

Abstract
Dissolution testing is a performance test for many dosage forms including tablets and capsules. The objective of this study was to evaluate if computer simulations can predict the in vitro dissolution of two model drugs for which different dissolution data were available. Published montelukast sodium and glyburide dissolution data was used for the simulations. Different pharmacopeial and biorelevant buffers, volumes, and rotations speeds were evaluated. Additionally, a pH change protocol was evaluated using these buffers. DDDPlus™ 3, Beta version (Simulation Plus, Inc.), was used to simulate the in vitro dissolution data. The simulated data were compared with the in vitro data. A regression coefficient between predicted and observed data was used to assess the simulations. The statistical analysis of Montelukast sodium showed that there was a significant correlation between the in vitro release data and the predicted data for all cases except for one buffer. For glyburide, there was also a significant correlation between the experimental data and the predicted data using single pH conditions. Using the dynamic pH protocol, a correlation was significant for one biorelevant media. The simulations showed that both in vitro drug releases were sensitive to solubility effects which confirmed their BCS class II category. Computer simulations of the in vitro release using DDDPlus™ have the potential to estimate the in vivo dissolution at an early stage in the drug development process. This might be used to choose the most appropriate dissolution condition to establish IVIVC and to develop biorelevant in vitro performance tests to capture critical product attributes for quality control procedures in quality by design environments.
KEY WORDS: biorelevant media, DDDPlus, dissolution testing, QbD, QC
INTRODUCTION
Drug absorption depends on physiological factors and a drug’s physiochemical properties to dissolve and to get absorbed. Dissolution from a dosage form depends both on the solubility of the drug by itself and the release pattern of the dosage form in which it is administered (1). A drug’s solubility is determined by many factors, such as molecular weight, lipophilicity, crystal or amorphous state, and pKa (2–4). Studies showed that a poor drug solubility might lead to limited drug absorption, but formulation processes can overcome such issues if the dissolution mechanism is understood (5–8). The Biopharmaceutics Classification System (BCS) classifies drugs according to their solubility and permeability to four classes. Class 1 and 3 drugs have high solubility, whereas class 2 and 4 have low solubility (8).
Drug dissolution testing is used both in the early and the late stages of drug development for many dosage forms, including tablets and capsules (9). In the early drug development stage, dissolution test helps researchers to find the best formulation to tailor the in vitro behavior of oral dosage form to the desired profile (1,6–8,10). Later, dissolution profiles can be used to establish an in vitro/in vivo correlation (IVIVC) which can reduce the need for costly bioequivalence studies (6,11).
In the final stage of drug development, the dissolution testing is used for quality control, that is, to test batch to batch consistency, stability, and to detect manufacturing defects which might lead to the rejection of an entire lot. Dissolution tests are required by the US Food and Drug Administration (FDA) and listed in the US Pharmacopeia (USP) as an in vitro performance test. The dissolution test was introduced into the USP over 50 years ago and is described in detail in chapter <711> (6,12,13).
Early estimation of dissolution behavior and investigation of the influence of formulation factors on solubility is essential for pharmaceutical formulation development (1,2,13). Using In silico methods in the drug discovery stage to estimate drug dissolution can eliminate many trial and error experiments and save time and money (2–5). The objective of this study was to evaluate the performance of computer simulations that predict the in vitro dissolution of two model drugs.
METHOD
Montelukast
Published montelukast dissolution data was used for the simulations (14). In vitro tests were performed in the USP apparatus II (paddle) at 37 ± 0.5°C in 900 mL with a rotation of 75 or 100 rpm. Four different biorelevant media were used for the test: USP simulated intestinal fluid (SIF), pH 6.8 buffer, blank fasted state simulated intestinal fluid (BFaSSIF), and fasted state simulated intestinal fluid (FaSSIF) (14). The FaSSIF was used in volumes of 500 and 900 mL. Blank FaSSIF did not contain lecithin or sodium taurocholate. The composition of the media is presented in Table I. DDDPlus™ 3 (Dose Disintegration and Dissolution Plus), beta version (Simulation Plus, Inc.) was used to simulate the in vitro release of the drugs using the above mentioned dissolution test conditions. DDDPlus™ is a computer program that models and simulates the in vitro dissolution of Active Pharmaceutical Ingredients (API) or its preparations. The software has three main tabs: formulation, experimental setup, and simulation. In the Formulation tab, a drug’s physiochemical parameters are used to define key parameters for the simulation as presented in Table II. In the experimental setup tab, the apparatus type, instrument speed, medium volume, and medium type is specified. In this study, a separate database record was generated for each medium. In blank FaSSIF, the solubility enhancement factor (SEF) of the lecithin and sodium taurocholate was set to 1 since this medium contains no solubilization enhancers. In 500 mL FaSSIF at 75 rpm, the SEF was optimized, so the software model fitted the experimental data: lecithin SEF = 36702.14; sodium taurocholate SEF = 96200.54. The same SEF values were used in the remaining FaSSIF media (500 mL at 100 rpm and 900 mL at 75 rpm). The particle size distribution of the API experimental data used was listed in Fig. 1. This distribution was used as an input parameter in DDDPlus™ to perform the simulations.
Table I.
Media Composition Used In the Simulation
| Media | USP SIF | (FaSSIF) | (BFaSSIF) |
|---|---|---|---|
| Composition (14) | (0.023 M) sodium hydroxide (0.005 M) potassium phosphate monobasic | (0.029 M) sodium phosphate monobasic (0.0095 M) sodium hydroxide (0.105 M) sodium chloride (0.0035 M) sodium taurocholate. (0.00075 M) lecithin. | Same composition as for FaSSIF but without bile salt (lecithin or sodium taurocholate) |
Table II.
Input Data Used for the Simulations
| Drug | Glyburide | Montelukast |
|---|---|---|
| Dose (mg) | 3.5 (15) | 10 (14) |
| Dosage form | Immediate release (15) | Immediate release (14) |
| Solubility (mg/mL) | 0.043 at pH 7.4 (15) | 0.0007 at pH 6.5 (16) |
| Mean particle radius (μm) | 6.28 (15) | 6.572 (14) |
| Particle density (g/mL) | 1.38 (15) | 1.2 (ADMET 5 default setting) |
| Diffusion coefficient (cm2/s × 10−5) | 0.5878 (15) | 0.54 (ADMET 5) |
| pKa | 5.5, (ADMET 5) 11.62,10.64 | 2.8, 5.7 (16) |
Fig. 1.
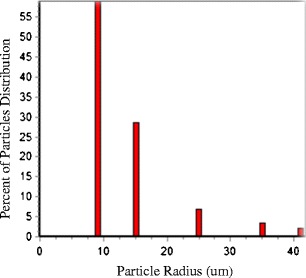
Particle size distribution of montelukast sodium used in the simulation (adopted from Okumu et al. 2008 (14))
Glyburide
Published glyburide in vitro dissolution data was used for the simulations (15,16). For the purpose of this study, the reference product of the study was chosen because more information was available about this product. The study was performed in three different media: SIF, BFaSSIF, and FaSSIF using the USP apparatus II (paddle). The experiments were carried out at 37 ± 0.5°C in 900 mL at 75 rpm at a single pH of 6.5 and at a dynamic pH range from 6.0, 6.5, 7.0, 7.5, and 5.0. The pH conditions were changed over time to simulate the intestinal passage of the dosage form. As presented in Table III, FaSSIF contained 3 mM sodium taurocholate and 0.75 mM lecithin. In the experimental study, two chemical grades of sodium taurocholate and lecithin were used: low quality (crude sodium taurocholate and 60% lecithin) and high quality (97% sodium taurocholate and 99% lecithin). In the program, no option was available for changing the chemical grade of the sodium taurocholate and lecithin. Therefore, the inputs to estimate the dissolution in LQ FaSSIF and HQ FaSSIF were identical.
Table III.
Glyburide Input Data for Dynamic pH Changes
| Experiment Phase | Start time | End time | Medium pH |
|---|---|---|---|
| 1 | 0 | 30 | 6 |
| 2 | 30 | 90 | 6.5 |
| 3 | 90 | 150 | 7.5 |
| 4 | 150 | 270 | 7 |
| 5 | 270 | 300 | 5 |
Input parameters were applied in the same manner as for montelukast sodium using the Formulation tab (Table II). Apparatus type, instrument speed, medium volume, and medium type were used as specified from the study (15,16). In blank FaSSIF the solubility enhancement factor (SEF) of lecithin and sodium taurocholate was equal to 1; however, in FaSSIF the SEF was optimized for low quality FaSSIF and the dynamic pH change protocol as following: Lecithin SEF = 1374.91; sodium taurocholate SEF = 96200.54. The same values were applied to the remaining FaSSIF media. To simulate the constant pH conditions the experiment phase was set to pH 6.5 throughout the simulation. To simulate the dynamic pH change protocol, the experimental phase was divided into 5 phases to represent the five different pH values (6.0, 6.5, 7.0, 7.5 and 5.0) as shown in Table III.
After running the simulations, a regression coefficient between predicted and observed data was calculated to assess the predictive power of the simulations using the statistical program SPSS 17.
RESULTS
Montelukast Sodium
The statistical analysis showed that there was a correlation between the in vitro release data and the predicted release data in all cases except for BFaSSIF. Figures 2 and 3, and Table IV show the correlation. In the experimental study, the measured drug release was zero for the BFaSSIF, and the predicted data was 5.9%; therefore, no correlation was found for this media. The correlations between in vitro release in the experimental data and the predicted values including the P values are presented in Table IV.
Fig. 2.
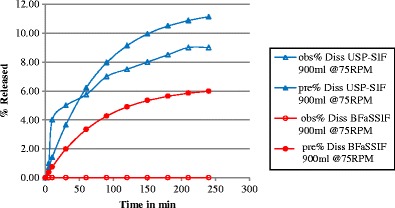
Observed (obs.) and predicted (pre.) dissolution release profiles (Diss) for montelukast sodium using USP simulated intestinal fluid (SIF) and blank fasted simulated intestinal fluid (BFaSSIF) in 900 mL at 75 rpm using USP apparatus II at 37°C
Fig. 3.
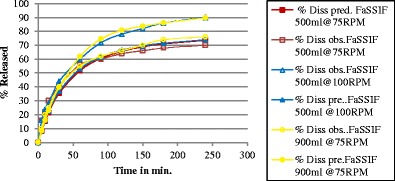
Observed (obs.) and predicted (pre.) dissolution profiles for montelukast sodium using fasted state simulated intestinal fluid (FaSSIF) using USP apparatus II at 37°C in the following condition: 500 mL at 75 rpm, 500 mL at 100 rpm, and 900 mL at 75 rpm
Table IV.
P values and Regression Coefficients for Predicted vs. Experimental Data of Montelukast Sodium and Glyburide
| Drug | Media | P values | Regression |
|---|---|---|---|
| Montelukast | USP-SIf | P < .001 | 0.92 |
| BFaSSIF | NA | NA | |
| FaSSIS 500 mL at 75 rpm | P < .001 | 0.99 | |
| FaSSIF 500 mL at 100 rpm | P < .001 | 0.99 | |
| FaSSIF 900 mL at 75 rpm | P < .001 | 0.99 | |
| Glyburide | USP-SIf at pH 6.5 | P = .003 | 0.69 |
| BFaSSIF at pH 6.5 | P = .005 | 0.64 | |
| FaSSIF-LQ at pH 6.5 | P < .001 | 0.93 | |
| FaSSIF-HQ at pH 6.5 | P = .001 | 0.83 | |
| USP-SIf at dynamic pH | P = .19 | 0.38 | |
| BFaSSIF at dynamic pH | P = .19 | 0.38 | |
| FaSSIF-LQ at dynamic pH | P < .001 | 0.99 | |
| FaSSIF-HQ at dynamic pH | P = .06 | 0.79 |
Glyburide
There was a statistically significant correlation between the experimental data and predicted release profiles when using a single pH condition (Table IV, Fig. 4). However, a dynamic pH changes protocol increased the correlation significantly using FaSSIF media. The best correlation was found when low quality lecithin and sodium taurocholate were used as shown in Fig. 5 and Table IV. In contrast, the correlations were insignificant in SIF and BFaSSIF.
Fig. 4.
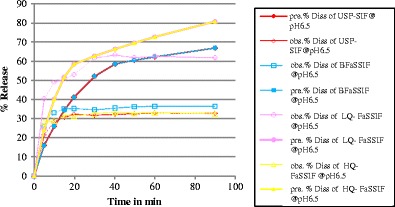
Observed (obs.) and predicted (pred.) dissolution release profiles (Diss) for glyburide at pH 6.5 using USP apparatus II at 37°C in 900 ml at 75 rpm and the following media: USP simulated intestinal fluid (USP-SIF), blank fasted state simulated intestinal fluid (BFaSSIF), low quality fasted state simulated intestinal fluid (LQ-FaSSIF), and high quality fasted state simulated intestinal fluid (HQ-FaSSIF)
Fig. 5.
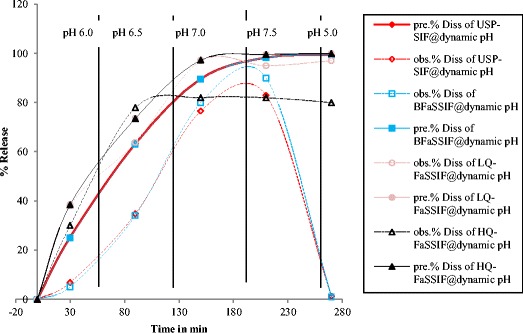
Observed (obs.) and predicted (pre.) dissolution release profiles (Diss) for glyburide at dynamic pH changes (6.0, 6.5, 7.0, 7.5, and 5.0) using USP apparatus II at 37°C in 900 ml at 75 rpm in the following media: USP simulated intestinal fluid (USP-SIF), blank fasted state simulated intestinal fluid (BFaSSIF), low quality fasted state simulated intestinal fluid (LQ-FaSSIF), and high quality fasted state simulated intestinal fluid (HQ-FaSSIF)
DISCUSSION
Simulations and computer modeling in drug development have gained interest and importance throughout the pharmaceutical industry. In slico tools are used in all phases of the drug development process from the synthesis of the active molecule to the optimization of the drug delivery system. The evaluation of the performance of such software programs is crucial to establish their suitability and acceptance for regulatory environment. FDA for example has licenses for different simulation software packages, and DDDPlus™ is one of them. DDDPlus™ is a commercially available program that can mimic in vitro dissolution of drug molecules using pharmacopeial apparatuses (21) However, there are limited data published, which validate its performance.
Dissolution testing is a valuable performance test for pharmaceutical preparations. Dissolution testing utilizes different media, such as water, or more sophisticated media, such as biorelevant media to test the performance of dosage forms (6,20).
Montelukast sodium is a lipophilic drug; hence, using solubilizing agents such as bile salts in FaSSIF media improve its solubility. In contrast, this drug did not dissolve in blank-SIF where no solubilizing agents are present. The predicted data using DDDPlus™ forecasted good and limited drug solubility in media, which indicates that the software uses solid prediction models.
In the experimental study, the highest percent of dissolved drug was found in FaSSIF-500 ml at 100 rpm (14). These experimental data were used to correlate the in vitro dissolution with an in vivo profile observed in a clinical study using GastroPlus™. This software can estimate drug plasma concentrations by using physiochemical properties or dissolution data for its calculations. In the study, the best correlation (r2 = 0.83) was shown in FaSSIF-500 ml at 100 rpm. In the current study, the correlation between the observed and the predicted in vitro dissolution profiles for this media was r2 = 0.99. DDDPlus™ was able to simulate the in vitro dissolution of the drug in FaSSIF. Such predictions can then be used by other software programs, such as Gastroplus, to estimate the in vivo performance of new drug molecules.
Glyburide is a weakly acidic drug. The experimental data showed that increasing the pH increased the drug’s solubility (16). Also, using solubilizing agent in the dissolution media (such as bile salts) increased the drug’s dissolution. Therefore, the highest percentage of dissolved drug was found in experimental media LQ-FaSSIF at pH 6.5 using DDDPlus™. The regression coefficients between the predicted dissolution profiles for LQ-FaSSIF and HQ-FaSSIF were quite high, 0.87 and 0.83, respectively. However, DDDPlus™ does not give an option to change the chemical grades of lecithin and sodium taurocholate. As mentioned in the “Method” section, the input value was the same for the LQ and HQ FaSSIF media, and the results represent the setting in DDDPlus™ using the LQ-chemical grades of lecithin and sodium taurocholate. The experimental data for glyburide had shown that low quality FaSSIF was the best media to establish an in vivo/in vitro correlation (IVIVC) and was, therefore, used in this study only (15,16).
In a quality by design (QbD) approach, dissolution should reflect ideally the in vivo product performance (17-19). Starting out with simulated in vitro dissolution data as an input to an in vivo prediction software such as GastroPlus™ may help to establish IVIVC in the later stages of the drug development process. The formulation scientists can use such data for the proper formulation design and selection of biorelevant performance testing.
The simulations in this study showed that in vitro release of both drugs was sensitive to solubility effects, which confirmed the API’s BCS class II classification (8). IVIVCs can be established for BCS class II drugs when the in vitro dissolution represents the in vivo dissolution (7,15,16). Using computer programs to predict the in vitro drug release can assist in choosing a suitable dissolution set-up for IVIVC and presumably link critical product attributes to meaningful quality control and performance testing procedures (20-22). By comparing the predictions of DDDPlus™ with those of experimental studies, the study showed that DDDPlus™ can predict API dissolution of poorly soluble drugs in a variety of media and apparatuses. Being able to predict dissolution profiles for BCS class II drugs indicates that DDDPlus™ is a suitable tool for estimating dissolution profiles of APIs, especially in the early discovery phase.
CONCLUSION
DDDPlus™ was capable in predicting the in vitro release pattern of montelukast sodium and glyburide under different experimental conditions. Computer simulations of in vitro release pattern using DDDPlus™ have the potential to estimate in vitro dissolution behaviors at an early stage in the drug development process. This might be used to choose the most appropriate dissolution condition to establish on the one hand IVIVC and to develop biorelevant in vitro performance tests which capture critical product attributes or proper quality control procedures in Quality by Design approaches.
Acknowledgments
The authors would like to thank Simulation Plus and Princess Nora Bint Abdulrahman University for their support.
References
- 1.Tong C, Lozano R, Mirza YMT, Loebenberg R, Nickerson B, Gray V, et al. The value of dissolution in the drug development continuum. Pharm Technol. 2009;33(4):52–64. [Google Scholar]
- 2.Hewitt M, Cronin MTD, Enoch SJ, Madden JC, Roberts DW, Dearden JC. In silico prediction of aqueous solubility: the solubility challenge. J Chem Inf Model. 2009;49(11):2572–87. doi: 10.1021/ci900286s. [DOI] [PubMed] [Google Scholar]
- 3.Norinder U, Bergström CAS. Prediction of ADMET properties. ChemMedChem. 2006;1(9):920–37. doi: 10.1002/cmdc.200600155. [DOI] [PubMed] [Google Scholar]
- 4.Oyarzabal J, Pastor J, Howe TJ. Optimizing the performance of in silico ADMET general models according to local requirements: MARS approach. Solubility estimations as case study. J Chem Inf Model. 2009;49(12):2837–50. doi: 10.1021/ci900308u. [DOI] [PubMed] [Google Scholar]
- 5.Göller AH, Hennemann M, Keldenich J, Clark T. In silico prediction of buffer solubility based on quantum-mechanical and HQSAR-and topology-based descriptors. J Chem Inf Model. 2006;46(2):648–58. doi: 10.1021/ci0503210. [DOI] [PubMed] [Google Scholar]
- 6.Almukainzi M, et al. Biorelevant Dissolution Testing, in Therapeutic Delivery Solutions, Wiley. 2014. p. 335–365.
- 7.Löbenberg R, Amidon GL, et al. Mechanism of gastrointestinal drug absorption and application in therapeutic drug delivery. Therapeutic Delivery Methods: A Concise Overview of Emerging Areas, Future Science Ltd. 2013; 8-22.
- 8.Löbenberg R, Amidon GL. Modern bioavailability, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards. Eur J Pharm Biopharm. 2000;50(1):3–12. doi: 10.1016/S0939-6411(00)00091-6. [DOI] [PubMed] [Google Scholar]
- 9.Azarmi S, Roa W, Löbenberg R. Current perspectives in dissolution testing of conventional and novel dosage forms. Int J Pharm. 2007;328(1 SPEC. ISS):12–21. doi: 10.1016/j.ijpharm.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Tong C, D’Souza SS, Parker JE, Mirza T. Commentary on AAPS workshop—dissolution testing for the twenty-first century: linking critical quality attributes and critical process parameters to clinically relevant dissolution. Pharm Res. 2007;24(9):1603–7. doi: 10.1007/s11095-007-9280-x. [DOI] [PubMed] [Google Scholar]
- 11.Dickinson PA, Lee WW, Stott PW, Townsend AI, Smart JP, Ghahramani P, et al. Clinical relevance of dissolution testing in quality by design. AAPS J. 2008;10(2):380–90. doi: 10.1208/s12248-008-9034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.USP United States pharmacopeia. U S Pharmacopeial Connvention. 2010;1(33):610. [Google Scholar]
- 13.Gray V, Kelly G, Xia M, Butler C, Thomas S, Mayock S. The science of USP 1 and 2 dissolution: present challenges and future relevance. Pharm Res. 2009;2626(6):1289–302. doi: 10.1007/s11095-008-9822-x. [DOI] [PubMed] [Google Scholar]
- 14.Okumu A, DiMaso M, Löbenberg R. Dynamic dissolution testing to establish in vitro/in vivo correlations for montelukast sodium, a poorly soluble drug. Pharm Res. 2008;25(12):2778–85. doi: 10.1007/s11095-008-9642-z. [DOI] [PubMed] [Google Scholar]
- 15.Wei H, Löbenberg R. Biorelevant dissolution media as a predictive tool for glyburide a class II drug. Eur J Pharm Sci. 2006;29(1):45–52. doi: 10.1016/j.ejps.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Wei H, Dalton C, Di Maso M, Kanfer I, Löbenberg R. Physicochemical characterization of five glyburide powders: a BCS based approach to predict oral absorption. Eur J Pharm Biopharm. 2008;6969(3):1046–56. doi: 10.1016/j.ejpb.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 17.Yu LX. Pharmaceutical quality by design: product and process development, understanding, and control. Pharm Res. 2008;25(4):781–91. [DOI] [PubMed]
- 18.D’Souza SS, Lozano R, Mayock S, Gray V. AAPS workshop on the role of dissolution in QbD and drug product life cycle: a commentary. Dissolution Technol. 2010;17(4):41–5. doi: 10.14227/DT170410P41. [DOI] [Google Scholar]
- 19.Dissolution testing and quality by design (QbD) [homepage on the Internet]. 2008, July 31. Available from: http://www.scitopics.com/Dissolution_Testing_and_Quality_by_Design_QbD.html.
- 20.Marques M, Löbenberg R, Almukainzi M. Simulated Biological Fluids with Possible Application in Dissolution Testing Dissolution Technology. 2011; 15–28.
- 21.Almukainzi M. Evaluation of Performance Testing and Computer Simulations for Quality by Design Approaches of Oral Dosage Forms. Pharmaceutical Sciences, University of Alberta. Master In Pharmaceutical Sciences. 2011.
- 22.Kanfer I, Walker R, Löbenberg R, Bou-Chacra NA. Experimental Formulation Development. In: Kanfer I, Shargel L, editors. Generic Drug Product Development. CRC Press. 2013.