

Abstract
Epalrestat (EPS) is the only aldose reductase inhibitor that is currently available for the treatment of diabetic neuropathy. Recently, we found that EPS at near-plasma concentration increases the intracellular levels of glutathione (GSH) in rat Schwann cells. GSH plays a crucial role in protecting endothelial cells from oxidative stress, thereby preventing vascular diseases. Here we show that EPS increases GSH levels in not only Schwann cells but also endothelial cells. Treatment of bovine aortic endothelial cells (BAECs), an in vitro model of the vascular endothelium, with EPS caused a dramatic increase in intracellular GSH levels. This was concomitant with the up-regulation of glutamate cysteine ligase, an enzyme catalyzing the first and rate-limiting step in de novo GSH synthesis. Moreover, EPS stimulated the expression of thioredoxin and heme oxygenase-1, which have important redox regulatory functions in endothelial cells. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key transcription factor that regulates the expression of antioxidant genes. EPS increased nuclear Nrf2 levels in BAECs. Nrf2 knockdown by siRNA suppressed the EPS-induced glutamate cysteine ligase, thioredoxin-1, and heme oxygenase-1 expression. Interestingly, LY294002, an inhibitor of phosphatidylinositol 3-kinase, abolished the EPS-stimulated GSH synthesis, suggesting that the kinase is associated with Nrf2 activation induced by EPS. Furthermore, EPS reduced the cytotoxicity induced by H2O2 and tert-butylhydroperoxide, indicating that EPS plays a role in protecting cells from oxidative stress. Taken together, the results provide evidence that EPS exerts new beneficial effects on endothelial cells by increasing GSH, thioredoxin, and heme oxygenase-1 levels through the activation of Nrf2. We suggest that EPS has the potential to prevent several vascular diseases caused by oxidative stress.
Keywords: Epalrestat, Endothelial cell, Glutathione, Heme oxygenase-1, Thioredoxin, Nuclear factor erythroid 2-related factor 2
Graphical abstract
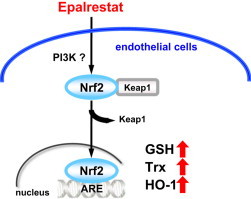
Highlights
-
•
Epalrestat increases GSH levels in endothelial cells through transcription regulation.
-
•
Epalrestat increases Trx and HO-1 levels.
-
•
Nrf2 activation by epalrestat is involved in increases in GSH, Trx, and HO-1 levels.
-
•
Epalrestat enhances endothelial cell resistance to oxidative stress.
Introduction
Epalrestat (5-[(1Z,2E)-2-methyl-3-phenyl propenylidene]-4-oxo-2-thioxo-3-thiazolidine acetic acid; EPS; Ono Pharmaceuticals, Osaka, Japan), which received approval for use in Japan in 1992, is currently being used for the treatment of diabetic neuropathy. EPS is an inhibitor of aldose reductase, a rate-limiting enzyme in the polyol pathway. Under hyperglycemic conditions, EPS reduces intracellular sorbitol accumulation, which is implicated in the pathogenesis of diabetic complications [1]. EPS is easily absorbed by neural tissue and inhibits aldose reductase with minimum adverse effects [2]. A recent study showed that treatment with EPS at an early stage delayed the progression of diabetic neuropathy and prevented the onset/progression of retinopathy and nephropathy [3].
The vascular endothelium, which regulates the passage of macromolecules and circulating cells from blood to tissues, is the major target of oxidative stress and plays a critical role in the pathophysiology of several diseases and disorders [4]. Endothelial dysfunction is an early event in atherosclerotic disease. Impaired endothelial function is noted in patients with coronary artery disease, diabetes mellitus, hypertension, and hypercholesterolemia. Inflammations and infections, which are often characterized by the excessive production of reactive oxygen species (ROS), impair endothelial function. Future research will focus on ways to prevent oxidative damage to the endothelium. Reduced glutathione (GSH) plays a crucial role in protecting endothelial cells from ROS, thereby preventing endothelial dysfunction in arteries exposed to oxidative stress [5]. It is important to find ways to increase the intracellular GSH level in order to prevent and/or minimize oxidative damage to the endothelium.
Glutamate cysteine ligase (GCL) is an enzyme that catalyzes the first and rate-limiting step in de novo GSH synthesis [6]. The regulation of GCL expression and activity is critical for GSH homeostasis. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key transcription factor that plays a central role in regulating the expression of antioxidant genes, including GCL [7–9]. Nrf2 usually binds to Kelch-like ECH associated protein-1 (Keap1) in the extranuclear space, and after suitable stimulation, Nrf2 translocates into the nucleus where it acts as a transcription factor, regulating the expression of many cytoprotective genes. Therefore, Nrf2 is important for the maintenance of intracellular GSH levels and redox homeostasis. Moreover, Nrf2 controls not only GCL gene but also the genes of many antioxidative proteins, such as thioredoxin (Trx) [10] and heme oxygenase-1 (HO-1) [11–13]. Trx, which is ubiquitously expressed in endothelial cells, regulates cellular redox status and protects cells from oxidative stress, in a similar manner to GSH [14]. Trx-1 has multiple functions in the cell, including antioxidant, anti-inflammatory, and anti-apoptotic activities. A recent study has shown that Trx-1 promotes anti-inflammatory macrophages of the M2 phenotype and antagonizes atherosclerosis [15]. HO-1, a representative Nrf2 target gene product [16], has important redox regulatory functions in endothelial cells [17,18]. There is evidence that the induction of HO-1 leads to several vascular-cell-specific protective activities in the setting of inflammatory atherosclerotic diseases [19].
Recently, we found that EPS increased GSH levels in rat Schwann cells by up-regulating GCL via Nrf2 activation [20]. We hypothesized that if EPS could increase GSH levels in endothelial cells, EPS would help prevent or minimize oxidative damage to the endothelium. The purpose of the present study was to determine (1) whether EPS increases GSH levels, (2) whether EPS affects HO-1 and Trx-1, which have redox regulatory functions, (3) whether the Nrf2 pathway is involved in the effects of EPS on GSH synthesis and the redox regulating proteins, and (4) whether EPS protects oxidative cell damage, using a culture system of bovine aortic endothelial cells (BAECs) as an in vitro model of the vascular endothelium.
Materials and methods
Endothelial cell culture and treatment with EPS
BAECs were purchased from Dainippon Sumitomo Pharma Co., Ltd. (Osaka, Japan). Cells were grown to 80–90% confluence in DMEM containing 10% fetal bovine serum (FBS), l-glutamine (4 mM), penicillin (100 U/ml), and streptomycin (100 µg/ml) at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Then, the cells were passaged by trypsinization.
Before treating the cells with EPS (Wako Pure Chemical Industries, Ltd., Osaka, Japan), the culture medium was replaced with DMEM containing 2% FBS because serum may include antioxidants, chelates of transition metal ions, and high-density lipoproteins [21]. EPS (10, 50, and 100 µM) was subsequently added to the medium.
Cell viability
Cell viability was assessed by measuring acid phosphatase activity. Acid phosphatase activity, which is an accurate indicator of the number of endothelial cells in culture, was assayed using the method of Connolly et al. [22]. BAECs on 96-well plates were treated with EPS. After the treatment with EPS, acid phosphatase activity was measured. The medium containing detached BAECs was removed. Cells remaining on the 96-well plates were washed with DPBS and incubated with 100 µl of 0.1 M sodium acetate buffer (pH 5.5) containing 0.1% Triton X-100 and 10 mM p-nitrophenyl phosphate at 37 °C for 20 min. The reaction was stopped by adding 10 µl of 1 M NaOH. Produced p-nitrophenol was measured at 405 nm using a Bio-Rad iMark microplate reader (Tokyo, Japan). Acid phosphatase activity was expressed as the ratio of the number of surviving cells to that of control without EPS.
Determination of nuclear Nrf2 translocation
Nuclear extracts of BAECs were prepared using an Active Motif Nuclear Extract Kit (Tokyo, Japan) according to the manufacturer's protocol. The amount of active Nrf2 in the nuclear extracts was determined by subjecting 20 µg of protein sample to assay with a TransAM Nrf2 DNA Binding ELISA Kit (Active Motif). The assay was performed according to the manufacturer's protocol. As a positive control, BAECs were incubated with a known Nrf2-activator sulforaphane. In addition, COS-7 cells (Nrf2 transfected) nuclear extracts provided by the manufacturer were used as a positive control in each experiment.
Knockdown of Nrf2 with small interfering RNA (siRNA)
Oligonucleotides directed against bovine Nrf2 (Sigma-Aldrich Co., St. Louis, MO, USA) and control siRNA (Ambion, Austin, TX, USA) were transfected into BAECs using Lipofectamine RNAiMAX (Invitrogen, Eugene, OR, USA) according to the manufacturer's protocol. Briefly, both Nrf2 siRNA and control siRNA were diluted with Opti-MEM medium and then, diluted Lipofectamine RNAiMAX was added. The transfection mixture was incubated at room temperature for 20 min. When BAECs reached 30–50% confluence, the culture medium was replaced with DMEM (without FBS) and the transfection mixture was added to each well. The final concentration of siRNA was 20 nM.
Measurement of GCLM, Nrf2, HO-1, and Trx-1 mRNA levels
GCL is the rate-limiting enzyme in de novo GSH synthesis and is a heterodimeric protein composed of catalytic (GCLC) and modifier (GCLM) subunits. We examined the effect of EPS on GCLM, which is limiting in most cell types and tissues [23]. Quantitative RT-PCR analysis was used to measure mRNA levels. Total RNA from treated cells was extracted with RNAspin Mini (GE Healthcare, Buckinghamshire, UK) according to the manufacturer's protocol. mRNAs were reverse-transcribed into cDNA with a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Quantitative RT-PCR was performed with a 7500 Fast Real-Time PCR System (Applied Biosystems). Primers for bovine GCLM (Bt03232353-m1), bovine Nrf2 (Bt03251879-m1), bovine HO-1 (Bt03218621-m1), and bovine Trx-1 (Bt03222877-g1) were purchased from Applied Biosystems. mRNA levels were acquired from the value of the threshold cycle (Ct) of GCLM, Nrf2, HO-1, or Trx-1 normalized to that of GAPDH. Relative mRNA levels were compared and expressed as percentage of control levels. Data are representative of three experiments.
Measurement of GCLM, HO-1, and Trx-1 protein levels
GCLM, HO-1, and Trx-1 protein levels were analyzed by Western blotting. After BAECs were treated with EPS, the cells were washed with DPBS and lysed in radioimmunoprecipitation assay (RIPA) buffer (Pierce, Rockford, IL, USA) containing protease inhibitors. The lysate was centrifuged at 10,000g for 15 min and 15 µg of protein in the supernatant was resolved by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were blotted onto a nitrocellulose or PVDF membrane. The membrane was incubated with primary antibodies for GCLM, HO-1, Trx-1, or β-actin and with horseradish-peroxidase-conjugated secondary antibodies. Chemiluminescence was detected with an ECL Plus Western blot detection kit (GE Healthcare, Buckinghamshire, UK). Band intensities were quantified using ImageJ software.
In addition, we measured HO-1 and Trx-1 protein levels by fluorescence microscopy studies and flow cytometry, respectively. Briefly, BAECs treated with EPS were fixed with 4% p-formaldehyde. HO-1 and Trx-1 proteins were detected by reacting with phycoerythrin (PE)-conjugated anti-rabbit HO-1 monoclonal antibody (Cell Signaling Technology, Cambridge, UK) and PE-conjugated anti-mouse Trx-1 monoclonal antibody (GenWay Biotech, Inc., San Diego, CA, USA), respectively. Following incubation with the antibody, the cells were washed with DPBS and analyzed by fluorescence microscopy (Carl Zeiss, Jena, Germany) or flow cytometry (Beckman Coulter, Fullerton, CA, USA). Fluorescence was detected with fluorescence channel 2 (FL2).
Determination of mitochondrial damage
Mitotracker Red CMXRos (Molecular Probes, Eugene, OR, USA) was used to estimate mitochondrial damage in BAECs. After exposing BAECs to oxidizing agent, the cells were incubated in the medium containing Mitotracker Red CMXRos (25 nM) for 30 min. Then, the cells were washed with DPBS and fixed with 4% p-formaldehyde. Changes of the mitochondrial membrane potential were visualized as red fluorescence by using a confocal microscope (Carl Zeiss). Fluorescence intensities were quantified using Zeiss ZEN software.
Other procedures
Intracellular GSH levels were measured by spectrophotometric methods, as described previously [24]. Aldose reductase activity was measured according to the method described by Kawasaki et al. [25] with dl-glyceraldehyde as the substrate. Lactate dehydrogenase activity was measured using lithium dl-lactate as the substrate. Protein concentrations were determined using the Bradford method with bovine serum albumin as the standard.
Statistical analysis
All experiments were performed independently at least three times. Data were combined and expressed as means±SD. Statistical significance between two groups was evaluated using Student's t-test after analysis of variance or the Scheffé test after the Kruskal–Wallis test. A P value of <0.05 was considered to be significant.
Results
Effect of EPS on GSH in BAECs
BAECs were treated with EPS at 10, 50, and 100 µM for 24 h. EPS, at the concentrations used, had little influence on cell viability as estimated by monitoring acid phosphatase activity (control, 100±5%; 10 µM, 100±6%; 50 µM, 100±6%; and 100 µM, 90±6%). Likewise, no release of lactate dehydrogenase was observed at those conditions (data not shown). Fig. 1A shows the intracellular GSH levels. Treatment of BAECs with EPS at 50 and 100 µM caused an increase in intracellular GSH levels. At those EPS concentrations, the increases were 2.7- and 8.4-fold, respectively, compared with control. When BAECs were treated with EPS at 50 µM, a significant increase in GSH levels was noted after 8 h (Fig. 1B). Fig. 1C and D demonstrates that EPS at 50 and 100 µM increased the protein and mRNA levels of GCLM. The treatment with 10 µM EPS did not cause a significant increase in GSH and GCLM levels. The results indicate that EPS increases intracellular GSH levels in BAECs through transcription regulation. Two other aldose reductase inhibitors, sorbinil [26] and alrestatin [27], failed to increase GSH levels (Fig. 1E), implying that the inhibition of aldose reductase does not contribute to the ability of EPS to increase GSH levels.
Fig. 1.
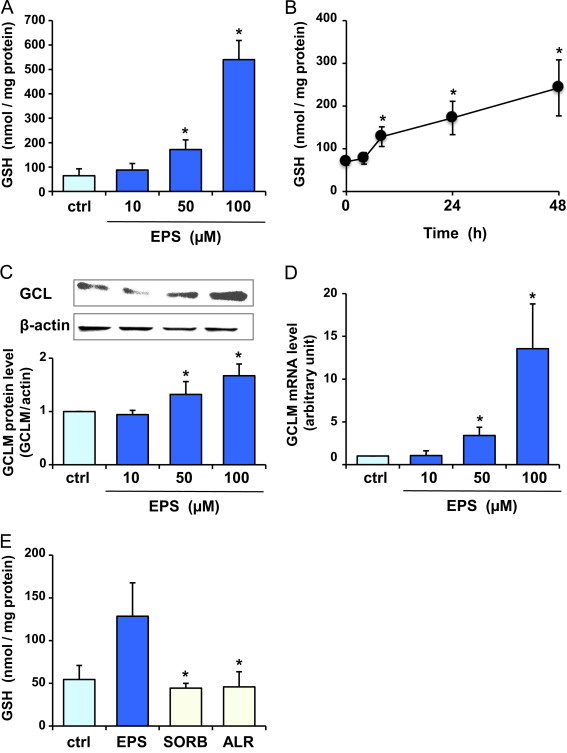
Effect of EPS on GSH and GCLM levels in BAECs. (A) Intracellular GSH levels. BAECs were treated with 10, 50 or 100 µM EPS for 24 h. (B) Time-dependent changes in GSH levels. BAECs were treated with 50 µM EPS for the indicated times. GCLM protein (C) and mRNA (D) levels were estimated by Western blot analysis and quantitative RT-PCR analysis, respectively, after treatment with 10, 50 or 100 µM EPS for 24 h (E) GSH levels were measured after treatment with 50 µM aldose reductase inhibitors, EPS, sorbinil (SORB), and alrestatin (ALR), for 24 h. Values are means±SD of three experiments. *Significant difference from the value of control (P<0.05).
Effect of EPS on Nrf2 in BAECs
Next, we examined how EPS increased the levels of GCL. Recent studies have reported that Nrf2 plays a pivotal role in inducing the expression of genes encoding detoxifying/defensive proteins, including GCL, by binding to the antioxidant response element (ARE) [7–9]. Nuclear translocation is an important mechanism for the activation of the transcription factor Nrf2 [28]. Fig. 2A demonstrates that EPS caused an increase in the nuclear level of active Nrf2, which was estimated by measuring the DNA binding activity of Nrf2. The nuclear levels of active Nrf2 were increased by 1.6- and 1.9-fold by treatment with 50 and 100 µM EPS, respectively. EPS at 10 µM did not significantly increase the nuclear levels of active Nrf2. The results in Fig. 2A were similar to those shown in Fig. 1A–D. As can be seen from Fig. 2B, EPS failed to increase Nrf2 mRNA level.
Fig. 2.
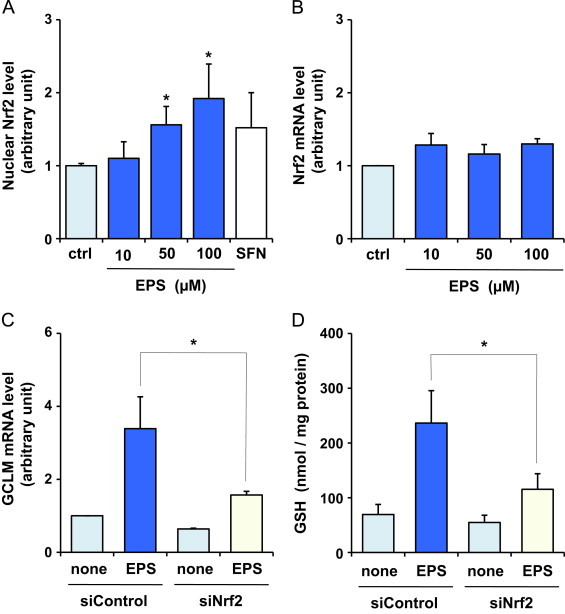
Effect of EPS on Nrf2 in BAECs. Nuclear levels of active Nrf2 (A) and Nrf2 mRNA levels (B) were measured after BAECs were treated with 10, 50 or 100 µM EPS for 4 h. Values are means±SD of three experiments. *Significant difference from the value of control (P<0.05). In (A), BAECs were incubated with a known Nrf2-activator sulforaphane (SFN, 10 µM) for 4 h, as a positive control experiment. COS-7 cells (Nrf2 transfected) nuclear extracts provided by the manufacturer were used as a different positive control; the value was 1.9±0.2. (C and D) BAECs transfected with control siRNA (siControl) or Nrf2 siRNA (siNrf2) were used. The cells were treated or not treated with 50 µM EPS for 24 h. Subsequently, GCLM mRNA levels (C) and GSH levels (D) were measured. Values are means±SD of three experiments. *Significant difference from the value of siControl treated with EPS (P<0.05).
We examined whether Nrf2 levels could alter the increases in GCL and GSH levels in cells treated with 50 µM EPS, by means of Nrf2 knockdown in BAECs. BAECs were transfected with control siRNA (siControl) or Nrf2 siRNA (siNrf2). Nrf2 mRNA expression levels in the cells transfected with Nrf2 siRNA were reduced by approximately 85% relative to those in control siRNA transfected cells (data not shown). As shown in Fig. 2C and D, the increase in GCLM mRNA and GSH levels after EPS treatment was inhibited by the knockdown of Nrf2 expression using siRNA. These results suggest that EPS induces GSH biosynthesis by up-regulating GCL via the activation of Nrf2 in BAECs.
Effect of EPS on HO-1 and Trx-1 in BAECs
Nrf2 controls not only GCL gene but also the genes of many cytoprotective enzymes, such as HO-1 and Trx. To determine whether EPS could alter the levels of cytoprotective proteins other than GCL regulated by Nrf2, we examined the effect of EPS on HO-1 and Trx-1 in BAECs. HO-1 protein levels in BAECs treated with EPS were estimated by fluorescence microscopy studies with PE-conjugated anti-HO-1 monoclonal antibody (Fig. 3A) and by Western blot analysis (Fig. 3B). Fluorescence microscopy studies demonstrated that 50 µM EPS, which induced nuclear levels of active Nrf2, increased HO-1 protein levels. Western blot analysis revealed a dose-dependent increase in HO-1 protein levels in BAECs treated with EPS. This was concomitant with the up-regulation of HO-1 mRNA (Fig. 3C). In BAECs treated with 10 µM EPS, the concentration that had no influence on Nrf2, no significant change was observed in HO-1 protein and mRNA levels. The knockdown of Nrf2 by siRNA suppressed the increase in HO-1 mRNA levels after EPS treatment (Fig. 3D).
Fig. 3.
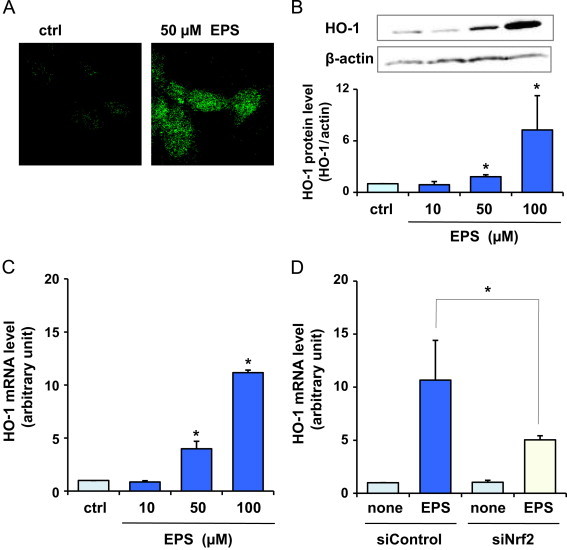
Effect of EPS on HO-1 in BAECs. BAECs were treated with EPS at the indicated concentrations for 24 h. HO-1 protein levels estimated by fluorescence microscopy studies (A) and by Western blot analysis (B). (C) HO-1 mRNA levels. Values in (B) and (C) are means±SD of three experiments. *Significant difference from the value of control (P<0.05). (D) BAECs were transfected with control siRNA (siControl) or Nrf2 siRNA (siNrf2) and were treated or not treated with 50 µM EPS for 24 h. Subsequently, HO-1 mRNA levels were measured. Values are means±SD of three experiments. *Significant difference from the value of siControl treated with EPS (P<0.05).
Trx-1 protein levels were measured by PE-conjugated anti-Trx-1 monoclonal antibody staining, followed by flow cytometry, which can be distinguished from small changes in the amount of the protein because it measures the amount of a protein within each individual cell [29]. The fluorescence intensity of BAECs treated with 50 µM EPS was shifted to the right side of the panel compared with control, suggesting that EPS can increase Trx-1 protein levels (Fig. 4A). As shown in Fig. 4B, Western blot analysis revealed that EPS at 50 and 100 µM stimulated Trx-1 protein expression in BAECs (Fig. 4B). This was concomitant with the up-regulation of Trx-1 mRNA (Fig. 4C). The up-regulation of Trx-1 mRNA after EPS treatment was inhibited by the knockdown of Nrf2 by siRNA (Fig. 4D). It seems that EPS can induce some cytoprotective proteins, including HO-1 and Trx-1, via the Nrf2 pathway.
Fig. 4.
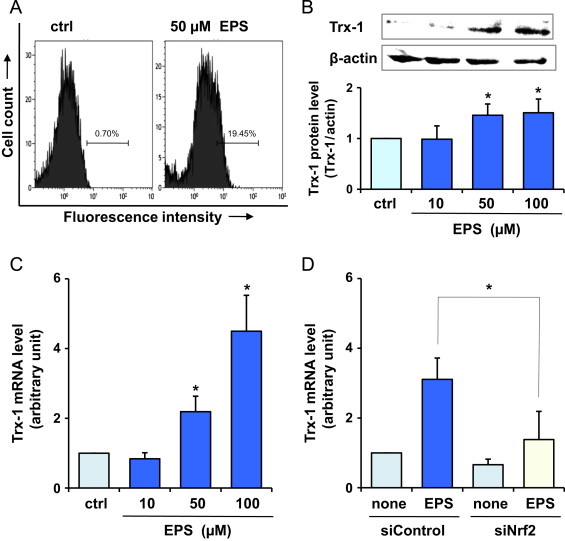
Effect of EPS on Trx-1 in BAECs. BAECs were treated with EPS at the indicated concentrations for 24 h. Trx-1 protein levels estimated by flow cytometry (A) and by Western blot analysis (B). (C) Trx-1 mRNA levels. Values in (B) and (C) are means±SD of three experiments. *Significant difference from the value of control (P<0.05). (D) BAECs were transfected with control siRNA (siControl) or Nrf2 siRNA (siNrf2) and were treated or not treated with 50 µM EPS for 24 h. Subsequently, Trx-1 mRNA levels were measured. Values are means±SD of three experiments. *Significant difference from the value of siControl treated with EPS (P<0.05).
Effect of phosphatidylinositol 3-kinase (PI3K) inhibitor on EPS-stimulated GSH synthesis and Nrf2 activation in BAECs
PI3K is a key molecule in the Nrf2-mediated regulation of GCL [30]. In order to determine whether PI3K was involved in the effect of EPS, we used a specific inhibitor of PI3K, LY294002 [31]. As shown in Fig. 5A and 5B, LY294002 abolished the increase in GCLM mRNA and GSH levels in BAECs treated with EPS. Inhibition of PI3K by LY294002 acutely reduced the capacity of EPS to increase the nuclear levels of active Nrf2 (Fig. 5C). These results indicate that PI3K promotes EPS-induced GSH biosynthesis by activating Nrf2.
Fig. 5.
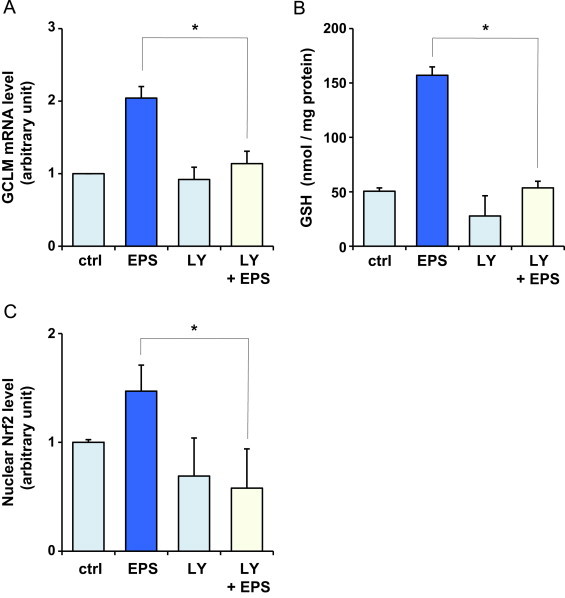
Effect of PI3K inhibitor on EPS-stimulated GSH synthesis and Nrf2 activation in BAECs. BAECs were pretreated with 20 µM LY294002 (LY) for 3 h and then treated with 50 µM EPS. GCLM mRNA (A), intracellular GSH (B) levels, and nuclear levels of active Nrf2 (C) were measured after 4 h, 24 h, and 4 h treatment, respectively. Values are means±SD of three experiments. *Significant difference from the value of control (P<0.05). (C) COS-7 cells (Nrf2 transfected) nuclear extracts provided by the manufacturer were used as a positive control; the value was 2.0±0.3.
Effect of EPS on oxidative stress in BAECs
Finally, we examined whether EPS could protect BAECs from oxidative stress. BAECs were pretreated with EPS (50 µM) for 16 h and then exposed to peroxides. After exposure for 24 h, cell viability was assessed by measuring acid phosphatase activity. The exposures to H2O2 and tert-butylhydroperoxide (t-BHP) resulted in approximately 80% cytotoxicity (Fig. 6A and B). EPS dramatically reduced the cytotoxicity induced by the exposure to the peroxides. dl-Buthionine-(S,R)-sulfoximine, an inhibitor of GCL, abolished the protective effect of EPS on the cytotoxicity (data not shown). It is known that the peroxides cause oxidative damage to mitochondria [32]. Then, the oxidative damage to mitochondria during exposure to t-BHP was estimated by using MitoTracker Red CMXRos, a red fluorescent dye that stains mitochondria in live cells. As shown in Fig. 6C and D, pretreatment of BAECs with EPS protected mitochondria from the t-BHP-induced oxidative damage. Meanwhile, LY294002 abolished the protective effect of EPS on the t-BHP-induced oxidative damage. LY294002 alone, at the concentration used, did not cause any significant decrease in fluorescence intensity (data not shown).
Fig. 6.
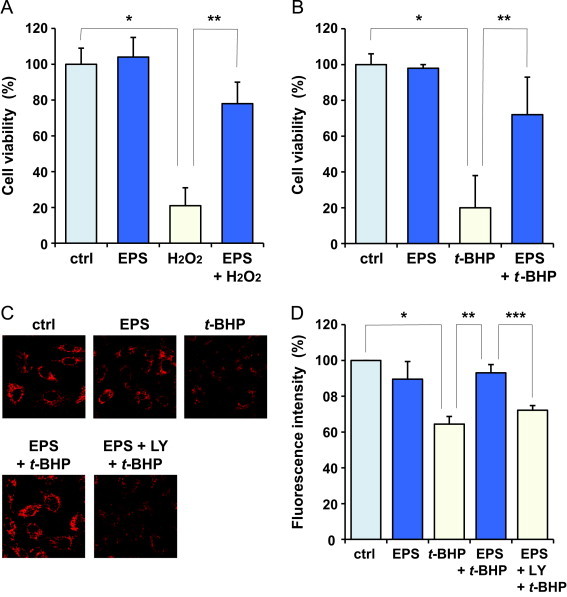
Effect of EPS on oxidative stress in BAECs. BAECs were pretreated with 50 µM EPS for 16 h. Subsequently, the untreated or EPS-treated cells were exposed to 100 µM H2O2 (A) and 50 µM tert-butylhydroperoxide (t-BHP) (B) for 24 h. Cell viability was assessed by measuring acid phosphatase activity. Values are means±SD of six experiments. (C) BAECs were pretreated with 50 µM EPS and 20 µM LY294002 (LY) and then exposed to 50 µM t-BHP for 4 h. Oxidative damage to mitochondria was estimated by using MitoTracker Red CMXRos and a confocal microscope. (D) Quantitative analysis of data shown in C. Values are means±SD of three experiments. Significant differences between the compared groups are indicated: *P<0.05, **P<0.05, and ***P<0.05 versus control, oxidizing agent alone, and oxidizing agent and EPS, respectively.
Discussion
EPS is the only aldose reductase inhibitor currently available for the treatment of diabetic neuropathy. Long-term treatment with EPS is well tolerated; it can delay the progression of diabetic neuropathy and ameliorate symptoms associated with the disease, with minimum adverse effects [2]. The usual dosage of EPS for oral use is 50 mg three times a day. The plasma EPS concentration of 3.9 µg/ml (12 µM) was observed 1 h after a single oral dose of 50 mg [33]. In this study, we examined the effects of EPS at near-plasma concentration on BAECs, which are a commonly used and well-characterized model for studying vascular endothelial dysfunction. Our new findings are that (1) treatment of BAECs with EPS increases intracellular GSH through transcription regulation, (2) EPS can also induce HO-1 and Trx-1 expression, (3) the Nrf2 pathway is involved in EPS-induced GSH biosynthesis and the expression of antioxidative proteins, and (4) EPS reduces the cytotoxicity induced by H2O2 and t-BHP.
Recently, we have shown that EPS affects GSH levels in rat Schwann cells [20]. In the present study, we demonstrated that EPS increased intracellular GSH and GCLM levels in BAECs (Fig. 1A–D). Aldose reductase activity in BAECs was very low and less than one-fifth of that in rat Schwann cells (data not shown). Indeed, two other aldose reductase inhibitors, sorbinil [26] and alrestatin [27], failed to increase GSH levels (Fig. 1E), suggesting that the ability of EPS to increase GSH levels is independent of its ability to inhibit aldose reductase. On the other hand, the knockdown of Nrf2, which regulates GSH levels by controlling the expression of GCL gene [7–9], suppressed the increase in GSH and GCLM mRNA levels after EPS treatment (Fig. 2C and D). Moreover, the activation/nuclear translocation of Nrf2 was observed after treatment with EPS, although EPS did not affect the mRNA level of Nrf2 (Fig. 2A and B). These findings indicate that EPS stimulates GSH biosynthesis in endothelial cells by up-regulating GCL via Nrf2. Activation of Nrf2 involves regulation of a number of kinase pathways like protein kinase cascades (MAPK), PI3K/Akt pathway, protein kinase C (PKC), GSK-3β pathway and ERK signaling pathways [34]. Interestingly, LY294002, a specific inhibitor of PI3K [31], almost completely abolished the EPS-stimulated GSH biosynthesis and Nrf2 activation in BAECs (Fig. 5). The PI3K-mediated phosphorylation of Nrf2 leads to an increase in its stability and subsequently, its transactivation activity [35]. Many studies have reported PI3K mediates the downstream activation of Akt in Nrf2 phosphorylation and nuclear translocation [34,36]. Therefore, we suggest that the PI3K/Akt-Nrf2 pathway is associated with the EPS-stimulated GSH up-regulation. PI3K is activated by 15-deoxy-Δ12,14-prostaglandin J2 (15D-PGJ2), which bears two α,β-unsaturated ketone moieties [37]. Because EPS contains an α,β-unsaturated ketone moiety within its structure, EPS might act as a PI3K activator.
Trx and GSH are the major thiol antioxidants that protect cells from oxidative-stress-induced cytotoxicity. Trx is ubiquitously expressed in endothelial cells and protects the cells from oxidative stress [38]. Recently, Trx and GSH systems were found to be able to provide electrons and to serve as a backup system for each other [39]. In addition, the Trx system regulates the induction of HO-1 expression in BAECs [40]. The magnitude of HO-1 activation in a pro-oxidant environment is critical for protection from the damaging effects of oxidative stress [40]. In this work, we demonstrated that the treatment of BAECs with EPS induces HO-1 and Trx-1 expression (Figs. 3 and 4). Knockdown of Nrf2 by siRNA suppressed the induction of HO-1 and Trx-1 by EPS treatment, indicating that the Nrf2 activation by EPS leads to HO-1 and Trx-1 up-regulation. PI3K is a key molecule in the Nrf2-mediated regulation of HO-1 and Trx proteins [41]. We suggest that PI3K is an upstream regulator of HO-1 and Trx-1, as well as GSH, via Nrf2 activation. Meanwhile, it was demonstrated that 15D-PGJ2 forms a covalent adduct with Keap1 and induces GSH and HO-1 expression in BAECs [42,43]. Electrophiles often interact with Keap1, leading to Nrf2 activation [44]. However, at present, the contribution of electrophilic EPS to Keap1 is unclear, and its evaluation will require further studies.
Oxidative stress impairs endothelial cells, thereby leading to numerous pathological conditions, such as atherosclerosis, diabetes, neurodegeneration, inflammation, and infection [45–48]. It is important to find ways to increase antioxidative ability in order to prevent and/or minimize ROS-induced cellular damage. We assumed that EPS played a role in protecting cells from oxidative stress. To determine whether EPS indeed protects BAECs from oxidative stress, we performed experiments by using H2O2 and t-BHP as the source of oxidative stress. Pretreatment with EPS clearly protected BAECs from toxicity induced by those oxidizing agents (Fig. 6), suggesting that EPS acts to suppress oxidative damage to cells. dl-Buthionine-(S,R)-sulfoximine (an inhibitor of GCL) and zinc protoporphyrin IX (an inhibitor of HO-1) promoted the proxide-induced cytotoxicity, though PX-12 (1-methylpropyl 2-imidazolyl disulfide, an inhibitor of Trx-1) had no effect on the cytotoxicity (data not shown). Nrf2 controls not only GCL, HO-1, and Trx genes but also the genes of many antioxidative enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase [12,13,49]. It is possible that the EPS-induced resistance to oxidative stress in BAECs is associated with the increased expression of some other cytoprotective enzymes. It was shown that EPS improved impaired superoxide generation in streptozotocin-induced diabetic rats [50]. In addition, EPS reduced plasma thiobarbituric acid reactive substances [51] or lipid peroxide level [52], an index of oxidative stress, in type 2 diabetic patients. Taken together, these results provide strong evidence that EPS has new beneficial properties: it may prevent not only diabetic neuropathy but also several vascular diseases caused by oxidative stress.
Drug re-profiling has emerged as a new strategy for drug discovery and development and a way to identify new treatments for diseases [53]. In this strategy, the pharmacological action of existing medicines, whose safety and pharmacokinetics have already been confirmed clinically and whose use has been approved, is examined comprehensively at the molecular level, and the results are adopted for the development of new medicines. The results can also be applied to the development of existing drugs for use as medicines for the treatment of other diseases. Our present study may lead to breakthroughs in drug discovery and development. We showed here that EPS induces GCL, Trx-1, and HO-1 expression in endothelial cells, suggesting the beneficial effect of EPS. GSH, the most abundant antioxidant, plays an essential role in maintaining the cellular redox state [54]. Trx, a key regulator of cardiovascular homeostasis, is an important future target for the development of clinical therapies for cardiovascular disorders associated with oxidative stress [14]. HO-1, which modulates the generation of IL-1β, IL-6, and soluble intercellular adhesion molecule-1, is regarded as an anti-inflammatory enzyme in human endothelial cells [55]. It is reported that studies on the regulation and amplification of HO-1 by pharmacological approaches may lead to the discovery of novel drugs for the treatment of a variety of diseases [56]. Therefore, the therapeutic EPS dose might be a new strategy against related diverse diseases, including vascular disorders and inflammation. Meanwhile, it is reported that the activation of the PI3K-Nrf2 system is a potential therapeutic strategy for Parkinson's disease and other neurodegenerative diseases [35]. EPS is likely to be beneficial for the development of neuroprotective therapies for Parkinson's disease. Further extensive investigations are required to clarify the protective mechanism of EPS against oxidative stress. In addition, because N-acetylcysteine as a GSH precursor is used to treat acute heavy metal poisoning from a suicide or an accident, EPS might be available for the treatment of acute toxicity.
In summary, we demonstrated for the first time that EPS at near-plasma concentration increases intracellular GSH levels in endothelial cells through transcription regulation by stimulating the Nrf2 pathway. Nrf2 activation by EPS leads to HO-1 and Trx up-regulation. Moreover, EPS enhances endothelial cell resistance to oxidative stress. As oxidative stress is the key contributor to aging and many diseases, EPS may be effective in preventing and/or attenuating the progress of those processes.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported in part by an Education and Research Grant from Hokkaido Pharmaceutical University School of Pharmacy and The Akiyama Life Science Foundation.
References
- 1.Steele J.W., Faulds D., Goa K.L. Epalrestat: a review of its pharmacology, and therapeutic potential in late-onset complications of diabetes mellitus. Drugs Aging. 1993;3(6):532–555. doi: 10.2165/00002512-199303060-00007. 8312678 [DOI] [PubMed] [Google Scholar]
- 2.Hotta N., Akanuma Y., Kawamori R., Matsuoka K., Oka Y., Shichiri M., Toyota T., Nakashima M., Yoshimura I., Sakamoto N., Shigeta Y. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: the 3-year, multicenter, comparative aldose reductase inhibitor-diabetes complications trial. Diabetes Care. 2006;29(7):1538–1544. doi: 10.2337/dc05-2370. 16801576 [DOI] [PubMed] [Google Scholar]
- 3.Hotta N., Kawamori R., Fukuda M., Shigeta Y., Aldose Reductase Inhibitor-Diabetes Complications Trial Study Group Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on progression of diabetic neuropathy and other microvascular complications: multivariate epidemiological analysis based on patient background factors and severity of diabetic neuropathy. Diabetic Medicine. 2012;29(12):1529–1533. doi: 10.1111/j.1464-5491.2012.03684.x. 22507139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lum H., Roebuck K.A. Oxidant stress and endothelial cell dysfunction. American Journal of Physiology—Cell Physiology. 2001;280(4):C719–C741. doi: 10.1152/ajpcell.2001.280.4.C719. 11245588 [DOI] [PubMed] [Google Scholar]
- 5.Kugiyama K., Ohgushi M., Motoyama T., Hirashima O., Soejima H., Misumi K., Yoshimura M., Ogawa H., Sugiyama S., Yasue H. Intracoronary infusion of reduced glutathione improves endothelial vasomotor response to acetylcholine in human coronary circulation. Circulation. 1998;97(23):2299–2301. doi: 10.1161/01.cir.97.23.2299. 9639372 [DOI] [PubMed] [Google Scholar]
- 6.Meister A. Selective modification of glutathione metabolism. Science. 1983;220(4596):472–477. doi: 10.1126/science.6836290. 6836290 [DOI] [PubMed] [Google Scholar]
- 7.Kensler T.W., Wakabayashi N., Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annual Review of Pharmacology and Toxicology. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. 16968214 [DOI] [PubMed] [Google Scholar]
- 8.Kalyanaraman B. Teaching the basics of redox biology to medical and graduate students: oxidants, antioxidants and disease mechanisms. Redox Biology. 2013;1(1):244–257. doi: 10.1016/j.redox.2013.01.014. 24024158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kansanen E., Kuosmanen S.M., Leinonen H., Levonen A.L. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biology. 2013;1(1):45–49. doi: 10.1016/j.redox.2012.10.001. 24024136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashino T., Yamamoto M., Yoshida T., Numazawa S. Redox-sensitive transcription factor Nrf2 regulates vascular smooth muscle cell migration and neointimal hyperplasia. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(4):760–768. doi: 10.1161/ATVBAHA.112.300614. 23413426 [DOI] [PubMed] [Google Scholar]
- 11.Eckersley L. Role of the Schwann cell in diabetic neuropathy. International Review of Neurobiology. 2002;50:293–321. doi: 10.1016/s0074-7742(02)50081-7. 12198814 [DOI] [PubMed] [Google Scholar]
- 12.Vincent A.M., Kato K., McLean L.L., Soules M.E., Feldman E.L. Sensory neurons and Schwann cells respond to oxidative stress by increasing antioxidant defense mechanisms. Antioxidants & Redox Signaling. 2009;11(3):425–438. doi: 10.1089/ars.2008.2235. 19072199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim H.J., Vaziri N.D. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. American Journal of Physiology—Renal Physiology. 2010;298(3):F662–F671. doi: 10.1152/ajprenal.00421.2009. 20007347 [DOI] [PubMed] [Google Scholar]
- 14.Yamawaki H., Haendeler J., Berk B.C. Thioredoxin: a key regulator of cardiovascular homeostasis. Circulation Research. 2003;93(11):1029–1033. doi: 10.1161/01.RES.0000102869.39150.23. 14645133 [DOI] [PubMed] [Google Scholar]
- 15.El Hadri K., Mahmood D.F., Couchie D., Jguirim-Souissi I., Genze F., Diderot V., Syrovets T., Lunov O., Simmet T., Rouis M. Thioredoxin-1 promotes anti-inflammatory macrophages of the M2 phenotype and antagonizes atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32(6):1445–1452. doi: 10.1161/ATVBAHA.112.249334. 22516068 [DOI] [PubMed] [Google Scholar]
- 16.Qi H., Chen B., Le X.C., Rong J. Concomitant induction of heme oxygenase-1 attenuates the cytotoxicity of arsenic species from lumbricus extract in human liver HepG2 cells. Chemistry & Biodiversity. 2012;9(4):739–754. doi: 10.1002/cbdv.201100133. 22492492 [DOI] [PubMed] [Google Scholar]
- 17.Joo Choi R., Cheng M.S., Shik Kim Y. Desoxyrhapontigenin up-regulates Nrf2-mediated heme oxygenase-1 expression in macrophages and inflammatory lung injury. Redox Biology. 2014;2:504–512. doi: 10.1016/j.redox.2014.02.001. 24624340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samuel S.M., Thirunavukkarasu M., Penumathsa S.V., Koneru S., Zhan L., Maulik G., Sudhakaran P.R., Maulik N. Thioredoxin-1 gene therapy enhances angiogenic signaling and reduces ventricular remodeling in infarcted myocardium of diabetic rats. Circulation. 2010;121(10):1244–1255. doi: 10.1161/CIRCULATIONAHA.109.872481. 20194885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stocker R., Perrella M.A. Heme oxygenase-1: a novel drug target for atherosclerotic diseases? Circulation. 2006;114(20):2178–2189. doi: 10.1161/CIRCULATIONAHA.105.598698. 17101869 [DOI] [PubMed] [Google Scholar]
- 20.Sato K., Yama K., Murao Y., Tatsunami R., Tampo Y. Epalrestat increases intracellular glutathione levels in Schwann cells through transcription regulation. Redox Biology. 2013;2:15–21. doi: 10.1016/j.redox.2013.11.003. 24363998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parthasarathy S., Barnett J., Fong L.G. High-density lipoprotein inhibits the oxidative modification of low-density lipoprotein. Biochimica et Biophysica Acta. 1990;1044(2):275–283. doi: 10.1016/0005-2760(90)90314-n. 2344447 [DOI] [PubMed] [Google Scholar]
- 22.Connolly D.T., Knight M.B., Harakas N.K., Wittwer A.J., Feder J. Determination of the number of endothelial cells in culture using an acid phosphatase assay. Analytical Biochemistry. 1986;152(1):136–140. doi: 10.1016/0003-2697(86)90131-4. 3954035 [DOI] [PubMed] [Google Scholar]
- 23.Chen Y., Shertzer H.G., Schneider S.N., Nebert D.W., Dalton T.P. Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. Journal of Biological Chemistry. 2005;280(40):33766–33774. doi: 10.1074/jbc.M504604200. 16081425 [DOI] [PubMed] [Google Scholar]
- 24.Tsukamoto M., Tampo Y., Sawada M., Yonaha M. Paraquat-induced oxidative stress and dysfunction of the glutathione redox cycle in pulmonary microvascular endothelial cells. Toxicology and Applied Pharmacology. 2002;178(2):82–92. doi: 10.1006/taap.2001.9325. 11814328 [DOI] [PubMed] [Google Scholar]
- 25.Kawasaki N., Tanimoto T., Tanaka A. Characterization of aldose reductase and aldehyde reductase from rat testis. Biochimica et Biophysica Acta. 1989;996(1–2):30–36. doi: 10.1016/0167-4838(89)90090-3. 2500152 [DOI] [PubMed] [Google Scholar]
- 26.Beyer-Mears A., Cruz E. Reversal of diabetic cataract by sorbinil, an aldose reductase inhibitor. Diabetes. 1985;34(1):15–21. doi: 10.2337/diab.34.1.15. 3917257 [DOI] [PubMed] [Google Scholar]
- 27.Gabbay K.H., Spack N., Loo S., Hirsch H.J., Ackil A.A. Aldose reductase inhibition: studies with alrestatin. Metabolism. 1979;28(4 Suppl. 1):S471–S476. doi: 10.1016/0026-0495(79)90059-3. 122298 [DOI] [PubMed] [Google Scholar]
- 28.Steele M.L., Fuller S., Patel M., Kersaitis C., Ooi L., Münch G. Effect of Nrf2 activators on release of glutathione, cysteinylglycine and homocysteine by human U373 astroglial cells. Redox Biology. 2013;1(1):441–445. doi: 10.1016/j.redox.2013.08.006. 24191238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.T.D. Friedrich F.A. Ray, J.A. Laffin, J.M. Lehman, Flow cytometric quantitation of cellular proteins, in: J.M. Walker (Ed.), The Protein Protocols Handbook Part I, 2002, pp. 45–50. doi:10.1385/1-59259-169-8:45
- 30.Langston W., Circu M.L., Aw T.Y. Insulin stimulation of γ-glutamylcysteine ligase catalytic subunit expression increases endothelial GSH during oxidative stress: influence of low glucose. Free Radical Biology and Medicine. 2008;45(11):1591–1599. doi: 10.1016/j.freeradbiomed.2008.09.013. 18926903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vlahos C.J., Matter W.F., Hui K.Y., Brown R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) Journal of Biological Chemistry. 1994;269(7):5241–5248. 8106507 [PubMed] [Google Scholar]
- 32.Dhanasekaran A., Kotamraju S., Kalivendi S.V., Matsunaga T., Shang T., Keszler A., Joseph J., Kalyanaraman B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. Journal of Biological Chemistry. 2004;279(36):37575–37587. doi: 10.1074/jbc.M404003200. 15220329 [DOI] [PubMed] [Google Scholar]
- 33.Ono Pharmaceutical Co., Ltd., Kinedak (Epalrestat) Package Insert, Osaka, Japan, 2009.
- 34.Bryan H.K., Olayanju A., Goldring C.E., Park B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochemical Pharmacology. 2013;85(6):705–717. doi: 10.1016/j.bcp.2012.11.016. 23219527 [DOI] [PubMed] [Google Scholar]
- 35.Nakaso K., Nakamura C., Sato H., Imamura K., Takeshima T., Nakashima K. Novel cytoprotective mechanism of anti-parkinsonian drug deprenyl: PI3K and Nrf2-derived induction of antioxidative proteins. Biochemical and Biophysical Research Communications. 2006;339(3):915–922. doi: 10.1016/j.bbrc.2005.11.095. 16325767 [DOI] [PubMed] [Google Scholar]
- 36.Chen H.H., Chen Y.T., Huang Y.W., Tsai H.J., Kuo C.C. 4-Ketopinoresinol, a novel naturally occurring ARE activator, induces the Nrf2/HO-1 axis and protects against oxidative stress-induced cell injury via activation of PI3K/AKT signaling. Free Radical Biology and Medicine. 2012;52(6):1054–1066. doi: 10.1016/j.freeradbiomed.2011.12.012. 22245092 [DOI] [PubMed] [Google Scholar]
- 37.Yu X., Egner P.A., Wakabayashi J., Wakabayashi N., Yamamoto M., Kensler T.W. Nrf2-mediated induction of cytoprotective enzymes by 15-deoxy-Δ12,14-prostaglandin J2 is attenuated by alkenal/one oxidoreductase. Journal of Biological Chemistry. 2006;281(36):26245–26252. doi: 10.1074/jbc.M604620200. 16857669 [DOI] [PubMed] [Google Scholar]
- 38.Yamawaki H., Haendeler J., Berk B.C. Thioredoxin: a key regulator of cardiovascular homeostasis. Circulation Research. 2003;93(11):1029–1033. doi: 10.1161/01.RES.0000102869.39150.23. 14645133 [DOI] [PubMed] [Google Scholar]
- 39.Lu J., Holmgren A. The thioredoxin antioxidant system. Free Radical Biology and Medicine. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. 23899494 [DOI] [PubMed] [Google Scholar]
- 40.Trigona W.L., Mullarky I.K., Cao Y., Sordillo L.M. Thioredoxin reductase regulates the induction of haem oxygenase-1 expression in aortic endothelial cells. Biochemical Journal. 2006;394(1):207–216. doi: 10.1042/BJ20050712. 16209660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakaso K., Yano H., Fukuhara Y., Takeshima T., Wada-Isoe K., Nakashima K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Letters. 2003;546(2–3):181–184. doi: 10.1016/s0014-5793(03)00517-9. 12832036 [DOI] [PubMed] [Google Scholar]
- 42.Levonen A.L., Landar A., Ramachandran A., Ceaser E.K., Dickinson D.A., Zanoni G., Morrow J.D., Darley-Usmar V.M. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochemical Journal. 2004;378(2):373–382. doi: 10.1042/BJ20031049. 14616092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oh J.Y., Giles N., Landar A., Darley-Usmar V. Accumulation of 15-deoxy-Δ12,14-prostaglandin J2 adduct formation with Keap1 over time: effects on potency for intracellular antioxidant defence induction. Biochemical Journal. 2008;411(2):297–306. doi: 10.1042/bj20071189. 18237271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi A., Kang M.I., Watai Y., Tong K.I., Shibata T., Uchida K., Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Molecular and Cellular Biology. 2006;26(1):221–229. doi: 10.1128/MCB.26.1.221-229.2006. 16354693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaw A., Doherty M.K., Mutch N.J., MacRury S.M., Megson I.L. Endothelial cell oxidative stress in diabetes: a key driver of cardiovascular complications? Biochemical Society Transactions. 2014;42(4):928–933. doi: 10.1042/BST20140113. 25109981 [DOI] [PubMed] [Google Scholar]
- 46.Kelleher R.J., Soiza R.L. Evidence of endothelial dysfunction in the development of Alzheimer's disease: is Alzheimer's a vascular disorder? American Journal of Cardiovascular Disease. 2013;3(4):197–226. 24224133 [PMC free article] [PubMed] [Google Scholar]
- 47.El Assar M., Angulo J., Rodríguez-Mañas L. Oxidative stress and vascular inflammation in aging. Free Radical Biology and Medicine. 2013;65:380–401. doi: 10.1016/j.freeradbiomed.2013.07.003. 23851032 [DOI] [PubMed] [Google Scholar]
- 48.Cepinskas G., Wilson J.X. Inflammatory response in microvascular endothelium in sepsis: role of oxidants. Journal of Clinical Biochemistry and Nutrition. 2008;42(3):175–184. doi: 10.3164/jcbn.2008026. 18545638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okita Y., Kamoshida A., Suzuki H., Itoh K., Motohashi H., Igarashi K., Yamamoto M., Ogami T., Koinuma D., Kato M. Transforming growth factor-β induces transcription factors MafK and Bach1 to suppress expression of the heme oxygenase-1 gene. Journal of Biological Chemistry. 2013;288(28):20658–20667. doi: 10.1074/jbc.M113.450478. 23737527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kashima K., Sato N., Sato K., Shimizu H., Mori M. Effect of epalrestat, an aldose reductase inhibitor, on the generation of oxygen-derived free radicals in neutrophils from streptozotocin-induced diabetic rats. Endocrinology. 1998;139(8):3404–3408. doi: 10.1210/endo.139.8.6152. 9681489 [DOI] [PubMed] [Google Scholar]
- 51.Hamada Y., Nakamura J., Naruse K., Komori T., Kato K., Kasuya Y., Nagai R., Horiuchi S., Hotta N. Epalrestat, an aldose reductase inhibitor, reduces the levels of Nepsilon-(carboxymethyl)lysine protein adducts and their precursors in erythrocytes from diabetic patients. Diabetes Care. 2000;23(10):1539–1544. doi: 10.2337/diacare.23.10.1539. 11023149 [DOI] [PubMed] [Google Scholar]
- 52.Ohmura C., Watada H., Azuma K., Shimizu T., Kanazawa A., Ikeda F., Yoshihara T., Fujitani Y., Hirose T., Tanaka Y., Kawamori R. Aldose reductase inhibitor, epalrestat, reduces lipid hydroperoxides in type 2 diabetes. Endocrine Journal. 2009;56(1):149–156. doi: 10.1507/endocrj.k08e-237. 18997444 [DOI] [PubMed] [Google Scholar]
- 53.Mizushima T. Drug discovery and development focusing on existing medicines: drug re-profiling strategy. Journal of Biochemistry. 2011;149(5):499–505. doi: 10.1093/jb/mvr032. 21436140 [DOI] [PubMed] [Google Scholar]
- 54.Valko M., Leibfritz D., Moncol J., Cronin M.T., Mazur M., Telser J. Free radicals and antioxidants in normal physiological functions and human disease. International Journal of Biochemistry & Cell Biology. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. 16978905 [DOI] [PubMed] [Google Scholar]
- 55.Taha H., Skrzypek K., Guevara I., Nigisch A., Mustafa S., Grochot-Przeczek A., Ferdek P., Was H., Kotlinowski J., Kozakowska M., Balcerczyk A., Muchova L., Vitek L., Weigel G., Dulak J., Jozkowicz A. Role of heme oxygenase-1 in human endothelial cells: lesson from the promoter allelic variants. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(8):1634–1641. doi: 10.1161/ATVBAHA.110.207316. 20508205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Motterlini R., Foresti R. Heme oxygenase-1 as a target for drug discovery. Antioxidants & Redox Signaling. 2014;20(11):1810–1826. doi: 10.1089/ars.2013.5658. 24180608 [DOI] [PubMed] [Google Scholar]