

Abstract
As part of the overall product development and manufacturing strategy, pharmaceutical companies routinely change formulation and manufacturing site. Depending on the type and level of change and the BCS class of the molecule, dissolution data and/or bioequivalence (BE) may be needed to support the change for immediate release dosage forms. In this report, we demonstrate that for certain weakly basic low-solubility molecules which rapidly dissolve in the stomach, absorption modeling could be used to justify a BE study waiver even when there is failure to show dissolution similarity under some conditions. The development of an absorption model for etoricoxib is described here, which was then used to a priori predict the BE outcome of tablet batches manufactured at two sites. Dissolution studies in 0.01 N HCl media (pH 2.0) had demonstrated similarity of etoricoxib tablets manufactured at two different sites. However, dissolution testing at pH 4.5 and pH 6.8 media failed to show comparability of the tablets manufactured at the two sites. Single simulations and virtual trials conducted using the 0.01 N HCl dissolution showed similarity in AUC and Cmax for all tablet strengths for batches manufactured at the two manufacturing sites. These predicted results were verified in a definitive bioequivalence study, which showed that both tablet batches were bioequivalent. Since the development of traditional in vitro–in vivo correlations (IVIVC) for immediate release (IR) products is challenging, in cases such as etoricoxib, absorption modeling could be used as an alternative to support waiver of a BE study.
KEY WORDS: bioequivalence, dissolution, modeling, pharmacokinetics, SUPAC
INTRODUCTION
Formulations and manufacturing changes are common place during drug development. As many companies have adopted fit-for-purpose formulation strategies for early clinical studies such as first-in-human (FIH) (1), in the early stages of development, the formulation is mainly optimized to achieve acceptable bioavailability and stability with less focus on the final product. As drug candidates progress through the development process, additional formulation changes are often considered to facilitate scale up of the formulation and to allow for production of consistent drug product in a commercial manufacturing setting. Finally, post-approval changes also occur such as change of equipment or further refinement of manufacturing process, changes of manufacturing sites, etc. While understanding of the impact of such changes on bioavailability is important throughout all the development stages to ensure that safety and efficacy of the compound is not compromised between studies, this becomes even more critical in late development, as well as after drug approval, when pivotal clinical data have been generated. At that stage, any formulation changes translate to specific regulatory agency requirements and often require the demonstration of bioequivalence between the test and reference formulations. The scale-up and post-approval changes (SUPAC) guidance by the Food and Drug Administration (FDA) addresses in detail the expectations for comparison of formulations based on the level of change that is implemented (2). For immediate release (IR) products, bioequivalence data are required for either significant formulation changes that are expected to have an impact on formulation quality and performance (referred to as level 3 SUPAC changes) as well as for less significant changes (level 2 SUPAC changes) if the necessary dissolution criteria are not met.
Dissolution testing is the primary tool for the evaluation of formulation changes. Thus, dissolution-based drug product release methods are put in place to ensure the consistency of manufacturing processes and sites for the different product batches. However, it is generally acknowledged that a direct link between traditional dissolution methods and in vivo bioavailability is not always possible. When such a link exists and a robust in vitro–in vivo correlation (IVIVC) can be established, dissolution data become a direct surrogate of in vivo dissolution and under certain circumstances can be used to waive bioequivalence studies (3). Development of IVIVCs, however, has traditionally been focused on modified release (MR) formulations, and development of such correlations for IR products is much more challenging. For IR products, the introduction of the Biopharmaceutics Classification System (BCS) (4) and the adoption by regulatory agencies provided a means to link in vitro dissolution data for IR formulations to their in vivo absorption and allowed for the use of dissolution data as surrogates of bioequivalence for BCS I (highly soluble, highly permeable) compounds (5). More recently, the application of biowaivers has been extended to BCS class III (highly soluble, low permeable compounds) (6).
The vast majority of drug compounds that are in development are low-solubility, i.e., BCS class II or IV (7) and thus under current guidelines are not eligible for biowaivers. In the recent years, researches have suggested that it may be possible to expand the concept of BCS biowaivers to certain classes of BCS class II compounds as well (8). For example, it has been suggested that for BCS II weak acids (also termed BCS IIa) that fully and rapidly solubilize in the intestine and for BCS II weak bases (also termed BCS IIb) that fully and rapidly solubilize in the stomach, in vitro dissolution in media of the corresponding “favorable” pH may be used as a surrogate of in vivo bioavailability. In addition, advancements in the physiologically based pharmacokinetic (PBPK) absorption modeling approaches have enabled the translation of in vitro dissolution data to in vivo performance of drug products (9–11). These approaches are now routinely used to guide formulation development particularly in the case of low-solubility molecules. Such models are also helpful to predict bioequivalence study outcomes based on dissolution data, in cases where formulation or manufacturing site changes happen either late in the development process or post-approval of the product. Tubic-Grozdanis et al. used a PBPK-based approach using the GastroPlus software to demonstrate how rapid in vitro dissolution translates to bioequivalence assurance for weak bases and weak acids (8). Tsume et al. utilized similar simulations to demonstrate the applicability of BCS biowaviers to ibuprofen and ketoprofen, two BCS IIa compounds (12). WHO has adopted this recommendation for BCS IIa compounds (13). Similar concepts and use of PBPK models have been used to argue for further modifications to the BCS system including expansion of the solubility criteria for BCS I compounds (14) or the application of the biowaivers for BCS III compounds (15).
Etoricoxib [5-chloro-2-(6-methylpyridin-3-yl)-3-(4-methylsulfonylphenyl) pyridine] is a BCS II weakly basic compound with pH-dependent solubility (pKa of 4.6) (Table I). In a previous publication, Okumu et al. (16) had discussed the development of an oral absorption model in GastroPlus as the basis of a biowaiver argument for etoricoxib formulation. Similar arguments have been made in the literature for other weak bases (8,12). However a detailed validation of the concept against experimental data for formulations with different dissolution rates has not been reported. In this manuscript, we discuss the use of a similar mechanistic absorption model, validated with additional clinical data at different doses as well against clinical data after antacid administration to ensure that the model accurately reflects clinical experience. In addition, we report virtual trial simulations to forecast the bioequivalence study outcome of two batches of Arcoxia tablets from different manufacturing sites that did not meet the multi-media dissolution F2 criteria. The predicted results are compared to the outcome of the definitive bioequivalence study comparing the two tablet batches. We demonstrate that for a weak base like etoricoxib, the complete dissolution in the stomach governing absorption and absorption modeling is a useful tool to predict bioequivalence study outcomes when F2 multi-media dissolution comparisons demonstrate a difference at the higher pH values.
Table I.
Etoricoxib Physicochemical Properties
| Molecular weight = 358.85 |
| Log D = 2.28 (pH 7.0) |
| pKa = 4.5 |
| Caco-2 permeability = 5.23 × 10−5 cm/s |
| Calculated human permeability (from Caco-2 data) = 4.75 × 10−4 cm/s |
| Drug particle density = 1.2 g/mL |
| Precipitation time = 900 s |
| Drug particle size = 40 μm |
| pH solubility profile |
| pH 2.0 (0.01 N hydrochloric acid) = 25.1 mg/mL |
| pH 3.07 (0.1 M glycine buffer) = 2.01 mg/mL |
| pH 3.54 (0.1 M glycine buffer) = 0.7 mg/mL |
| pH 4.01 (0.1 M sodium acetate buffer) = 0.3 mg/mL |
| pH 4.54 (0.1 M sodium acetate buffer) = 0.14 mg/mL |
| pH 5.03 (0.1 M sodium acetate buffer) = 0.09 mg/mL |
| pH 5.47 (0.1 M sodium acetate buffer) = 0.08 mg/mL |
| pH 6.9 (water) = 0.05 mg/mL |
MATERIALS AND METHODS
Etoricoxib Physicochemical Properties
The key physicochemical properties of etoricoxib used to build the model are summarized in Table I. Etoricoxib is a weak base exhibiting pH-dependent solubility with high solubility at pH <3. Due to the lower solubility at the pH 4–7 range, etoricoxib is classified as a low-solubility compound. When dosed orally, etoricoxib is completely and rapidly absorbed, with an oral bioavailability of up to 100% (17). Hence, it is classified as a BCS class II molecule.
Dissolution Tests
Dissolution of 30, 60, 90, and 120 mg etoricoxib, immediate-release tablets were conducted in 0.01 N HCl (pH 2.0) as well as in pH 4.5 (50 mM sodium acetate buffer) and pH 6.8 (50 mM potassium phosphate buffer) media. The dissolution was conducted in a USP-2 apparatus using 900 mL of medium at 50 rpm, and temperature was maintained at 37°C. At pre-determined time intervals, samples were drawn from the dissolution vessel, filtered, and analyzed in HPLC. The mean dissolution data of the etoricoxib tablets are summarized in Figs. 1 and 2 and in Table II.
Fig. 1.
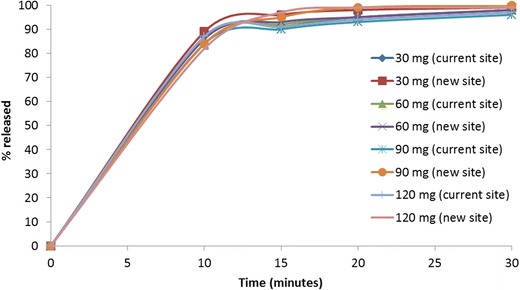
Mean dissolution data in 0.01 N HCl medium comparing the four etoricoxib tablet strengths manufactured at the current and new sites
Fig. 2.
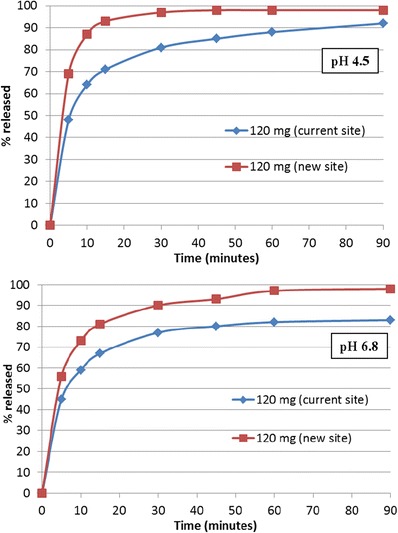
Mean dissolution data in pH 4.5 and pH 6.8 medium comparing the 120 mg etoricoxib tablet strengths manufactured at the current and new sites
Table II.
Dissolution of 120 mg Etoricoxib Tablets in 0.01 N HCl
| Time (min) | % Dissolution | ||
|---|---|---|---|
| Dissolution data used to model study protocol numbers 43 and 48 | Dissolution data used to model study protocol number 70 | ||
| Tablet MR-4312 |
Tablet MR-4629 |
||
| 10 | 90 | 90 | 87 |
| 15 | 96 | 96 | 94 |
| 20 | 98 | 98 | 96 |
| 30 | 100 | 100 | 99 |
Development of an Oral Absorption Model for Etoricoxib
All simulations were conducted using GastroPlus™ v6.1 and v8.5 using the human fasted physiological model, which was slightly modified to fit the observed human data (details are described below). The relevant inputs in the model are described here:
Etoricoxib Physicochemical Properties
Etoricoxib properties used in building the model are summarized in Table I.
Dissolution Data Input
The dissolution data for the corresponding clinical batches shown in Table II were used to simulate the PK profiles and PK parameters shown in Fig. 3, Tables III and IV. These dissolution data were primarily used for building and validating the etoricoxib absorption model against historical data. The dissolution data for the 30-, 60-, 90-, and 120-mg tablets in 0.01 N HCl (pH 2.0) shown in Fig. 1 were used to simulate the data in Fig. 4 and Tables VI and VII. Dissolution data generated in pH 4.5 and pH 6.8 media for the 120-mg strength tablets (Fig. 2) were used as input to simulate the data shown in Table VIII. To allow for a more mechanistic modeling of the dissolution process in the gastrointestinal (GI) tract, instead of incorporation of the dissolution curve directly in the simulation, the in vitro dissolution data were fit in GastroPlus™ using the built in Johnson dissolution model to obtain a representative diffusion coefficient value. The resulting diffusion coefficient (e.g., approximately 0.02 and 3 × 10−5 cm2/s at pH 2.0 and pH 6.8, respectively, for 120-mg tablet), which was used in subsequent simulations, represents a correction value taking into account the contribution of formulation to the release rate over the simple drug particle-based dissolution. In order to do the dissolution fitting, a separate drug record was created and the reference solubility and pH in the compound tab were adjusted as per the dissolution condition. In the physiology tab, the stomach pH was adjusted to the pH of the dissolution media, e.g., pH 2.0 and the volume was changed to 900 mL. Subsequently, a simulation was run for 30 or 90 min and the simulated amount dissolved was compared to the in vitro dissolution data; the diffusion coefficient was changed iteratively to match the simulated amount dissolved and the in vitro dissolution data.
Fig. 3.
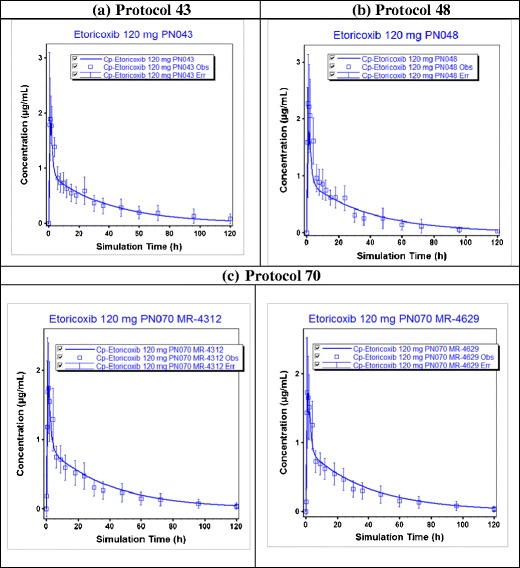
Predicted (line) and observed (squares) pharmacokinetic profiles of etoricoxib at a dose of 120 mg for three clinical studies a protocol number 43, b protocol number 48, and c protocol number 70 (for this study, predicted and observed data of two different tablet batches MR-4312 and MR-4619 are shown). All the observed clinical data are on Merck file
Table III.
Mean Observed and Predicted AUC0–120 h and C max for 120 mg Etoricoxib Tablets for Three Clinical Studies
| Clinical study | AUC0–120 h (μg*h/mL) | C max (μg/mL) | ||||
|---|---|---|---|---|---|---|
| Observed | Predicted | % PE | Observed | Predicted | % PE | |
| Protocol 43 | 39.5 | 35.8 | −9.4 | 1.88 | 1.83 | −2.7 |
| Protocol 48 | 36.7 | −2.5 | 2.27 | −19.4 | ||
| Protocol 70 | 32.9 (MR-4312) | 8.8 | 1.74 (MR-4312) | 5.2 | ||
| 33.5 (MR-4629) | 6.9 | 1.72 (MR-4629) | 6.4 | |||
For protocol number 70, predicted and observed data for two different tablet batches MR-4312 and MR-4619 are shown. The % prediction errors (% PE) for these simulations were calculated as ((predicted–observed)/observed) × 100. All the observed clinical data are on Merck file
AUC area under curve, C max maximum plasma concentration
Table IV.
Mean Observed and Predicted AUC0–∞ and C max for Etoricoxib at 30- and 60-mg Doses
| Dose (mg) | AUC0–∞ (μg*h/mL) | C max (μg/mL) | ||||
|---|---|---|---|---|---|---|
| Observed | Predicted | % PE | Observed | Predicted | % PE | |
| 30 | 9.08 | 9.39 | 3.4 | 0.53 | 0.49 | −7.5 |
| 60 | 18.57 | 18.77 | 1.1 | 1.27 | 0.99 | −22 |
The % prediction errors (% PE) for these simulations were calculated as ((predicted–observed)/observed) × 100. The observed data were taken from reference (17)
AUC area under curve, C max maximum concentration
Fig. 4.
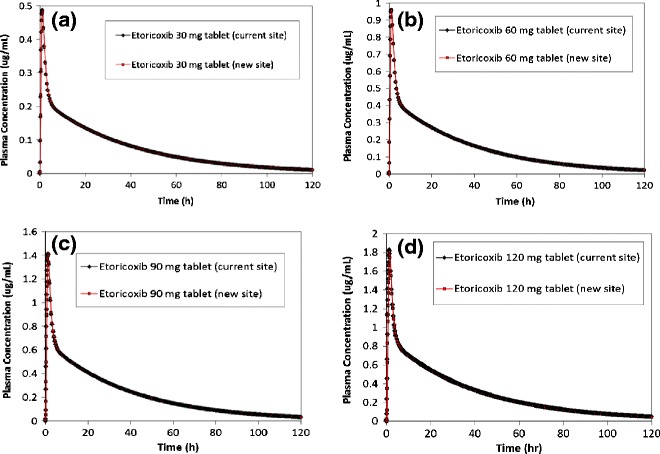
Predicted plasma concentration vs. time profiles for a 30, b 60, c 90, and d 120 mg etoricoxib tablets manufactured at the current site and new site
Table VI.
Predicted AUC0–120 h and C max for 30, 60, 90, and 120 mg Etoricoxib Tablets Manufactured at the Current Site and the New Site
| Tablets (mg) | AUC0–120 h (μg*h/mL) | C max (μg/mL) |
|---|---|---|
| 30 (current site) | 8.96 | 0.49 |
| 30 (new site) | 8.95 | 0.48 |
| 60 (current site) | 17.9 | 0.95 |
| 60 (new site) | 17.8 | 0.95 |
| 90 (current site) | 26.8 | 1.39 |
| 90 (new site) | 27.1 | 1.41 |
| 120 (current site) | 35.4 | 1.77 |
| 120 (new site) | 35.8 | 1.79 |
AUC area under curve, C max maximum concentration
Table VII.
Predicted AUC0–120 h and C max from Virtual Trial Simulations for 120 mg Etoricoxib Tablets Manufactured at the Current Site and the New Site, Using Dissolution in 0.01 N HCl
| AUC0–120 h (% CV) | C max (% CV) | Relative AUC0–120 h |
Relative C max |
|
|---|---|---|---|---|
| 120 mg (current site) |
35.9 (15.8%) | 1.81 (14.8%) | – | – |
| 120 mg (new site) |
37.1 (15.3%) | 1.85 (14.4%) | 1.03 | 1.02 |
AUC area under curve, C max maximum concentration, % CV coefficient of variation
Table VIII.
Predicted AUC0–120 h and C max from Virtual Trial Simulations for 120 mg Etoricoxib Tablets Manufactured at the Current Site and the New Site, Using Dissolution in pH 4.5 and pH 6.8 Medium
| AUC0–120 h (% CV) | C max (% CV) | Relative AUC0–120 h | Relative C max | |
|---|---|---|---|---|
| Dissolution in pH 4.5 | ||||
| 120 mg (current site) | 34.4 (16.3%) | 1.65 (15.3%) | – | – |
| 120 mg (new site) | 35.8 (15.3%) | 1.82 (14.4%) | 1.04 | 1.10 |
| Dissolution in pH 6.8 | ||||
| 120 mg (current site) | 30.8 (17.2%) | 1.50 (18.6%) | – | – |
| 120 mg (new site) | 34.1 (15.1%) | 1.71 (19.1%) | 1.11 | 1.14 |
AUC area under curve, C max maximum concentration, % CV coefficient of variation
Physiology
The default human fasted physiological model in GastroPlus™ v6.1 and v8.5 was slightly modified to increase the absorption scale factors (ASF) in duodenum (ASF changed to 3.794 from the default value of 2.794) and jejunum 1 (ASF changed to 3.750 from the default value of 2.750) to fit the observed clinical data from protocol 43 (Merck data on file) as described in the “RESULTS” section. These changes are considered within possible ranges for in vivo deviation of parameters solely relying on in vitro measurements. The ASF values help translate the projected permeability value to an absorption rate constant; in this case, initial permeability estimates obtained based on Caco-2 data suggesting that in vivo permeability may be slightly higher. For the simulation of PK in the presence of antacids, the stomach pH was set at 3.0 (18,19). All other gastrointestinal compartmental data (ASF, pH, transit time, and fluid volumes) were used as default in the human fasted physiological model.
Pharmacokinetic (PK) Parameters
Human PK parameters were estimated by fitting the individual subject IV data at a 25-mg dose from protocol number 43 (Merck data on file) in WinNonLin™ v5.2 to a two-compartment model. The mean PK parameters used in these simulations were CL = 3.19 L/h ((percent coefficient of variation) % CV = 44%), Vc = 35.49 L (% CV = 21%), k12 = 0.62 1/h (% CV = 80%), and k21 = 0.28 1/h (% CV = 80%).
Virtual Bioequivalence Trials.
Virtual trials were conducted in 36 healthy subjects randomly selected by GastroPlus™ in a crossover design to compare bioperformance of the tablets manufactured at the two sites. The default population in GastroPlus was used in these simulations. However, the mean values and % CV for parameters specific to etoricoxib such as dose, permeability, solubility, drug particle size, fraction unbound in plasma, and the PK parameters were changed to the measured values, as listed in the previous sections and in Table I. These simulations allowed the assessment of the combined effects of variations in population physiology and formulation variables, thus enabling assessment of a bioequivalence study outcome.
Definitive Bioequivalence (BE) Study
The BE study was an open-label, 2-period, randomized, crossover study in 24 healthy male and female subjects. In each period, subjects received either treatment A (120 mg etoricoxib tablet, manufactured at the new site) or treatment B (120 mg etoricoxib tablet, manufactured at the current site) as a single dose following an overnight fast of at least 10 h in a randomized order. There was a minimum of 7 days between dosing in each treatment period. Blood samples were collected at pre-determined time points for up to 120 h post dosing. Samples were analyzed for etoricoxib using a validated LC-MS/MS method. The study was conducted in accordance with the principles of good clinical practice and was approved by the appropriate institutional review board (IRB) and regulatory agency. All subjects provided written informed consent before any screening activity.
RESULTS
Etoricoxib Oral Absorption Model Development and Assessment of Model Performance
The input parameters listed in the “MATERIALS AND METHODS” section were used to build the absorption model for etoricoxib to simulate the fasted state exposure of etoricoxib in human following administration of a single oral dose (120 mg). The predicted mean plasma concentration profiles, AUC, and Cmax from three clinical studies were then compared to the observed data (Fig. 3 and Table III). Data from protocol number 43 (a crossover absolute bioavailability study using 120-mg oral dose and 25-mg IV dose) (Merck data on file) were primarily used to assess the performance of the model in predicting the 120-mg oral data. Due to the crossover nature of this study, any lack of prediction of the observed oral data due to PK variability can be ruled out, and hence, that study provides the most accurate validation of the absorption model. Additional simulations at 30- and 60-mg doses were conducted to further assess the robustness of the model. The predicted and observed data (17) are summarized in Table IV. As can be seen from these simulations and the % prediction errors, the model predicts the observed data with reasonable accuracy for all the clinical studies simulated here. The comparison of bioperformance of the oral tablet to oral solution was assessed to evaluate whether dissolution in stomach (i.e., at low pH) was key for adequate bioperformance of etoricoxib tablets. Comparison of 120-mg oral tablet to that with an oral solution showed similar pharmacokinetic profiles indicating that there was no difference in the absorption of tablet and solution formulations (assuming gastric emptying times were not different for the solution and the tablet formulation) (data not shown). Based on these simulations, it was concluded that this model using the 0.01 N HCl dissolution data could be used to predict the fasted state bioperformance of the tablets manufactured in the current site and those manufactured in the proposed new site.
In order to further assess the robustness of the model, the impact of stomach pH on etoricoxib PK was simulated and compared to previously reported results (20). These simulations were conducted at 120-mg dose under normal stomach pH of 1.3 and stomach pH of 3.0, which mimics gastric pH in the presence of antacid (18,19). These simulations (Table V) demonstrated that stomach pH of 3.0 has minimal effect on etoricoxib AUC and Cmax. These predictions were in agreement with previously published report, where the authors had reported that co-dosing with antacids had no effect on etoricoxib PK (20). The calculated dose number (dose/solubility/250 mL) (21) of 0.24 at pH 3 suggests that the whole etoricoxib dose (120 mg) will be completely soluble even at the higher stomach pH. This supports the observed and predicted results showing a lack of effect of antacid on etoricoxib PK.
Table V.
Predicted AUC0–120 h and C max for Etoricoxib Tablet at 120 mg Under Normal Stomach pH (1.3) and High Stomach pH (3.0) Conditions
| Stomach pH | AUC0–120 h (μg*h/mL) | C max (μg/mL) |
|---|---|---|
| 1.3 | 35.8 | 1.83 |
| 3.0 | 35.3 | 1.81 |
AUC area under curve, C max maximum concentration
Prediction of Bioequivalence of Etoricoxib Tablets Manufactured at Two Manufacturing Sites
The formulation bioperformance of etoricoxib tablets manufactured at the current and new manufacturing sites were simulated using their respective 0.01 N HCl dissolution data (as shown in Fig. 1). The predicted mean plasma concentration profiles and PK parameters for 30-, 60-, 90-, and 120-mg tablet strengths are shown in Fig. 4 and Table VI, respectively. Subsequently, virtual trial simulations were conducted at a dose of 120 mg in 36 healthy subjects to assess the outcome of a bioequivalence study comparing the tablets manufactured at the two sites. The virtual trial results (Table VII) demonstrate that the 120-mg tablets manufactured at the two sites are expected to be bioequivalent. Since the highest strength tablets, i.e., 120 mg, are predicted to be bioequivalent, it can be expected that the 30-, 60-, and 90-mg tablets manufactured at the two sites would also show a similar bioperformance as all these tablet strengths are weight multiples of each other. Based on the results from the single simulations and virtual trials, it can be concluded that the tablets manufactured at the two sites are expected to be bioequivalent, as was later confirmed in the definitive bioequivalence clinical study.
Simulations Using Tablet Dissolution Data in pH 4.5 and pH 6.8 Medium
The dissolution data for the 120 mg etoricoxib tablets at pH 4.5 and pH 6.8 are shown in Fig. 2. F2 values calculated using these dissolution profiles were 35 and 44, respectively, at pH 4.5 and pH 6.8, which suggested small differences in dissolution of the tablets manufactured at the two sites. Similar dissolution differences were also observed for the other tablet strengths—with F2 of 35 and 40 for 60 mg, and 34 and 38 for 90 mg, at pH 4.5 and pH 6.8, respectively. To investigate whether the dissolution differences observed for the 120-mg tablets would have any effect on bioequivalence outcome, virtual trial simulations were conducted. These simulation results (Table VIII) suggest that the AUC and Cmax of the tablets manufactured at the new site would trend higher than the tablets manufactured at the current site, with especially Cmax potentially deviating more than 10%. While the predicted difference was still moderate, it is clear that the predictions based on these media do not accurately capture the stomach solubilization and does not fully reflect the outcome of the clinical study where tablets were shown to be bioequivalent.
Bioequivalence (BE) Study Outcome
The BE study results are summarized in Table IX. The results show that the two tablets met the study hypothesis that the 90% confidence interval of the true GMRs for AUC and Cmax for etoricoxib after dosing of 120-mg tablets from two different manufacturing sites will be contained within 0.80 and 1.25. Hence, based on these results, the two tablets are considered to be bioequivalent.
Table IX.
Bioequivalence Study Results and Statistical Analysis of the Data Comparing Etoricoxib Tablets Manufactured at Two Sites at a Dose of 120 mg
| PK parameters | Treatment | Geometric mean ratio (A vs. B) | 90% confidence internal (A vs. B) | |
|---|---|---|---|---|
| A | B | |||
| AUC0–∞ (μg*h/mL)1 | 32.3 ± 13.1 | 32.1 ± 14.6 | 1.01 | 0.97, 1.06 |
| C max (μg/mL)1 | 1.94 ± 0.47 | 1.98 ± 0.41 | 0.97 | 0.89, 1.06 |
| T max (h) 2 | 1.25 (0.5–2.0) | 1.00 (0.5–4.0) | – | – |
Treatment A tablets manufactured at the new site, Treatment B tablets manufactured at the current site, PK parameters pharmacokinetic parameters
aAUC and C max reported as geometric mean ± SD
b T max reported as median with range
DISCUSSION
As part of the overall product development and manufacturing strategy, pharmaceutical companies routinely change formulation and manufacturing site. Depending on the type and level of change, dissolution can be used as a surrogate for BE studies to support the change for IR dosage forms (2). For example, level 2 SUPAC changes in components and composition can be supported by dissolution comparison on different media, dictated by the BCS class of the molecule. For BCS II molecules, dissolution similarity in water, 0.1 N HCl, USP media pH 4.5, pH 6.5 and pH 7.5 should be shown to justify waiver for a BE study. Level 3 components and composition changes generally require bioequivalence studies unless a biowaiver can be granted either due to BCS classification (i.e., BCS I or BCS III) or the existence of an acceptable IVIVC. For this level of change, low-solubility molecules typically would require a BE study to support the change. However, in the case of manufacturing site changes of immediate-release dosage forms, all levels of changes can be supported by showing dissolution similarity in filed the dissolution method, irrespective of BCS classification. In this case, BE study will be needed only if differences are observed in the dissolution data. It should also be noted that although the FDA SUPAC guidance is cited in this manuscript for discussion, specific health authority guidances may differ across different regions where a product is registered and may dictate additional testing.
In this paper, the development of an absorption model for etoricoxib is described to a priori predict the bioequivalence (BE) outcome of tablet batches manufactured at two sites. Based on the proposed manufacturing site change, this would qualify as a level 3 SUPAC change (2). Additionally, given the F2 failure at higher pH dissolution, BE study was needed to support the manufacturing site change for etoricoxib tablets by the corresponding health authority in the country of registration. However, we demonstrate here that for certain weakly basic low-solubility molecules which rapidly dissolve in the stomach, absorption modeling could be used to justify a BE study waiver. Etoricoxib is a weak base exhibiting pH-dependent solubility with high solubility at pH <3. Due to the lower solubility at the pH 4–7 range (0.05 mg/mL in water), etoricoxib is classified as a BCS II molecule. Etoricoxib tablets exhibit very fast and complete dissolution in pH 2.0 media while dissolution, as expected, is slower at pH 4.5 and pH 6.8 (Figs. 1 and 2). The lower solubility at pH 4.5 and 6.8 also resulted in different dissolution profiles for tablets manufactured at different sites (tablets did not meet the F2 criteria for dissolution similarity at either pH 4.5 or 6.8).
Given the high solubility at lower pH, it is anticipated that absorption and bioavailability of fast-dissolving formulations will be dictated by the initial dissolution in the normal stomach environment (pH < 3). This is supported by the high absolute bioavailability of etoricoxib (∼100%) indicating no solubility limitations to absorption (17). Furthermore, in a previously published report, Okumu et al. (16) has demonstrated that the dissolution of etoricoxib in the stomach is the determining factor for bioavailability by the use of carefully designed in vitro studies to probe intestinal precipitation coupled with absorption modeling was provided. Here, we have adopted the etoricoxib absorption model, validated it further against data across multiple studies and for different tablet strengths potencies, and applied it to the prediction of bioequivalence of the tablet batches that failed F2 similarity in the dissolution comparison.
As described in the methods section, the etoricoxib model was built in GastroPlus™ using the available dissolution data for etoricoxib tablets in pH 2.0 medium and the predictions were compared to the observed data from several previous clinical studies and at three different doses—30, 60, and 120 mg (Fig. 3, Tables III and IV). These simulations were conducted to include the top etoricoxib dose (120 mg) such that the whole dose range, i.e., 30–120 mg is covered and to provide validation of the predictions against a wider dataset of historical clinical studies. Based on these results, the model was deemed to be robust enough for the prediction of BE study outcome for the etoricoxib tablets manufactured at two different sites. Simulations conducted to compare etoricoxib PK for the tablets manufactured at the two sites predicted that tablets would have similar bioperformance for all four strengths (30, 60, 90, and 120 mg) (Fig. 4 and Table VI). Subsequently, virtual trial simulations were conducted to assess bioequivalence between the 120-mg tablets from the two sites using the dissolution data in 0.01 N HCl. These simulations predicted that etoricoxib tablets would be bioequivalent (Table VI). The outcome of these simulations were verified by the BE study results (Table IX), which demonstrated that the 120-mg tablets manufactured at the two sites were bioequivalent.
Several additional simulations were conducted in order to mechanistically investigate whether dissolution in the stomach (i.e., dissolution at low pH) was the key variable to achieve adequate bioperformance of the tablets and that dissolution F2 failure at the higher pH (pH 4.5 and 6.8) were not relevant to the tablet performance in vivo in fasted state. First, simulations conducted to compare PK of the tablets with that of an oral solution showed that the PK profiles were similar for the tablets and the solution, thus indicating that the tablet formulation (even at the highest strength, 120 mg) would undergo rapid dissolution at the normal stomach pH and minimal precipitation in the small intestine, if any. Thus, the primary driver to achieve adequate exposure from the tablets is to ensure rapid dissolution at the stomach pH. These predictions are also in agreement with the in vitro precipitation data reported by Okumu et al. (16), where the authors have shown that etoricoxib dissolved in simulated gastric fluid (SGF) and then transferred to fasted state simulated intestinal fluid (FaSSIF) showed no precipitation in FaSSIF even though etoricoxib is a weak base. Second, simulations to predict the impact of higher gastric pH conditions, e.g., co-dosing with antacid, on PK of the etoricoxib tablets demonstrated that the pH of 3.0 would not have any negative impact of bioperformance of the tablets. These simulated results were also in agreement with the pH solubility profile of etoricoxib (i.e., complete dissolution of 120-mg dose at pH 3.0) and with previous human PK studies which had shown minimal effect on AUC when etoricoxib was co-dosed with antacids (calcium carbonate suspension or magnesium–aluminum hydroxide suspension) (20). Finally, this model was also able to reasonably predict the effect of food on etoricoxib PK. Agrawal et al. (17) had reported that at 120-mg dose, high-fat meal had no effect on etoricoxib AUC (fed/fasted GMR = 0.97) but had reduced Cmax (fed/fasted GMR = 0.64) and median Tmax shifted from 1 h for fasted to 3 h for fed. In agreement with the observed data, this absorption model predicted AUC ratio of 0.92 (fed/fasted), Cmax ratio of 0.58 (fed/fasted), and Tmax of 1 h (fasted) vs. 2.8 h (fed). In this model, the gastric emptying time was increased to 2 h (from GastroPlus default value of 1 h) to better mimic high-fat and high-calorie meal conditions. The effect of food on the rate of absorption of etoricoxib is most likely due to the high stomach pH under fed condition (GastroPlus fed human physiology stomach pH = 4.9), which will impact the dissolution rate of etoricoxib tablets due to solubility limitation at that pH.
We further conducted simulations using dissolution data at pH 4.5 and pH 6.8 for the two batches. The simulations indicated that there could be slight differences in PK for the two tablets at 120 mg, with greater predicted PK difference when using pH 6.8 dissolution data that could result in 11% difference in AUC and 14% difference in Cmax. However, these projected differences did not materialize during clinical testing as the two tablets were found to be bioequivalent with a geometric mean ratio (GMR) of 1.01 and 0.97 for AUC and Cmax, respectively. Thus, the outcome of the BE study appears more in line with the pH 2.0 dissolution-based simulations compared to the 4.5 and pH 6.8 dissolution-based simulations, further providing support to the argument that in fasted state, stomach dissolution is more critical for the product performance for etoricoxib.
The data presented here demonstrate that for BCS class II weak bases such as etoricoxib, which are expected to have complete dissolution in the acidic environment of the stomach and also maintain supersaturation in the small intestine, absorption simulation can be used to justify waiver of BE study. Although in this case a BE study was conducted to satisfy the relevant health authority guidances, the absorption simulation results show that this type of modeling could be used as an alternative for IVIVC to support the in vivo bioequivalence of the etoricoxib IR tablets.
CONCLUSION
Dissolution studies of etoricoxib tablets manufactured at two different sites showed similarity for 30-, 60-, 90-, and 120-mg tablet strengths in 0.01 N HCl media (pH 2.0). However, dissolution testing at pH 4.5 and pH 6.8 media failed to show comparability of the tablets manufactured at the two sites, primarily due to poor aqueous solubility of etoricoxib at pH >3.0. In order to investigate the impact of dissolution differences at high pH on the bioequivalence of the tablets, an absorption model for etoricoxib was built in GastroPlus™ and was validated against several previous clinical studies. The simulations presented here demonstrate that dissolution in 0.01 N HCl media is the most relevant to assess the bioperformance of the etoricoxib tablets. Since etoricoxib is highly soluble in pH 2.0 (25.1 mg/mL), so even the highest strength tablet (120 mg) will be completely soluble in normal gastric pH conditions. Furthermore, previous publication had demonstrated that the solubilized etoricoxib does not precipitate out in the intestinal pH. Single simulations and virtual trials conducted using the 0.01 N HCl dissolution showed similarity in AUC and Cmax for all tablet strengths for batches manufactured at the two manufacturing sites. These predicted results were verified in a definitive bioequivalence study, which showed that both tablet batches were bioequivalent. Since development of traditional in vitro–in vivo correlations for immediate release (IR) products is challenging, in cases such as etoricoxib, absorption modeling could be used as an alternative to IVIVC to support waiver of in vivo bioequivalence of different formulation batches.
Acknowledgments
The authors would like to thank our colleagues from the analytical group for generating the dissolution data and colleagues from pharmacokinetics group for sharing the clinical data.
References
- 1.Wu Y and Kesisoglou F, Immediate release oral dosage forms: formulation screening in the pharmaceutical industry. In Dressman J, Reppas C, editors. Oral drug absorption: prediction and assessment, 2nd Ed, Informa Healthcare; 2010. p. 296–337.
- 2.Guidance for industry. Immediate release solid oral dosage forms. Scale-up and postapproval changes: chemistry, manufacturing, and controls, in vitro dissolution testing, and in vivo bioequivalence documentation. Food Drug Adm. 1995.
- 3.Lu Y, Kim S, Park K. In vitro-in vivo correlation: perspectives on model development. Int J Pharm. 2011;418:142–8. doi: 10.1016/j.ijpharm.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutics drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–20. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 5.Guidance for industry: waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. Food Drug Adm.
- 6.Guideline on the investigation of bioequivalence. Committee for medicinal products for human use. Eur Med Agency. 2010.
- 7.Benet LZ, Wu CY. Using a biopharmaceutics drug disposition classification system to predict bioavailability and elimination characteristics of new molecular entities. Somerset: NJDMDG; 2006. [Google Scholar]
- 8.Tubic-Grozdanis M, Bolger MB, Langguth P. Application of gastrointestinal simulation for extensions for biowaivers of highly permeable compounds. AAPS J. 2008;10(1):213–26. doi: 10.1208/s12248-008-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kostewicz ES, Aarons L, Bergstrand M, Bolger MB, Galetin A, Hatley O, Jamei M, Lloyd R, Pepin X, Rostami-Hodjegan A, Sjögren E, Tannergren C, Turner DB, Wagner C, Weitschies W, Dressman J. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2013 [Epub ahead of print]. [DOI] [PubMed]
- 10.Huang S-M, Abernethy DR, Wang Y, Zhao P, Zineh I. The utility of modeling and simulation in drug development and regulatory review. J Pharm Sci. 2013;102(9):2912–23. doi: 10.1002/jps.23570. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Lionberger RA, Davit BM, Yu LX. Utility of physiologically based absorption modeling in implementing quality by design in drug development. AAPS J. 2011;13(1):59–71. doi: 10.1208/s12248-010-9250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsume Y, Langguth P, Garcia-Arieta A, Amidon GL. In silico prediction of drug dissolution and absorption with variation in intestinal pH for BCS class II weak acid drugs: ibuprofen and ketoprofen. Biopharm Drug Dispos. 2012;33(7):366–77. doi: 10.1002/bdd.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Proposal to waive in vivo bioequivalence requirements for the WHO model list of essential medicines immediate release, solid oral dosage forms. World Health Organization. http://www.who.int/medicines/services/expertcommittees/pharmprep/QAS04_109Rev1_Waive_invivo_bioequiv.pdf
- 14.Fagerholm U. Evaluation and suggested improvements of the biopharmaceutics classification system (BCS) J Pharm Pharmacol. 2007;59(6):751–7. doi: 10.1211/jpp.59.6.0001. [DOI] [PubMed] [Google Scholar]
- 15.Tsume Y, Amidon GL. The biowaiver extension for BCS class III drugs: the effect of dissolution rate on the bioequivalence of BCS class III immediate-release drugs predicted by computer simulation. Mol Pharm. 2010;7(4):1235–43. doi: 10.1021/mp100053q. [DOI] [PubMed] [Google Scholar]
- 16.Okumu A, DiMaso M, Lobenberg R. Computer simulations using GastroPlus to justify a biowaiver for etoricoxib solid oral drug products. Eur J Pharm Biopharm. 2009;72:91–8. doi: 10.1016/j.ejpb.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 17.Agrawal NGB, Porras AG, Matthews CZ, Rose MJ, Woolf EJ, Musser BJ, et al. Single- and multiple-dose pharmacokinetics of etoricoxib, a selective inhibitor of cyclooxygenase-2, in man. J Clin Pharmacol. 2003;43:268–76. doi: 10.1177/0091270003251122. [DOI] [PubMed] [Google Scholar]
- 18.Decktor DL, Robinson M, Gottlieb S. Comparative effects of liquid antacids on esophageal and gastric pH in patients with heartburn. Am J Ther. 1995;2(7):481–6. doi: 10.1097/00045391-199506000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Monés J, Carrio I, Sainz S, Berná L, Clavé P, Liszkay M, et al. Gastric emptying of two radiolabelled antacids with simultaneous monitoring of gastric pH. Eur J Nucl Med. 1995;22(10):1123–8. doi: 10.1007/BF00800593. [DOI] [PubMed] [Google Scholar]
- 20.Schwartz JI, Agrawal NGB, Kher UA, DeSmet M, Cavanaugh PF, Guillaume M, et al. Lack of effect of antacids on single-dose pharmacokinetics of etoricoxib. J Clin Pharmacol. 2007;47:1342–6. doi: 10.1177/0091270007304777. [DOI] [PubMed] [Google Scholar]
- 21.Oh D-M, Curl R, Amidon G. Estimating the fraction of dose absorbed from suspensions of poorly soluble compounds in humans: a mathematical model. Pharm Res. 1993;10:264–70. doi: 10.1023/A:1018947113238. [DOI] [PubMed] [Google Scholar]