

Highlights
-
•
Nearly complete backbone assignments for monomeric SOD1 were obtained.
-
•
Monomeric SOD1 is unstructured in 9 M urea.
-
•
Zn2+-binding to monomeric SOD1 occurs through diffuse coordination to most His residues.
-
•
The binding geometry of Zn2+ is different than in the native, folded SOD1.
Abbreviations: SOD1, superoxide dismutase 1; HSQC, heteronuclear single-quantum coherence; TOCSY, total correlation spectroscopy; NOESY, nuclear Overhauser enhancement spectroscopy; IDP, intrinsically disordered protein
Keywords: NMR, SOD1, Folding, Metal-binding
Abstract
The stability and structural properties of the metalloprotein superoxide dismutase 1 (SOD1) are found to depend critically on metal ions. Native SOD1 monomers coordinate one structural Zn2+ and one redox-active Cu2+/1+ to the active site. To do this, the Zn2+ ions need to interact with the SOD1 protein on the denatured side of the folding barrier, prior to the formation of the folding nucleus. In this study, we have examined at residue level the nature of this early Zn2+ binding by NMR studies on the urea denatured-state of SOD1. Nearly complete backbone chemical shift assignments were obtained in 9 M urea at physiological pH, conditions at which NMR studies are scarce. Our results demonstrate that SOD1 is predominantly unstructured under these conditions. Chemical-shift changes upon Zn2+ titration show that denatured SOD1 retains a significant affinity to Zn2+ ions, even in 9 M urea. However, the Zn2+ interactions are not limited to the native metal-binding ligands in the two binding sites, but are seen for all His residues. Moreover, the native Cu2+/1+ ligand H46 seems not to bind as well as the other His residues, while the nearby non-native H43 does bind, indicating that the binding geometry is relaxed. The result suggests that the Zn2+-binding observed to catalyze folding of SOD1 in physiological buffer is initiated by diffuse, non-specific coordination to the coil, which subsequently funnels by ligand exchange into the native coordination geometry of the folded monomer. Altogether, this diffuse binding is a result with fundamental implications for folding of metalloproteins in general.
1. Introduction
Superoxide dismutases are ubiquitous enzymes that neutralize reactive oxygen species in the cells. Human Cu/Zn superoxide dismutase (SOD1) is a 153 amino acid residue homo-dimeric protein with two cofactors per subunit; one copper and one zinc (Fig. 1). The copper ion is, in its oxidized form, bound by four histidines (H46, H48, H63 and H120). All these histidines are essential for enzyme activity [1]. The zinc ion is bound by three histidines (H63, H71 and H80) and one aspartate (D83), all part of what is commonly called “the zinc binding loop” (loop IV, residues 48–84). This loop and its zinc ion stabilize the protein structure, including both metal centers [2–5] but Cu2+ is necessary for catalysis and native structure (Fig. 1). Both metals, as well as many other divalent cations, are able to bind to both metal sites in natively folded SOD1 in vitro [1].
Fig. 1.
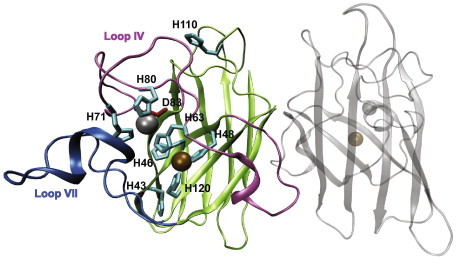
Metal binding sites in folded, dimeric, holo-SOD1. The zinc ion (gray) is bound by three histidines (cyan) and one aspartate (red). The copper ion (orange) is bound by four histidines. H63 is bridging both metals in the oxidized form. Loop IV (the so-called zinc-binding loop) is depicted in pink, loop VII (the “electrostatic loop”) in blue. The figure was made in VMD with the coordinates taken from the PDB (accession code 2GBT, www.pdb.org). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Zn2+ binds to both the zinc and copper sites, and this observation has been used to elucidate how metals affect folding of SOD1 [5–7]. These studies showed that Zn2+ catalyses protein folding by binding transiently to the SOD1 Cu2+/1+ ligands. Once folding is completed, the zinc ion is transferred to the thermodynamically favorable position in the Zn2+ site [7]. A question then arises concerning the nature of this Zn2+ coordination. There are many potential competing ligands (e.g. His residues, charged residues) in the protein and furthermore there is no tertiary structural context that would favor the native ligands. To investigate the nature of the early Zn2+ binding we have in this work focused on the metal-binding properties of unfolded SOD1. We have examined possible structures that may arise due to specific metal interaction of the urea-denatured state of the monomeric SOD1, F50E/G51E, C6A/C111A variant (SOD16/111/50/51). The first two substitutions hinder dimer formation [8], whereas the latter two abolish erroneous disulfide bond formation [9], which provides a way to study the folding and metal-binding properties of the monomeric protein. Our aim with the study was to examine the early folding events as introduced by Zn2+ coordination. For doing this, we need relatively high Zn2+ concentrations in order to capture what may be relevant Zn2+-interactions for the unfolded state even at physiological conditions. Urea-denatured SOD1 may be a simple model for the unfolded state, but nevertheless presents a way to capture possible ways for the protein to interact with metal ions. In this way we are able to capture the effect of metalation at the very start of the folding reaction, prior to the formation of a folding nucleus. We have furthermore characterized the residual structure in the denatured protein and examined the effect of urea at several urea concentrations. The results presented here demonstrate that Zn2+-binding to unfolded SOD1 occurs through diffuse and non-native coordination. This suggests that the ligands are shuffled towards the native coordination during the course of the folding reaction providing a favorable funnel-like restriction of the conformational entropy of the coil.
2. Results
2.1. SOD1 is unstructured in 9 M urea
The overall appearance of the 15N-HSQC is indicative of a protein with no or very little secondary structure (Fig. 2). We have assigned the resonances of backbone atoms (NH, Cα, C′ and Hα) in apo-SOD16/111/50/51 in 9 M urea and pH 6.3 to a completeness of 97%. The N-terminal residues A1 and T2 lack assignment since there were no detectable peaks in any of the spectra that could be assigned to them. C′ of H63, F64 and P66 were not assigned whereas the rest of the assignment for the backbone is complete.
Fig. 2.
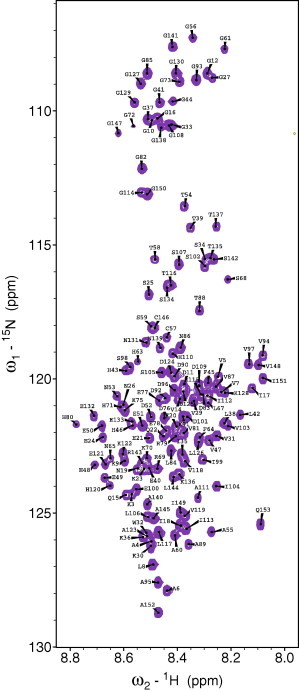
A 15N-HSQC indicating the backbone assignment of SOD16/111/50/51 in 9 M urea. Basically all residues have typical random coil chemical shifts, giving the spectrum the typical clustering appearance one normally see for unfolded proteins without residual elements.
To evaluate the extent of remaining structure of urea-denatured SOD16/111/50/51, we compared the backbone carbon atom chemical shifts to those previously measured for short peptides [10] using the chemical shift index method [11]. This simple analysis verified that only little structure existed in the protein (Fig. 3). Most of the very small deviations in the chemical shifts, which were only observed for Cα, were slightly positive, corresponding to α-helical structure. The analysis of Δδ(C′) supported this observation, indicating a random variation along the sequence. Since the deviations were so small (on average less than 0.4 ppm) and did not indicate β-sheet structure we concluded that neither the Δδ(Cα) nor the Δδ(C′) analyses support the presence of any significant degree of structure in SOD16/111/50/51. Additionally, and as expected for random coil conformations, no long-range NOEs were found. This further underlines that there are no secondary structure elements in SOD1 under the conditions studied. Therefore, we conclude that there is no residual β-sheet structure in the protein.
Fig. 3.
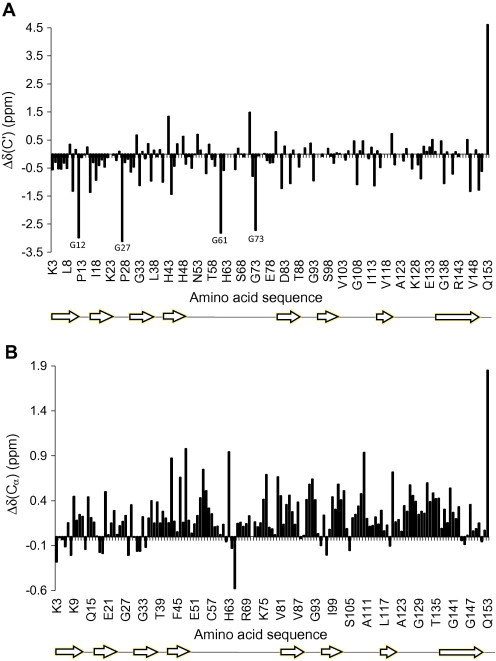
Secondary chemical shifts for SOD16/111/50/51 in 9 M urea. The position of the secondary structure elements as seen in the X-ray structure of monomeric SOD1 (accession code 3HFF, www.pdb.org) are shown under the sequence. In panel A Cα is shown, in panel B C′ secondary shifts according to Zhang et al. [8].
Since we assigned urea-denatured SOD1, we also compared the chemical shifts with data obtained for denatured GGXGG peptides by Schwarzinger et al. [12,13], which were measured at much lower pH (pH 2.3) than in the present study. No significant differences between this analysis and the one using the dataset by Wishart et al. were observed, except that all aspartic and glutamic acids in the amino acid sequence of SOD1 showed extreme values (data not shown), as expected for pH-dependent residues. Also, Δδ(Cα) for N65 displayed a large deviation from what is expected for an unfolded protein, most likely due to the proline situated subsequent to it [10,13]. This effect was also seen for all glycines followed by prolines (G12, G27, G61 and G73). Similar deviations in the Δδ(Cα) for proline-neighboring residues was seen when using the Wishart dataset but was compensated for, as described [10].
To determine the effect of the denaturant on the protein structure the urea concentration was lowered in steps of 0.5 M. Fig. 4 shows the effect on the chemical shifts of HN and NH when diluting the urea from 9 to 4 M. The changes (Δavgδ) were generally very small, on the order of 0.01–0.08 ppm. It is noteworthy that the region that displayed the largest shifts when lowering the urea concentration comprises amino acid residues H63–H71, which in turn is part of the zinc-binding loop. Especially residues S68 and R69 clearly shifted towards the expected chemical shifts in the natively folded protein, as seen in a previous assignment of apo-SOD1 [14]. Two other regions displayed small but consistent chemical shift differences that may be significant, I17-K23 and R143-I149. These regions correspond to β-strands 2 and 8 in the folded protein.
Fig. 4.
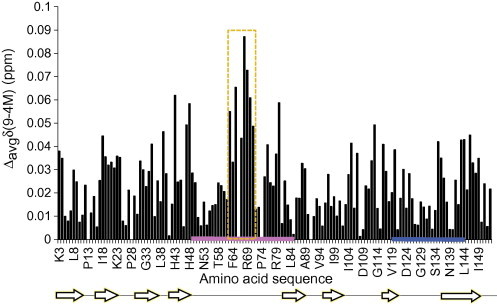
The weighted average difference in HN and NH chemical shifts when lowering the urea concentration from 9 to 4 M. The region H63-H71 (yellow), part of the zinc-binding loop (pink), is most affected. Loop VII is marked in blue. The positions of the secondary structure elements as seen in the X-ray structure of monomeric SOD1 (accession code 3HFF, www.pdb.org) are shown under the sequence. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.2. Zn2+ binds to all His residues in SOD1
To monitor Zn2+-binding of urea-denatured SOD16/111/50/51 we performed a series of 15N-HSQC experiments (Fig. 5A). The protein was exposed to 9 M urea and increasing Zn2+ concentrations and the resulting chemical shift changes, Δavgδ were plotted as a function of relative Zn2+ concentration (Fig. 5B). When zinc ions were added at lower ratio than 1:1 molar concentration there were no noticeable changes in the spectra (Δavgδ < 0.01 ppm, Fig. 5B). At equivalent concentrations of Zn2+ and protein (1:1 Zn2+:SOD1) the peaks of all eight His residues shifted slightly to higher values for HN and lower for NH (Δavgδ ∼ 0.01 ppm for all His residues, Fig. 5B), and this became even more pronounced at a 2:1 Zn2+:SOD1 ratio. No consistent line-broadening for the shifting His peaks and no additional peaks were observed. We therefore conclude that the interaction is most likely characterized by a fast exchange event. At higher concentrations, certain differences between the His residues could be discerned. The chemical shifts of H46 were significantly less affected than what was seen for the other residues; the maximum change was around 0.06 ppm, which can be compared to H71 for which the maximum was 0.16 ppm (Fig. 5B). Fig. 6A shows the chemical shift changes at a Zn2+:SOD1 ratio of 12:1, and from this plot it is evident that all His peaks but one, His46, has shifted their position significantly. At this concentration, also peaks neighboring the His residues (i.e. V47, E49, F64 and E121) display shifts that are larger than what is observed for the bulk of the protein (Fig. 6), supporting the observation that the effect is specific for His residues. At a very high concentration of Zn2+, corresponding to more than a 30:1 ratio, the resonances of most peaks shifted in a more uniform way and the specificity of the interaction was less pronounced. To ascertain that this effect at a high zinc concentration was purely ionic, measurement with a “charge equivalent” of NaCl corresponding to a 30:1 Zn2+:SOD1 ratio was performed. No shifts were, however, observed in the spectrum by adding NaCl. Hence, a high ZnSO4 concentration induces a general effect in the protein that was not observed using NaCl. Adding NaCl also demonstrated that the effect observed for the His residues is specific for Zn2+ and not a consequence of altering the sample conditions.
Fig. 5.
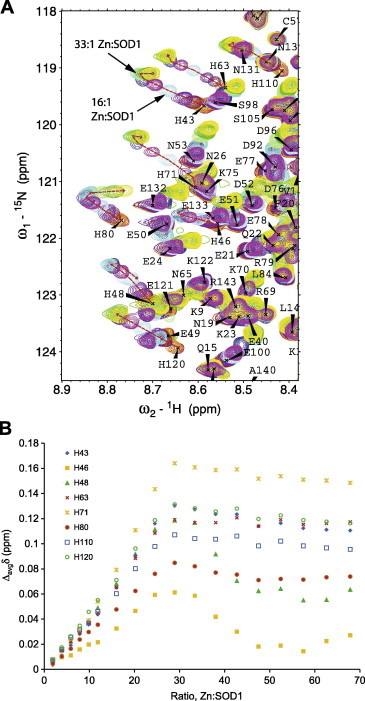
Overlaid 15N-HSQC spectra showing a subset of spectra obtained by adding 3 mM Zn2+–260 μM SOD1 (A). The Zn2+:SOD1 ratios were obtained by taking into account the (small) differences in sample volumes as a consequence of adding Zn2+.The color coding is as follows: apo-SOD1 (red), 2:1 Zn:SOD1 (orange), 6:1 Zn:SOD1 (maroon), 10:1 Zn:SOD1 (purple), 16:1 Zn:SOD1 (light blue), 25:1 Zn:SOD1 (blue), 33:1 Zn:SOD1 (turquoise), 43:1 Zn:SOD1 (light green), 52:1 Zn:SOD1 (green) and 68:1 Zn:SOD1 (yellow). (B) The weighted and averaged chemical shift difference for the His residues as a function of Zn2+:SOD1 ratio. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 6.
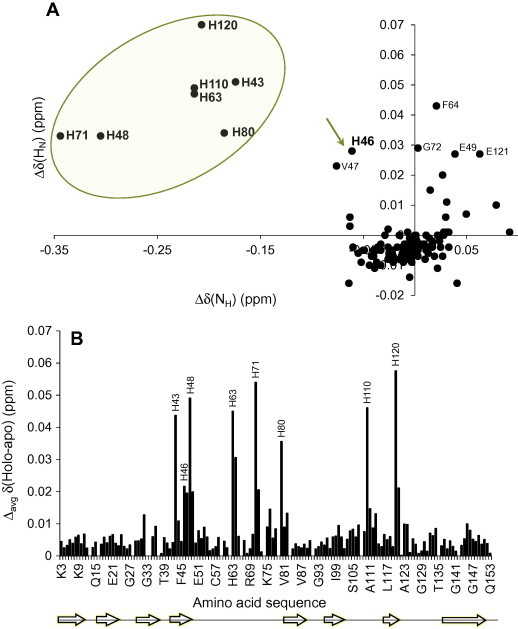
Zn2+-induced changes in SOD16/111/50/51 chemical shifts. Correlation between the chemical shift changes in SOD1 due to the addition of 3 mM Zn2+–260 μM SOD1 for HN and NH (A). The Zn2+:SOD1 ratios were obtained by taking into account the (small) differences in sample volumes as a consequence of adding Zn2+. The Zn2+-induced weighted average difference in 1HN and 15N chemical shifts as a function of amino acid sequence (B).
Next, the patterns of the shifts were analyzed (Fig. 5B). All of the His resonances were initially shifted in a linear manner with increasing concentrations of Zn2+. However, at the same ratio where we also observed more general shifts in the protein, there was a change in this linear pattern for several residues (H43, H46, H48 and H80), and the HN resonances were now shifted to lower values, indicating a possibility for one or several intermediate species along the titration series. This effect continued until a concentration around 60:1 Zn2+:SOD1. Here saturation was achieved, with no further changes. None of the other residues, including the native Zn2+-ligand D83 display large shifts even at these high Zn2+ concentrations (data not shown). In conclusion, we find that all His residues display chemical shift changes, but that for H46 the change is much smaller than for the others, indicating that this residue is less involved in metal coordination.
3. Discussion
NMR studies of intrinsically disordered proteins (IDPs) have provided a wealth of information about how to best describe the properties of them [15–19]. It has been established that there often is residual structure present and that there is variation in the dynamic behavior along the backbone [20,21]. There are, however, not that many examples of NMR studies of proteins in high concentrations of urea at physiological pH. Suggestions have been made that residual structure remains also in high amounts of urea, but these studies were conducted at low pH [22]. There have been arguments both for and against residual structure in urea-unfolded proteins, as reviewed by McCarney et al. [23]. The urea-unfolded barstar was demonstrated to contain little native structure [24], while studies in lower amounts of urea revealed residual helical structure [25]. For the tRNA methyltransferase YibK it was on the other hand suggested that residual structure remained in 8 M urea at pH 7.5, although the protein was mostly described as random coil [26]. Therefore, to understand the molecular properties related to folding of proteins, studies of unfolded states are necessary. Here we report on the backbone assignment of monomeric urea-denatured SOD16/111/50/51. Our analysis of the backbone chemical shifts shows that SOD1 in 9 M urea is unstructured, and remains largely unstructured to a urea concentration of less than 4 M. At low urea concentration evidence for slight structure induction in two of the β-strands (strands 2 and 8) and in the zinc-loop is observed. This is in agreement with an earlier study by Assfalg et al. that demonstrated that monomeric SOD1 is largely unstructured in guanidinium chloride (GdmCl) [27].
Although we needed high concentrations of Zn2+ to map out the binding sites in the urea-denatured state of SOD1, the results demonstrate the possible interaction sites also in unfolded SOD1 under more physiological conditions. Theoretically, the insertion of metal ions and the folding of a polypeptide chain may take place in any order. Either the metal ion is bound to the completely or partially folded structure, or to the unfolded polypeptide chain. In the case of active (holo) azurin, a blue-copper protein, the complete folding pathway has been shown to be 4000 times faster if copper is allowed to bind before the rest of the polypeptide folds [28,29]. A few years later it was shown that the metal ions could bind in several ways, but with one major population [30]. Here we have shown that in unfolded SOD1 most of the His residues, and not only the ligands in the two metal-binding sites, are sensitive to addition of Zn2+ ions. We also noted that high concentrations of Zn2+ were needed to induce significant changes in the NMR spectrum of urea-denatured SOD1. The relatively high concentration needed may indicate that all of the His residues are transiently involved in binding of several ions, but it should also be noted that urea is known to chelate Zn2+, and that the effective concentration present for interaction with the His residues may be much lower [31]. Under the conditions used in this study, with high Zn2+ concentrations, the effects that may very well be common to most proteins that are abundant in His residues, and not specific for SOD1. Another general observation is that the Zn2+-binding appears to proceed via one or several intermediate states. This is evidenced by the non-linear change in both HN and NH chemical shifts with increasing concentration. This has recently been explored in detail in a study of the ultrafast folding of the miniprotein Trp-cage [32]. Our data, however, point toward a more distinct change in the chemical shift pattern. The data indicates an onset of a different Zn2+ interaction at very high ion concentrations rather than an exchange between Zn2+-free unstructured SOD1, fully Zn2+-bound, and one or several intermediate states.
Zn2+ seems to coordinate, to some extent, to all the His residues in the unfolded protein, including the two non-native metal ligands H110 and H43. Our data reveal that there is no selectivity for any of the two native metal centers (Fig. 6). We can also conclude that only His residues, and no other common ligands such as negatively charged amino acid residues, bind the cationic Zn2+ ions in urea-denatured SOD1. Even D83, the only non-His metal binding ligand in the native protein, is largely unaffected by Zn2+, and no large shifts are observed (Fig. 6). This finding indicates a His-specific binding and not only an ionic effect. H120, one of the copper ligands, is affected to the greatest extent by the presence of zinc ions. We also note that binding to the non-native copper ligand H43 seems to be preferred over H46. This implies that it may not be favorable for Zn2+ to bind to both H46 and H48 simultaneously in unfolded SOD1, since these residues may be situated too close in the sequence. Therefore it appears that in the unfolded protein, zinc coordination to H43 and H48 is more favorable than to H46 and H48, i.e. in an extended structure this relaxes the binding geometry of the protein [33,34]. Together these results demonstrate that the initial binding of zinc to the protein occurs via a diffuse coordination, which in turn suggests that the ligands have to be rearranged during the course of the folding process to reach the native metal-bound state. This is in agreement with a study of SOD1 in GdmCl, which demonstrated structural rearrangements of the both native metal-binding sites [27]. The study in GdmCl further showed that the presence of metals induced a somewhat more compact protein, indicating (transient) metal interactions.
A recent study demonstrated that Zn2+ appears to be coordinated to the copper site in the protein in the early folding process, and that the zinc site forms late in the folding process [7]. Contrary to these results, it has been argued that the Zn2+ site is preformed in the presence of Zn2+, and that this stabilizes the folding nucleus [35]. Here we see that the diffuse binding of zinc, where the metal is coordinated to residues in both metal-binding sites, provides a means for SOD1 to explore several structural states at the very start of the folding reaction. Clearly, early zinc binding does not induce a pre-organized metal-binding site. This is consistent with the result that the zinc-binding site forms late in the folding process.
The region known as the zinc-binding loop in the folded protein (loop IV) appears to be the one that undergoes the most significant changes when slowly lowering the urea concentration. Small, but significant changes in chemical shifts for e.g. residues S68 and R69 in the zinc-binding loop are observed (Fig. 6), indicating that this region changes its structural characteristics. This implies that some sort of rearrangement of the loop takes place in the early stages of folding. NMR relaxation data for the apo protein has shown that loop IV is highly dynamic [36], and it has been demonstrated that the structural integrity of the protein relies on metal coordination [5]. The present finding indicating that the zinc-binding loop undergoes dynamic rearrangements, together with the unspecific metal-binding in the unfolded protein provides an indication that transient structural rearrangements in the zinc-binding loop may be important in the folding of human SOD1. For the protein to fold, several structural states are explored, which may provide a means for the protein to initiate folding. Finally, most proteins that contain His residues will most likely coordinate to Zn2+ in a diffuse and non-specific way, providing a means for metal-bound proteins to explore several conformational states. The diffuse metal binding in the early folding pathway observed here may well be a general mechanism by how metalloproteins initiate the folding process.
4. Materials and methods
4.1. Materials
EDTA was purchased from Merck (Whitehouse station, U.S.A.), guanidine hydrochloride (ultrapure) from AppliChem (Darmstadt, Germany) and bis-tris hydrochloride from Sigma (St. Louis, U.S.A.). Urea (ultra pure) was bought from MP Biomedicals (Illkirch, France). 15N-NH4Cl, 13C-glucose and D2O was purchased from Cambridge Isotope Laboratories (Andover, MA, USA). ZnSO4 was from Fluka (Buchs, Switzerland). Spectra/Por dialysis membranes were from Spectrum Laboratories (Rancho Dominguez, U.S.A.)
4.2. Expression and purification
The construct used, SOD16/111/50/51, was over-expressed in Escherichia coli grown in M9 minimal medium, containing 15N-labeled ammonium chloride and/or 13C-labeled d-glucose as sole nitrogen and carbon sources. The expression and purification of SOD1 were conducted as described in [37], but with no CuSO4 addition upon induction.
4.3. Metal depletion and unfolding of SOD1
Apo-SOD1 was prepared by dialysis against 500 mL 50 mM EDTA and 4 M guanidine hydrochloride, buffered by 10 mM bis-Tris (pH 6.3). The dialysis membranes had a pore size of 6-8000 MWCO and the dialysis continued in this solution for at least four hours in room temperature with stirring. The tubing and its content was then moved into a solution containing 250 mL 10 M urea and 10 mM bis-Tris (pH 6.3) and dialyzed again for 2 × 2 h in room temperature. This was done to keep the protein completely unfolded, remove EDTA and reduce the salt content in the sample. Addition of 10% D2O (for the field frequency lock stabilization in NMR experiments) resulted in a final urea concentration of 9 M and protein concentrations ranging between 0.2 and 0.5 mM for the different samples. The samples were handled at a maximum of 25 °C, since urea degrades more rapidly at higher temperature. The sample was always used within two weeks from preparation (unfolding and metal depletion) to avoid carbamylation of lysines and arginines or other effects due to urea breakdown and production of cyanate/isocyanic acid.
4.4. NMR experiments
All triple-resonance and titration experiments were done using a Bruker Avance 700 spectrometer equipped with cryoprobe, operating at 16.4 T. 15N-TOCSY-HSQC was measured with a Bruker Avance 600 spectrometer, operating at 14.1 T. The temperature was set to 25 °C for all NMR experiments.
To assign the backbone resonances of urea-denatured SOD1 a series of NMR experiments were measured on an uniformly 15N, 13C-labeled sample. HNCO, HNCA, HN(CO)CA, CANCO and CACO experiments were measured to achieve inter- and intra-residual connections. The 13C-detected experiments, CANCO and CACO, were conducted to attain greater dispersion of the peaks and also to assign amide 15N resonances for proline amino acid residues. Side-chain resonances obtained from HNCACB, 15N-TOCSY-HSQC and 15N-NOESY-HSQC spectra were also helpful in the assignment process. 15N-TOCSY-HSQC experiments were measured with mixing times of 60 and 90 ms, and a 15N-NOESY-HSQC with a mixing time of 120 ms. Finally, a total of eight multidimensional spectra were used to correlate the backbone nuclei, and to obtain sequential resonance assignments. The spectra were processed with TopSpin 2.1 (Bruker BioSpin), including, amongst other, linear prediction and zero-filling. The spectral analysis was performed using Sparky 3 [38].
Chemical shift difference analysis of Cα and Ć, Δδ(Cα) and Δδ(Ć) respectively, was done to evaluate the extent of residual structure in the protein. The differences in chemical shifts as compared to the data by Wishart et al., based on GGXAGG and GGXPGG peptides [10] were determined for this purpose. This dataset was chosen since it represents truly unstructured sequences, and not coil regions in otherwise folded proteins [39] or intrinsically disordered proteins [40].
4.5. Zinc titration
A series of 15N-HSQC spectra were measured with an increasing amount of metal ions present. A solution of ZnSO4 was titrated directly into the NMR tube. The added volumes were kept minimal (typically 2–7 μL into a volume of 600 μL) to abolish dilution effects and therefore stock solutions of different concentration (5, 10, 50 or 100 mM) were used. The pH of the sample and the ZnSO4 solutions was kept at pH 6.3. After zinc addition the tube was carefully shaken to allow for a homogenous mixing and then put back into the NMR spectrometer and incubated for at least 15 minutes before any new measurement was made. If measurements were continued after an over-night recess, a new spectrum was recorded with the same sample and compared to the last one measured the day before to make sure the longer incubation time did not change the sample in any way. The ZnSO4 concentration varied between 15 μM and 15 mM and the protein concentration was 260 μM. The weighted average difference in amide proton (ΔHN) and amide nitrogen (ΔNH) chemical shifts upon Zn2+ addition, Δavgδ, was calculated from [41,42]:
| (1) |
which takes into consideration the different gyromagnetic ratios and spectral widths of 15N and 1H.
4.6. Urea dilution experiments
The urea dilution was made by addition of 10 mM bis-tris buffer (pH 6.3) with 10% (v/v) D2O to a sample of SOD1 in 9 M urea. The dilution was made in 0.5 M steps, starting at 9 M and ending at 3.5 M. No correction was made for the protein concentration that was lowered from 380 to 148 μM during the measurements. In all other practical aspects the samples were treated as the samples in the zinc experiments during measurements.
Acknowledgements
This work was supported by grants from the Swedish Research Council (grant number 621-2011-5964). We thank Dr. Helena Kovacs for assistance with some of the NMR experiments. SS, MO and LM conceived and designed the project, SS acquired and analyzed the data, SS MO and LM interpreted the data, and SS and LM wrote the paper.
Footnotes
Database: The backbone chemical shift assignments for urea-denatured human SOD1 have been deposited with the Biological Magnetic Resonance Bank (BMRB) under the accession code 18968.
References
- 1.Ferraroni M., Rypniewski W., Wilson K., Viezzoli M., Banci L., Bertini I., Mangani S. The crystal structure of the monomeric human SOD mutant F50E/G51E/E133Q at atomic resolution. The enzyme mechanism revisited. J. Mol. Biol. 1999;288:413–426. doi: 10.1006/jmbi.1999.2681. [DOI] [PubMed] [Google Scholar]
- 2.Lynch S.M., Boswell S.A., Colón W. Kinetic stability of Cu/Zn superoxide dismutase is dependent on its metal ligands: implications for ALS. Biochemistry. 2004;43:16525–16531. doi: 10.1021/bi048831v. [DOI] [PubMed] [Google Scholar]
- 3.Roberts B.R., Tainer J.A., Getzoff E.D., Malencik D.A., Anderson S.R., Bomben V.C., Meyers K.R., Karplus A., Beckman J.S. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J. Mol. Biol. 2007;373:877–890. doi: 10.1016/j.jmb.2007.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mulligan V.K., Kerman A., Ho S., Chakrabartty A. Denaturational stress induces formation of zinc-deficient monomers of Cu, Zn superoxide dismutase: implications for pathogenesis in amyotrophic lateral sclerosis. J. Mol. Biol. 2008;383:424–436. doi: 10.1016/j.jmb.2008.08.024. [DOI] [PubMed] [Google Scholar]
- 5.Nordlund A., Leinartaite L., Saraboji K., Aisenbrey C., Gröbner G., Zetterström P., Danielsson J., Logan D.T., Oliveberg M. Functional features cause misfolding of the ALS-provoking enzyme SOD1. Proc. Natl. Acad. Sci. U.S.A. 2009;106:9667–9672. doi: 10.1073/pnas.0812046106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Estévez A.G., Crow J.P., Sampson J.B., Reiter C., Zhuang Y., Richardson G.J., Tarpey M.M., Barbeito L., Beckman J.S. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- 7.Leinartaite L., Saraboji K., Nordlund A., Logan D.T., Oliveberg M. Folding catalysis by transient coordination of Zn2 to the Cu ligands of the ALS-associated enzyme Cu/Zn superoxide dismutase 1. J. Am. Chem. Soc. 2010;132:13495–13504. doi: 10.1021/ja1057136. [DOI] [PubMed] [Google Scholar]
- 8.Bertini I., Piccioli M., Viezzoli M.S., Chiu C.Y., Mullenbach G.T. A spectroscopic characterization of a monomeric analog of copper, zinc superoxide dismutase. Eur. Biophys. J. 1994;23:167–176. doi: 10.1007/BF01007608. [DOI] [PubMed] [Google Scholar]
- 9.Lepock J., Frey H.E., Hallewell R. Contribution of conformational stability and reversibility of unfolding to the increased thermostability of human and bovine superoxide dismutase mutated at free cysteines. J. Biol. Chem. 1990;265:21612–21618. [PubMed] [Google Scholar]
- 10.Wishart D.S., Bigam C.G., Holm A., Hodges R.S., Sykes B.D. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 11.Wishart D.S., Sykes B.D. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 12.Schwarzinger S., Kroon G.J.A., Foss T.R., Chung J., Wright P.E., Dyson H.J. Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 2001;123:2970–2978. doi: 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- 13.Schwarzinger S., Kroon G.J.A., Foss T.R., Wright P.E., Dyson H.J. Random coil chemical shifts in acidic 8 M urea: implementation of random coil shift data in NMR view. J. Biomol. NMR. 2000;18:43–48. doi: 10.1023/a:1008386816521. [DOI] [PubMed] [Google Scholar]
- 14.Teilum K., Smith M.H., Schulz E., Christensen L.C., Solomentsev G., Oliveberg M., Akke M. Transient structural distortion of metal-free Cu/Zn superoxide dismutase triggers aberrant oligomerization. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18273–18278. doi: 10.1073/pnas.0907387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tompa P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 16.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 17.Tompa P. Unstructural biology coming of age. Curr. Opin. Struct. Biol. 2011;21:419–425. doi: 10.1016/j.sbi.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 18.Uversky V.N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 2011;43:1090–1103. doi: 10.1016/j.biocel.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Kosol S., Contreras-Martos S., Cedeno C., Tompa P. Structural characterization of intrinsically disordered proteins by NMR spectroscopy. Molecules. 2013;18:10802–10828. doi: 10.3390/molecules180910802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marsh J.A., Singh V.K., Jia Z., Forman-Kay J.D. Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: Implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camilloni C., De Simone A., Vranken W.F., Vendruscolo M. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry. 2012;51:2224–2231. doi: 10.1021/bi3001825. [DOI] [PubMed] [Google Scholar]
- 22.Neri D., Billeter M., Wider G., Wüthrich K. NMR determination of residual structure in a urea-denatured protein, the 434-repressor. Science. 1992;257:1559–1563. doi: 10.1126/science.1523410. [DOI] [PubMed] [Google Scholar]
- 23.McCarney E.R., Kohn J.E., Plaxco K.W. Is there or is not there? The case for (and against) residual structure in chemically denatured proteins. Crit. Rev. Biochem. Mol. Biol. 2005;40:181–189. doi: 10.1080/10409230591008143. [DOI] [PubMed] [Google Scholar]
- 24.Bhavesh N.S., Juneja J., Udgaonkar J.B., Hosur R.V. Native and nonnative conformational preferences in the urea-unfolded state of barstar. Protein Sci. 2004;13:3085–3091. doi: 10.1110/ps.04805204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong K., Freund S., Fersht A.R. Cold denaturation of barstar: 1H, 15N and 13C NMR assignment and characterisation of residual structure. J. Mol. Biol. 1996;259:805–818. doi: 10.1006/jmbi.1996.0359. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh S.M., Mallam A.L., Jackson S.E., Hsu S.D. Backbone NMR assignments of a topologically knotted protein in urea-denatured state. Biomol. NMR Assign. 2013;8:283–285. doi: 10.1007/s12104-013-9501-7. [DOI] [PubMed] [Google Scholar]
- 27.Assfalg M., Banci L., Bertini I., Turano P., Vasos P.R. Superoxide dismutase folding/unfolding pathway: role of the metal ions in modulating structural and dynamical features. J. Mol. Biol. 2003;330:145–158. doi: 10.1016/s0022-2836(03)00533-3. [DOI] [PubMed] [Google Scholar]
- 28.Pozdnyakova I., Wittung-Stafshede P. Copper binding before polypeptide folding speeds up formation of active (holo) Pseudomonas aeruginosa azurin. Biochemistry. 2001;40:13728–13733. doi: 10.1021/bi011591o. [DOI] [PubMed] [Google Scholar]
- 29.Pozdnyakova I., Wittung-Stafshede P. Biological relevance of metal binding before protein folding. J. Am. Chem. Soc. 2001;123:10135–10136. doi: 10.1021/ja016252x. [DOI] [PubMed] [Google Scholar]
- 30.Pozdnyakova I., Wittung-Stafshede P. Approaching the speed limit for Greek Key β-barrel formation: transition-state movement tunes folding rate of zinc-substituted azurin. Biochim. Biophys. Acta. 2003;1651:1–4. doi: 10.1016/s1570-9639(03)00240-1. [DOI] [PubMed] [Google Scholar]
- 31.Ozutsumi K., Taguchi Y., Kawashima T. Thermodynamics of formation of urea complexes with manganese (II), nickel (II) and zinc (II) ions in N, N-dimethylformamide. Talanta. 1995;42:535–541. doi: 10.1016/0039-9140(95)01437-g. [DOI] [PubMed] [Google Scholar]
- 32.Rovó P., Stráner P., Láng A., Bartha I., Huszár K., Nyitray L., Perczel A. Structural insights into the Trp-cage folding intermediate formation. Chem. Eur. J. 2013;19:2628–2640. doi: 10.1002/chem.201203764. [DOI] [PubMed] [Google Scholar]
- 33.Krantz B.A., Sosnick T.R. Engineered metal binding sites map the heterogeneous folding landscape of a coiled coil. Nat. Struct. Mol. Biol. 2001;8:1042–1047. doi: 10.1038/nsb723. [DOI] [PubMed] [Google Scholar]
- 34.Bosco G.L., Baxa M., Sosnick T.R. Metal binding kinetics of bi-histidine sites used in ψ analysis: evidence of high-energy protein folding intermediates. Biochemistry. 2009;48:2950–2959. doi: 10.1021/bi802072u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kayatekin C., Zitzewitz J.A., Matthews C.R. Zinc binding modulates the entire folding free energy surface of human Cu, Zn superoxide dismutase. J. Mol. Biol. 2008;384:540–555. doi: 10.1016/j.jmb.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Banci L., Bertini I., Cramaro F., Del Conte R., Viezzoli M.S. Solution structure of Apo Cu, Zn superoxide dismutase: role of metal ions in protein folding. Biochemistry. 2003;42:9543–9553. doi: 10.1021/bi034324m. [DOI] [PubMed] [Google Scholar]
- 37.Lindberg M.J., Tibell L., Oliveberg M. Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo state. Proc. Natl. Acad. Sci. U.S.A. 2002;99:16607–16612. doi: 10.1073/pnas.262527099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goddard T., Kneller D. University of California; San Francisco: 2004. SPARKY 3. [Google Scholar]
- 39.Zhang H., Neal S., Wishart D.S. RefDB: a database of uniformly referenced protein chemical shifts. J. Biomol. NMR. 2003;25:173–195. doi: 10.1023/a:1022836027055. [DOI] [PubMed] [Google Scholar]
- 40.Tamiola K., Acar B., Mulder F.A.A. Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J. Am. Chem. Soc. 2010;132:18000–18003. doi: 10.1021/ja105656t. [DOI] [PubMed] [Google Scholar]
- 41.Grzesiek S., Bax A., Clore G.M., Gronenborn A.M., Hu J.S., Kaufman J., Palmer I., Stahl S.J., Wingfield P.T. The solution structure of HIV-1 Nef reveals an unexpected fold and permits delineation of the binding surface for the SH3 domain of Hck tyrosine protein kinase. Nat. Struct. Biol. 1996;3:340–345. doi: 10.1038/nsb0496-340. [DOI] [PubMed] [Google Scholar]
- 42.Garrett D.S., Seok Y.J., Peterkofsky A., Clore G.M., Gronenborn A.M. Identification by NMR of the binding surface for the histidine-containing phosphocarrier protein HPr on the N-terminal domain of enzyme I of the Escherichia coli phosphotransferase system. Biochemistry. 1997;36:4393–4398. doi: 10.1021/bi970221q. [DOI] [PubMed] [Google Scholar]