

Abstract
Utilization of lipid-based drug delivery systems has recently gained focus for drugs characterized by poor aqueous solubility. The improved aqueous solubility overcomes one of the main barriers that limit their bioavailability. The objective of this work was to improve the solubility and oral bioavailability of Avanafil (AVA), a recently approved second generation type 5 phospodiesterase inhibitor used for erectile dysfunction.AVA was formulated as self-nanoemulsifying drug delivery system (SNEDDS) utilizing various oils, surfactants, and cosurfactants. The solubility of AVA in various oils, surfactants, and cosurfactants was determined. Ternary phase diagram was constructed to identify stable nanoemulsion region. The prepared AVA loaded SNEDDS were assessed for optical clarity, droplet size, conductivity, and stability studies. In vitro drug release and in vivo pharmacokinetic parameters using animal model were also investigated. Results revealed that stable AVA (SNEDDS) were successfully developed with a droplet size range of 65 to 190 nm. SNEDDS composed of 25% dill oil, 55% Tween 80, and 20% propylene glycol successfully improved solubilization of AVA (over 80% within 30 min) vis-a-vis the powder AVA (35% within 30 min). In vivo pharmacokinetic showed a significant (P < 0.05) increase in Cmax, reduction in Tmax, and SNEDDS enhanced the bioavailability in the rats by 1.4-fold when compared with pure drug.
Key words: avanafil, erectile dysfunction, dill oil, self-nanoemulsifying, SNEDDS
INTRODUCTION
Avanavil was recently approved by the international pharamceutical regulating agencies, United States Food and Drug Administration (U.S. FDA) and The European Medicines Agency (EMA) (1,2) as a second generation type 5 phospodiesterase inhibitor used for erectile dysfunction (3). Avanafil (AVA) was developed for its high selectivity for the PDE5 isoenzyme relative to other PDE5 inhibitors. AVA shows onset of action in the range of 30–45 min (1).
Improvement in AVA bioavailability relies heavily upon the improvement in its aqueous solubility. One of the approaches applied to improve drug aqueous solubility is achieved through the utilization of self-nanoemulsifying drug delivery systems (SNEDDS) (4–9). SNEDDS disperse in aqueous media as fine emulsion with globules in the nanosize range, so the drug remains in solution, consequently overcoming one of the main barriers for drug absorption, the dissolution step (10–14). In addition, formation of emulsion as a result of the emulsification process of SNEDDS in the aqueous gastrointestinal tract (GIT) medium improved permeability of the drug across the GIT membrane, which improves its bioavailablity.
This study aimed to develop AVA-SNEDDS formulation characterized by having small globule size and high emulsification and dissolution rates. The study also aimed at investigating pharmacokinetics parameters of AVA-SNEDDS formula compared with AVA powder in rats.
MATERIALS AND METHODS
Materials
AVA was purchased from Jinlan Pharm-Drugs Technology Co., Ltd. (Hangzhou, China). Dill oil, eucalyptus oil, and orange oil were purchased from Winlab Lab. Chem. (Leicestershire, UK). Labrafil M1944 and Labrasol were kind gifts from Gattefosse (Saint-Priest Cedex, France). Sefsol was a generous gift from Nikko chemicals Company, Ltd. (Chuoku, Tokyo, Japan). Isopropanol and propylene glycol were obtained from TEDIA Company, Inc. (OH, USA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
METHODS
Solubility Studies
The solubility of AVA in various oils (castor oil, Labrafil M1944, isopropyl myristate, dill oil, Sefsol, oleic acid, linoleic acid, eucalyptus oil, orange oil, and Triacetin), surfactants (Span 20, Span 80, Tween 20, Tween 80, and Labrasol), and cosurfactants (polyethylene glycol (PEG) 200, ethanol, propylene glycol (PG), and isopropanol) was determined by addition of excess amount of the drug to screw-capped glass vials containing 2 mL of each of the studied vehicles. The mixtures were placed in a thermostatically controlled shaking water bath at 25 ± 0.5°C for 48 h. After reaching equilibrium, the mixtures were centrifuged at 3,000 rpm for 15 min. The supernatant was diluted with methanol, and the concentration of AVA was quantified by HPLC, Agilent 1,200 series equipped with UV diode array detector. Reversed-phase column was 25 cm × 4.6 mm (i.d.) C18, 5 μm–Intersil® ODS-3(GL Sciences Inc, Japan). The column effluent was monitored by UV detector at 230 nm and the eluent flow was 1.3 ml/min. The mobile phase was acetonitrile:methanol:0.05 M ammonium acetate buffer pH 2.5 (20:20:60 v/v/v). AVA retention time was 5 min.
Construction of Ternary Phase Mixture
Based on the solubility studies, the selected oil (dill oil), surfactant (Tween 80), and cosurfactant (PG) were used to prepare the nanoemulsion (NE). For ternary phase, 12 ternary mixtures were prepared with varying concentration of oil, surfactant, and cosurfactant as shown in Table I. For any mixture, the total of the three components always added to 100%.
Table I.
UV Absorbance, Mean Globule Size, Zeta Potential, Conductivity, and Stability for Different AVA-SNEDDS Formulations
| Formula no. | Dill oil (%) | Tween 80 (%) | PG (%) | Globule size (nm) | Zeta potential (mV) | UV absorbance at 638 nm | Conductivity | Stability |
|---|---|---|---|---|---|---|---|---|
| F1 | 35 | 50 | 15 | 190 | 8.45 | 0.0096 | 0.122 | Fail |
| F2 | 35 | 55 | 10 | 175 | 9.87 | 0.0085 | 0.123 | Fail |
| F3 | 30 | 48 | 22 | 138 | 10.21 | 0.0061 | 0.124 | Fail |
| F4 | 30 | 55 | 15 | 150 | 9.92 | 0.0074 | 0.126 | Fail |
| F5 | 27.5 | 52.5 | 20 | 121 | 11.15 | 0.0055 | 0.131 | Fail |
| F6 | 25 | 55 | 20 | 95 | 12.22 | 0.0042 | 0.136 | Pass |
| F7 | 25 | 47.5 | 27.5 | 108 | 11.56 | 0.0049 | 0.135 | Pass |
| F8 | 20 | 55 | 25 | 80 | 13.71 | 0.0038 | 0.144 | Pass |
| F9 | 20 | 42.5 | 37.5 | 90 | 12.34 | 0.0039 | 0.141 | Pass |
| F10 | 15 | 47.5 | 37.5 | 70 | 14.23 | 0.0035 | 0.153 | Pass |
| F11 | 15 | 37.5 | 47.5 | 78 | 13.82 | 0.0037 | 0.154 | Pass |
| F12 | 10 | 35 | 55 | 65 | 15.43 | 0.0030 | 0.167 | Pass |
PG propylene glycol
Visual Assessment and Emulsification Ability of the Ternary Mixtures
The efficiency of the prepared NE was visually assessed for its ability to emulsify spontaneously and for the clarity of the final emulsion.
Spectroscopic Characterization of Optical Clarity
The optical clarity of the aqueous dispersions of the NE formulations was measured spectroscopically. Briefly, 100 μL of each NE was diluted with 20 mL distilled water. The absorbance of each solution was measured at 638 nm.
NE Droplet Size and Zeta Potential Analysis
To test the droplet size and zeta potential of the NE, 100 μL of each formulation was diluted with 20 mL distilled water. The mean droplet size and zeta potential of the resulting dispersion was determined by dynamic light scattering using a Zetatrac machine from Microtrac Inc. (PA, USA).
Conductivity
The conductivity was determined using the Zetatrac machine from Microtrac Inc. (PA, USA).
Stability Studies
The NE formulations were centrifuged three times every 24 h at 3,000 rpm for 30 min. Formulations that did not show any phase separations were selected for dilution study, this study include the measurement of particle size and zeta potential after dilution with buffer pH 1.2, sterile water, and buffer pH 7.4 immediately and after 24 h, and the formulation which show no change in particle size after dilution with different dispersion media will be selected for the in vitro release studies.
In Vitro Release Studies
SNEDDS samples containing 100 mg of AVA were packed into size (1) hard gelatine capsules. In vitro dissolution was done using USP dissolution tester, apparatus II, rotated at 100 rpm in 450 ml of 0.1 N HCl maintained at 37 ± 0.5°C for 2 h then the pH raised to 7.4, and the test completed to 4 h. Three-milliliter samples were taken at different time intervals (5, 10, 15, 30, 45, and 60 min; 2 h, 3 h, and 4 h), and AVA content was determined by HPLC.
In Vivo Pharmacokinetic Study
Male Wistar rats (weight 230 ± 20 g) were used for the pharmacokinetic study. The rats were acclimated for at least 5 days in their environmentally-controlled cages (23 ± 1°C and 12/12 h dark/light cycle) with free access to standard food and water. The rats were fasted overnight before the experiments. All experimental protocols were conducted after being approved by the Animal Ethics Committee of King Abdulaziz University, Jeddah, KSA who ensured the care and use of animals conformed to the Declaration of Helsinki and the Guiding Principle in Care and Use of Animals (DHEW publication NIH 80–23) and stick to the “Principles of Laboratory Animal Care” (NIH publication #85-23, revised in 1985). The animals were divided into two groups (six rats per group). The first group received 2 mL of AVA suspension in water orally, 10 mg/Kg dose. The second group received the same dose using 2 mL AVA-SNEDDS orally.
Pharmacokinetics data was based on AVA plasma concentrations and was analyzed by PK-SOLVER using non-compartmental analysis model. The plasma drug concentrations were analyzed by HPLC. An isocratic HPLC elution mobile phase was used, consisting of 60% acetonitrile and 40% ammonium acetate 10 mM (v/v), adjusted to pH 3 with acetic acid. A flow rate of 0.3 mL/min was used for sample analysis on an (ODS-A, C18, ACE Ltd, Scotland) analytical column (50 mm × 4.0 mm i.d.). The column was maintained at ambient temperature (23°C), the injection volume was 20 μL, and the total run time was set for 5 min. A PE Sciex API 3,000 triple quadrupole mass spectrometer interfaced with the HPLC via a turbo-ionspray source was used for the mass analysis and detection. The analytes were detected by monitoring the precursor product ion transition using multiple reactions monitoring (MRM) scan mode. The MRM was performed at m/z 484 → 383 for AVA.
The pharmacokinetic parameters, maximum plasma concentration (Cmax), corresponding time for maximum concentration (Tmax), area under the plasma concentration-time curve (AUC), and total body clearance (CLT) were performed on each individual rat. Data are presented as mean ± SD. Significant differences between the pharmacokinetic data were tested by ANOVA followed by Sidak’s multiple comparisons test and the confidence level was set at a value of P < 0.05 (GraphPad Prism 6, GraphPad Software, CA, USA).
RESULTS
Solubility Studies
Figure 1 shows AVA solubility in different oils, surfactants, and cosurfactants. The solubilization of AVA was highest in dill oil (27.15 ± 2.13 mg/mL) compared with other investigated oils. Of the surfactants screened, Tween 80 showed a superior solubilizing potential (13.43 ± 1.42 mg/mL). Additionally, PG was selected as a cosurfactant due to its efficient solubilizing effect (12.29 ± 1.23 mg/mL).
Fig. 1.
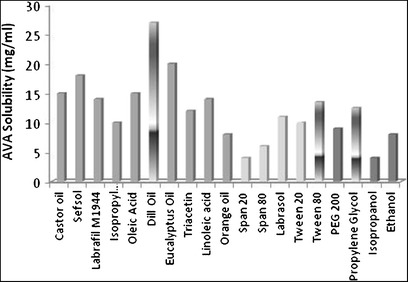
Graphical representation of solubility of AVA in various oils, surfactants, and cosurfactants
Ternary Phase Mixture
The detailed composition and characteristics of the ternary phase mixture is demonstrated in Table I and Fig. 2. It was observed that the maximum concentration of oil that could be solubilized was 35% using 50% of Tween 80 and 15% of PG. However, when the concentration of PG was increased with respect to Tween 80, the maximum concentration of oil that could be solubilized was decreased to 10%, and this concentration was increased by increasing the amount of Tween 80 with respect to PG.
Fig. 2.
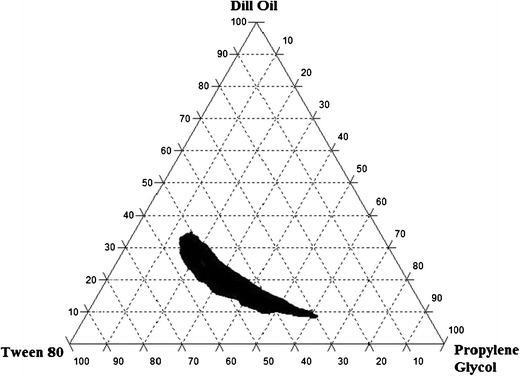
Ternary phase diagrams of AVA indicating the o/w nanoemulsion region at different dill oil, Tween 80, and PG ratios
Characterization of the NE
Table I shows NEs that had droplets in the nanosize range (65–190 nm). The zeta potential varied between (8.45–15.43 mV). The mean UV absorbance at 638 nm varied between 0.0032 and 0.0091, and the droplet size increased as the concentration of oil increased. However, the droplet size decreased as the concentration of the surfactant was increased and as zeta potential increased. The conductivity measurements (0.122–0.167 ms/cm) indicated that NEs formed were of the oil-in-water type (15).
Stability of the NEs
The stress tests, including centrifugation for three times, showed that formulae (F1–5) containing dill oil in concentrations more than 25% were unstable and showed phase separation. All remaining formulations (F6–12) were physically stable. The measurement of particle size after dilution with different dispersion media (buffer pH 1.2 and buffer pH 7.4) showed no changes in results from that represented in Table I, which include the particle size measurement after dilution with sterile water in all tested formulations (F6–12).
In Vitro Release
Figure 3 shows the in vitro release of AVA from different stable formulations (F6–12). All formulations showed rapid release, and more than 90% of AVA was released within 15 min. Accordingly, formula F6 which composed of 25% dill oil, 55% Tween 80, and 20% PG was selected for in vivo study. The selection was based on the maximum amount of oil in the formula compared to other formulations (F8–12). This increased amount of oil is essential in nanoemulsion formulation to enhance drug loading. In addition, F6 was superior in droplet size (Table I) compared with F7 that contain the same concentration of oil.
Fig. 3.
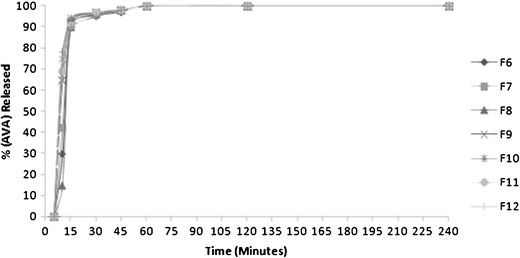
In vitro release of AVA from different SNEDDS formulations
In Vivo Pharmacokinetic Study
AVA is rapidly and relatively well absorbed from the gastrointestinal tract and have low to moderate bioavailability (16). The two main causes of low to moderate bioavailability are the lower solubility in alkaline pH and the second factor is AVA undergoes a first-pass effect. After estimation of pharmacokinetics parameters, there was significant increase in Cmax 40.36 ± 9.19 ng/ml compared with 25.37 ± 5.32 ng/ml for AVA-SNEDDS and control groups, respectively. In addition, Tmax was decreased significantly from 70 ± 29.4 min to 32.5 ± 11.29 min for control and AVA-SNEDDS groups, respectively. Two main factors can rationalize this significant increase in plasma maximum concentration and rapid absorption of AVA-SNEDDS. Firstly, AVA has low solubility in alkaline pH and Tween 80 is capable of enhancing its permeability in Caco-2 cells that mean enhancing AVA solubility in the intestine (17). Secondly, droplet size is an important factor in the absorption process that permits passive transport directly in addition to active transport mechanisms for drugs. It was reported that grisofulvin absorption was duplicated by decreasing particle size from 200 to 20 nm (18). About 21% of AVA is excreted in the urine (3), furthermore, AVA is p-glycoprotein(P-gp) substrate and Tween 80 have inhibition effect on P-gp; this mean increasing AVA residency time in case of AVA-SNEDDS and explains increasing half life by about 45% and AUC by about 18% as shown in Table II and Fig. 4.
Table II.
Pharmacokinetic Parameters of AVA after Administration of 10 mg/kg Oral SNEDDS of Formula F6 and 10 mg/kg of AVA Powder Suspension (n = 6). The Mean Difference is Significant at the 0.05 Level
| Parameter | Unit | AVA powder | AVA-SNEDDS |
|---|---|---|---|
| Ke | 1/min | 0.00575 ± 0.0011 | 0.0057 ± 0.0022 |
| t1/2 | min | 124.84 ± 26.84 | 139.40 ± 57.88 |
| Tmax | min | 65 ± 20.49 | 35.00 ± 7.74* |
| Cmax | ng/ml | 27.41 ± 5.88 | 42.38 ± 7.52* |
| AUC 0-inf_obs | ng/ml.min | 5,130.24 ± 717.42 | 7,150.50 ± 834.73* |
| Vz/F_obs | (mg)/(ng/ml) | 0.104 ± 0.017 | 0.1234 ± 0.039 |
| Cl/F_obs | (mg)/(ng/ml)/min | 0.00060 ± 0.00017 | 0.00053 ± 0.00020 |
AVA Avanafil, SNEDDS self-nanoemulsifying drug delivery system
*Significantly different from corresponding AVA powder at p < 0.05
Fig. 4.
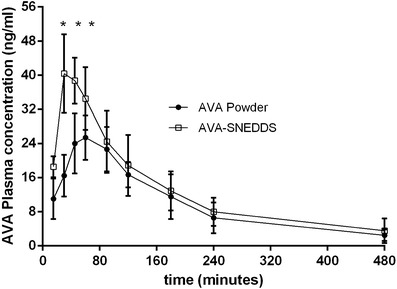
Plasma concentration-time curve of AVA after administration of10 mg/kg oral SNEDDS and 10 mg/kg of powder suspension (n = 6)
DISCUSSION
Nanoemulsions are thermodynamically stable systems and are formed when a particular concentration of oil, Smix, and water are combined. When the concentration of Tween 80 was increased with respect to PG, a higher nanoemulsion region was observed, and about 35% of the dill oil could be solubilized. This may be due to a further reduction in the interfacial tension, thereby increasing the fluidity of the interface; this could result in greater penetration of the oil phase in the hydrophobic region of the Tween 80 monomers (19). The presence of PG as cosurfactant decreases the ending stress of the interface and makes the interfacial film sufficiently flexible to exhibit different curvatures that are required to form nanoemulsion over a wide range (15). However, when the concentration of PG was increased with respect to Tween 80, the nanoemulsion area decreased; this could be due to the formation of a smaller amount of micelles (20).
The NE formulations were then evaluated for their physical stability. There was no sign of phase separation or turbidity observed for all the formulations during their preparation, indicating the formulations were physically stable. Seven formulations (F6–12) passed stability stress tests, including centrifugation and freeze thaw cycles; the five formulation 1–5 became turbid, which may be due to the coagulation of the internal phase which leaded to phase separation, especially that those formulations contain higher percent of dill oil.
Spectroscopic absorbance of the aqueous dispersions of the NEs varied between 0.0030 and 0.0096. The compositions with the lower absorbance values showed the smallest droplet size because aqueous dispersions with small absorbance values are optically clear, and oil droplets are thought to be in a state of finer dispersion (21). All the tested NEs had droplets within the nanosize range (between 65 and 190 nm). The droplet size increased as the concentration of oil in the formulations increased. The droplet size decreased as the concentration of Tween 80 with respect to PG increased at a same dill oil percent. The addition of a surfactant to the NE systems may cause the interfacial film to condense and stabilize, resulting in smaller droplet diameters, whereas the addition of the PG cosurfactant may cause the film to expand (22).
The conductivity measurements (0.122–0.167 ms/cm) indicated that the NEs were of the o/w type, and an increase in the dill oil concentration in the NE resulted in a decrease in conductivity.
The pharmacokinetic study results revealed that preparation of AVA as SNEDDS can significantly modify its pharmacokinetic profile and can increase its bioavailability by more than 1.4-fold in comparison with the pure drug. This was due to the fact that AVA is a lipophilic drug with poor aqueous solubility, and the preparation of this drug as a SNEDDS enhanced its solubility and tissue permeability which leads to shortening in the onset of action, because the onset start in case of powder after 30–45 min when the plasma level reach a certain concentration (20 ng/ml), this concentration was attained after 15 min only in case of AVA-SNEDDS as appears in Fig. 4, this means that SNEDDS solved the problem of delayed onset of action. Also, the pharmacokinetics of drugs upon delivery in nanoemulsion formulation are dictated by the properties of the nanoemulsions rather than by the physicochemical properties of the drug molecules. Additionally, AVA is p-glycoprotein(P-gp) substrate and Tween 80 have inhibition effect on (P-gp) this mean increasing AVA residency time in case of AVA-SNEDDS and explain increasing half life by about (45%) and AUC by about (1.4 folds) as shown in Table II and Fig. 4.
CONCLUSIONS
The formulation of avanafil as self-nanoemulsifying drug delivery system, which is a novel drug delivery system, provided the maximum in vitro drug release. The bioavailability of AVA was enhanced by more than 1.4-fold in relation to the pure powder. The improved SNEDDS formula was composed of 25% dill oil, 55% Tween 80, and 20% propylene glycol. Of course, it will not obviate the need for further clinical evaluation for this novel formula, which may inform clinicians of other important data.
Acknowledgments
Conflict of Interest
The authors state no conflicts of interest.
References
- 1.Sanford M. Avanafil: a review of its use in patients with erectile dysfunction. Drugs Aging. 2013;30:853–62. doi: 10.1007/s40266-013-0112-x. [DOI] [PubMed] [Google Scholar]
- 2.FDA approves Stendra for erectile dysfunction 2014.
- 3.European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP), CHMP assessment report, EMA, 321885, 2013.
- 4.Ahmed OAA, Afouna MI, El-Say KM, Abdel-Naim AB, Khedr A, Banjar ZM. Optimization of self nanoemulsifying systems for the enhancement of in vivo hypoglycemic efficacy of glimepiride transdermal patches. Expert Opin Drug Deliv 2014;11:In press. [DOI] [PubMed]
- 5.Ahmed OAA, Badr-Eldin SM, Tawfik MK, Ahmed TA, El-Say KM, Badr JM. Design and optimization of self-nanoemulsifying delivery system to enhance quercetin hepatoprotective activity in paracetamol-induced hepatotoxicity. J Pharm Sci. 2014;103:602–12. doi: 10.1002/jps.23834. [DOI] [PubMed] [Google Scholar]
- 6.Date AA, Desai N, Dixit R, Nagarsenker M. Self-nanoemulsifying drug delivery systems: formulation insights, applications and advances. Nanomedicine (London) 2010;5:1595–616. doi: 10.2217/nnm.10.126. [DOI] [PubMed] [Google Scholar]
- 7.Dixit RP, Nagarsenker MS. Self-nanoemulsifying granules of ezetimibe: design, optimization and evaluation. Eur J Pharm Sci. 2008;35:183–92. doi: 10.1016/j.ejps.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 8.Janga KY, Jukanti R, Sunkavalli S, Velpula A, Bandari S, Kandadi P, et al. In situ absorption and relative bioavailability studies of zaleplon loaded self-nanoemulsifying powders. J Microencapsul. 2013;30:161–72. doi: 10.3109/02652048.2012.714408. [DOI] [PubMed] [Google Scholar]
- 9.Ren F, Gao Y, Chen J, Jing Q, Yu Y. New self-nanoemulsifying drug delivery system (SNEDDS) with amphiphilic diblock copolymer methoxy poly (ethylene glycol)-block-poly (−caprolactone) Pharm Dev Technol. 2013;18:745–51. doi: 10.3109/10837450.2012.734517. [DOI] [PubMed] [Google Scholar]
- 10.Singh B, Bandyopadhyay S, Kapil R, Singh R, Katare O. Self-emulsifying drug delivery systems (SEDDS): formulation development, characterization, and applications. Crit Rev Ther Drug Carrier Syst. 2009;26:427–521. doi: 10.1615/CritRevTherDrugCarrierSyst.v26.i5.10. [DOI] [PubMed] [Google Scholar]
- 11.Singh B, Khurana L, Bandyopadhyay S, Kapil R, Katare OO. Development of optimized self-nano-emulsifying drug delivery systems (SNEDDS) of carvedilol with enhanced bioavailability potential. Drug Deliv. 2011;18:599–612. doi: 10.3109/10717544.2011.604686. [DOI] [PubMed] [Google Scholar]
- 12.Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12:1561–72. doi: 10.1023/A:1016268311867. [DOI] [PubMed] [Google Scholar]
- 13.Pouton CW. Formulation of self-emulsifying drug delivery systems. Adv Drug Deliv Rev. 1997;25:47–58. doi: 10.1016/S0169-409X(96)00490-5. [DOI] [Google Scholar]
- 14.El Maghraby GM. Self-microemulsifying and microemulsion systems for transdermal delivery of indomethacin: effect of phase transition. Colloids Surf B: Biointerfaces. 2010;75:595–600. doi: 10.1016/j.colsurfb.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Hosny KM, Banjar ZM. The formulation of a nasal nanoemulsion zaleplon in situ gel for the treatment of insomnia. Expert Opin Drug Deliv. 2013;10:1033–41. doi: 10.1517/17425247.2013.812069. [DOI] [PubMed] [Google Scholar]
- 16.Jung J, Choi S, Cho SH, Ghim JL, Hwang A, Kim U, et al. Tolerability and pharmacokinetics of avanafil, a phosphodiesterase type 5 inhibitor: a single- and multiple-dose, double-blind, randomized, placebo-controlled, dose-escalation study in healthy Korean male volunteers. Clin Ther. 2010;32:1178–87. doi: 10.1016/j.clinthera.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 17.Hugger ED, Novak BL, Burton PS, Audus KL, Borchardt RT. A comparison of commonly used polyethoxylated pharmaceutical excipients on their ability to inhibit P-glycoprotein activity in vitro. J Pharm Sci. 2002;91:1991–2002. doi: 10.1002/jps.10176. [DOI] [PubMed] [Google Scholar]
- 18.Türk M, Hils P, Helfgen B, Schaber K, Martin H-J, Wahl MA. Micronization of pharmaceutical substances by the Rapid Expansion of Supercritical Solutions (RESS): a promising method to improve bioavailability of poorly soluble pharmaceutical agents. J Supercrit Fluids. 2002;22:75–84. doi: 10.1016/S0896-8446(01)00109-7. [DOI] [Google Scholar]
- 19.Yuan JS, Ansari M, Samaan M, Acosta EJ. Linker-based lecithin microemulsions for transdermal delivery of lidocaine. Int J Pharm. 2008;349:130–43. doi: 10.1016/j.ijpharm.2007.07.047. [DOI] [PubMed] [Google Scholar]
- 20.Lawrence MJ, Rees GD. Microemulsion-based media as novel drug delivery systems. Adv Drug Deliv Rev. 2000;45:89–121. doi: 10.1016/S0169-409X(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 21.Basalious EB, Shawky N, Badr-Eldin SM. SNEDDS containing bioenhancers for improvement of dissolution and oral absorption of lacidipine. I: development and optimization. Int J Pharm. 2010;391:203–11. doi: 10.1016/j.ijpharm.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Djekic L, Primorac M. The influence of cosurfactants and oils on the formation of pharmaceutical microemulsions based on PEG-8 caprylic/capric glycerides. Int J Pharm. 2008;352:231–9. doi: 10.1016/j.ijpharm.2007.10.041. [DOI] [PubMed] [Google Scholar]