/02652040802312572. [DOI] [PubMed] [Google Scholar]
- 30.Croy SR, Kwon GS. Polymeric micelles for drug delivery. Curr Pharma Des. 2006;12:4669–84. doi: 10.2174/138161206779026245. [DOI] [PubMed] [Google Scholar]
- 31.Jones M, Leroux J. Polymeric micelles—a new generation of colloidal drug carriers. Eur J Pharm Biopharm. 1999;48:101–11. doi: 10.1016/S0939-6411(99)00039-9. [DOI] [PubMed] [Google Scholar]
- 32.Miyata K, Christie RJ, Kataoka K. Polymeric micelles for nano-scale drug delivery. React Funct Polym. 2011;71:227–34. doi: 10.1016/j.reactfunctpolym.2010.10.009. [DOI] [Google Scholar]
- 33.Kwon GS. Polymeric micelles for delivery of poorly water-soluble compounds. Crit Rev Ther Drug. 2003;20:357–403. doi: 10.1615/CritRevTherDrugCarrierSyst.v20.i5.20. [DOI] [PubMed] [Google Scholar]
- 34.Rapoport N. Physical stimuli-responsive polymeric micelles for anti-cancer drug delivery. Prog Polym Sci. 2007;32:962–90. doi: 10.1016/j.progpolymsci.2007.05.009. [DOI] [Google Scholar]
- 35.Kwon GS, Okano T. Polymeric micelles as new drug carriers. Adv Drug Deliver Rev. 1996;71:227–34. [Google Scholar]
- 36.Sutton D, Wang S, Nasongkla N, Gao J, Dormidontova EE. Doxorubicin and beta-lapachone release and interaction with micellar core materials: experiment and modeling. Exp Biol Med. 2007;232:1090–9. doi: 10.3181/0702-RM-31. [DOI] [PubMed] [Google Scholar]
- 37.Lavasanifar A, Samuel J, Kwon GS. Poly(ethylene oxide)-block-poly(L-amino acid) micelles for drug delivery. Adv Drug Deliv Rev. 2002;54:169–90. doi: 10.1016/S0169-409X(02)00015-7. [DOI] [PubMed] [Google Scholar]
- 38.Batrakova EV, Bronich TK, Vetro JA, Kabanov AV. Polymeric micelles as drug carriers. In: Torchilin VP, editor. Nanoparticulates as drug carriers. London: World Scientific; 2006. pp. 63–7. [Google Scholar]
- 39.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–49. doi: 10.1016/S1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 40.Kim S, Shi Y, Kim JY, Park K, Cheng JX. Overcoming the barriers in micellar drug delivery: loading efficiency, in vivo stability, and micelle-cell interaction. Expert Opin Drug Deliv. 2010;7:49–62. doi: 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
- 41.Matsumura Y, Kataoka K. Preclinical and clinical studies of anticancer agent-incorporating polymer micelles. Cancer Sci. 2009;100:572–9. doi: 10.1111/j.1349-7006.2009.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee KS, Chung HC, Im SA, Park YH, Kim CS, Kim SB, et al. Multicenter phase II trial of Genexol-PM, a Cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Res Treat. 2008;108:241–50. doi: 10.1007/s10549-007-9591-y. [DOI] [PubMed] [Google Scholar]
- 43.Matsumura Y. The drug discovery by nanomedicine and its clinical experience. Jpn J Clin Oncol. 2014;44:515–25. doi: 10.1093/jjco/hyu046. [DOI] [PubMed] [Google Scholar]
- 44.Cho H, Lai TC, Kwon GS. Poly(ethylene glycol)-block-poly(epsilon-caprolactone) micelles for combination drug delivery: evaluation of paclitaxel, cyclopamine and gossypol in intraperitoneal xenograft models of ovarian cancer. J Control Release. 2013;166:1–9. doi: 10.1016/j.jconrel.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hasenstein JR, Shin HC, Kasmerchak K, Buehler D, Kwon GS, Kozak KR. Antitumor activity of Triolimus: a novel multidrug-loaded micelle containing paclitaxel, rapamycin, and 17-AAG. Mol Cancer Ther. 2012;11:2233–42. doi: 10.1158/1535-7163.MCT-11-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shin HC, Alani AW, Cho H, Bae Y, Kolesar JM, Kwon GS. A 3-in-1 polymeric micelle nanocontainer for poorly water-soluble drugs. Mol Pharm. 2011;8:1257–65. doi: 10.1021/mp2000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin HC, Cho H, Lai TC, Kozak KR, Kolesar JM, Kwon GS. Pharmacokinetic study of 3-in-1 poly(ethylene glycol)-block-poly(d,l-lactic acid) micelles carrying paclitaxel, 17-allylamino-17-demethoxygeldanamycin, and rapamycin. J Control Release. 2012;163:93–9. doi: 10.1016/j.jconrel.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bae Y, Diezi TA, Zhao A, Kwon GS. Mixed polymeric micelles for combination cancer chemotherapy through the concurrent delivery of multiple chemotherapeutic agents. J Control Release. 2007;122:324–30. doi: 10.1016/j.jconrel.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 49.Bae Y, Alani AWG, Rockich NC, Lai TSZC, Kwon GS. Mixed pH-sensitive polymeric micelles for combination drug delivery. Pharm Res-Dordr. 2010;27:2421–32. doi: 10.1007/s11095-010-0234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 51.Greco F, Vicent MJ. Combination therapy: opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv Drug Deliver Rev. 2009;61:1203–13. doi: 10.1016/j.addr.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 52.Na HS, Lim YK, Jeong YI, Lee HS, Lim YJ, Kang MS, et al. Combination antitumor effects of micelle-loaded anticancer drugs in a CT-26 murine colorectal carcinoma model. Int J Pharm. 2010;383:192–200. doi: 10.1016/j.ijpharm.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 53.Wang H, Zhao Y, Wu Y, Hu YL, Nan K, Nie G, et al. Enhanced anti-tumor efficacy by co-delivery of doxorubicin and paclitaxel with amphiphilic methoxy PEG-PLGA copolymer nanoparticles. Biomaterials. 2011;32:8281–90. doi: 10.1016/j.biomaterials.2011.07.032. [DOI] [PubMed] [Google Scholar]
- 54.Han Y, He Z, Schulz A, Bronich TK, Jordan R, Luxenhofer R, et al. Synergistic combinations of multiple chemotherapeutic agents in high capacity poly(2-oxazoline) micelles. Mol Pharm. 2012;9:2302–13. doi: 10.1021/mp300159u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chitkara D, Singh S, Kumar V, Danquah M, Behrman SW, Kumar N, et al. Micellar delivery of cyclopamine and gefitinib for treating pancreatic cancer. Mol Pharm. 2012;9:2350–7. doi: 10.1021/mp3002792. [DOI] [PubMed] [Google Scholar]
- 56.Katragadda U, Teng Q, Rayaprolu BM, Chandran T, Tan C. Multi-drug delivery to tumor cells via micellar nanocarriers. Int J Pharm. 2011;419:281–6. doi: 10.1016/j.ijpharm.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Desale SS, Cohen SM, Zhao Y, Kabanov AV, Bronich TK. Biodegradable hybrid polymer micelles for combination drug therapy in ovarian cancer. J Control Release. 2013;171:339–48. doi: 10.1016/j.jconrel.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cho H, Kwon GS. Thermosensitive poly-(d, l-lactide-co-glycolide)-block-poly(ethylene glycol)-block-poly-(d, l-lactide-co-glycolide) hydrogels for multi-drug delivery. J Drug Target. 2014;22:669–77. doi: 10.3109/1061186X.2014.931406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–64. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jang SH, Wientjes MG, Lu D, Au JL. Drug delivery and transport to solid tumors. Pharm Res. 2003;20:1337–50. doi: 10.1023/A:1025785505977. [DOI] [PubMed] [Google Scholar]
- 61.Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65:71–9. doi: 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 62.Lu D, Wientjes MG, Lu Z, Au JL. Tumor priming enhances delivery and efficacy of nanomedicines. J Pharmacol Exp Ther. 2007;322:80–8. doi: 10.1124/jpet.107.121632. [DOI] [PubMed] [Google Scholar]
- 63.Ait-Oudhia S, Straubinger RM, Mager DE. Systems pharmacological analysis of paclitaxel-mediated tumor priming that enhances nanocarrier deposition and efficacy. J Pharmacol Exp Ther. 2013;344:103–12. doi: 10.1124/jpet.112.199109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cho H, Kwon GS. Polymeric micelles for neoadjuvant cancer therapy and tumor-primed optical imaging. ACS Nano. 2011;5:8721–9. doi: 10.1021/nn202676u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815–23. [DOI] [PubMed]
- 66.Yap TA, Omlin A, de Bono JS. Development of therapeutic combinations targeting major cancer signaling pathways. J Clin Oncol. 2013;31:1592–605. doi: 10.1200/JCO.2011.37.6418. [DOI] [PubMed] [Google Scholar]
- 67.Scripture CD, Figg WD. Drug interactions in cancer therapy. Nat Rev Cancer. 2006;6:546–58. doi: 10.1038/nrc1887. [DOI] [PubMed] [Google Scholar]
- 68.Rijcken CJ, Snel CJ, Schiffelers RM, van Nostrum CF, Hennink WE. Hydrolysable core-crosslinked thermosensitive polymeric micelles: synthesis, characterisation and in vivo studies. Biomaterials. 2007;28:5581–93. doi: 10.1016/j.biomaterials.2007.08.047. [DOI] [PubMed] [Google Scholar]