

Abstract
Autophagy regulates the metabolism, survival, and function of numerous cell types, including those comprising the cardiovascular system. In the vasculature, changes in autophagy have been documented in atherosclerotic and restenotic lesions and in hypertensive vessels. The biology of vascular smooth muscle cells appears particularly sensitive to changes in the autophagic program. Recent evidence indicates that stimuli or stressors evoked during the course of vascular disease can regulate autophagic activity, resulting in modulation of VSMC phenotype and viability. In particular, certain growth factors and cytokines, oxygen tension, and pharmacological drugs have been shown to trigger autophagy in smooth muscle cells. Importantly, each of these stimuli has a redox component, typically associated with changes in the abundance of reactive oxygen, nitrogen, or lipid species. Collective findings support the hypothesis that autophagy plays a critical role in vascular remodeling by regulating smooth muscle cell phenotype transitions and by influencing the cellular response to stress. In this graphical review, we summarize current knowledge on the role of autophagy in the biology of the smooth muscle cell in (patho)physiology.
Keywords: Autophagy, Metabolism, Oxidative stress, Restenosis, Atherosclerosis, Platelet-derived growth factor
Introduction
Vascular smooth muscle cells (VSMCs) comprise the medial layer of blood vessels. By their contraction or relaxation, VSMCs control vessel tone and blood flow, thereby playing a fundamental role in blood pressure regulation and in nutrient and oxygen delivery [1]. VSMCs also demonstrate significant plasticity and are capable of assuming synthetic, osteochondrogenic, and macrophage-like phenotypes, with such roles called upon during development [2], angiogenesis [3], or disease [4]. In diseases such as atherosclerosis, VSMCs can assume a foam cell phenotype, redolent of the sub-intimal macrophage-derived foam cell. Lesional VSMCs also commonly show increased proliferative, migratory, and/or extracellular matrix-synthesizing capacities, indicative of their conversion to the synthetic phenotype. This form of VSMC is commonly associated with vascular injury and leads to a (re)stenosis of the vessel lumen. In hypertensive vessels, VSMCs commonly hypertrophy and increase their contractile tone (Fig. 1).
Fig. 1.
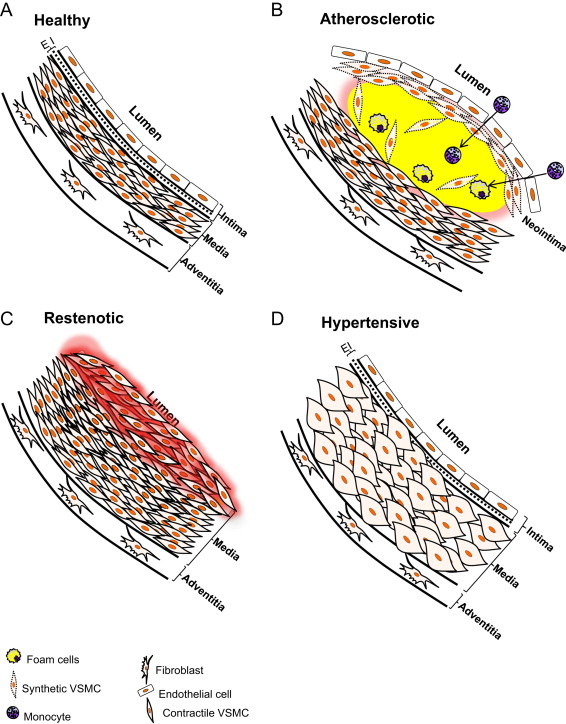
Illustration of VSMCs in healthy and diseased arteries: (A) the mature, healthy mammalian artery is composed of three principal layers: the intima, which comprises endothelial cells; the media, which is occupied primarily by differentiated VSMCs; and the adventitia, which is composed of fibroblasts and connective tissue. (B) The sub-intimal and medial layers of the artery are heavily involved in the development of vascular diseases. In atherosclerosis, LDL and its oxidized forms accumulate in the sub-intimal space, which recruits monocytes and provokes the proliferation and migration of VSMCs. Monocytes differentiate into macrophages which attempt to remove the excess lipids, resulting in the formation of foam cells. The VSMCs also possess an ability to take up lipid and similarly develop into foam-like cells. In addition, synthetic VSMCs migrate to the subintimal space, proliferate, and secrete extracellular matrix, which are thought to help stabilize the atherosclerotic lesion by forming a protective cap around the plaque. Failure to clear excess lipid, debris, and lipid-laden cells, coupled with increased vascular inflammation, could lead to plaque rupture and thrombosis. (C) Severe obstruction of blood flow may occur due to VSMC hyperproliferation after angioplasty or in other vascular injuries where significant tracts of endothelium are removed. The etiology and progression of stenosis is due in part to the phenotypic transition of VSMCs to a synthetic phenotype, which renders VSMCs excessively proliferative and migratory. (D) In hypertension, VSMCs commonly undergo hypertrophy, secrete extracellular matrix, and increase contractile tone. EI=elastic intima.
The unique phenotypic flexibility of VSMCs requires central integration of transcriptional, metabolic, and ultrastructural programs. Autophagy is a principal coordinator of cell homeostasis that could integrate these programs and is finely tuned to respond to stimuli to regulate cell function. Importantly, autophagy is affected in numerous vascular disease states, including restenosis, atherosclerosis, and hypertension [4]. The molecular activation of autophagy is primed via phosphorylation of ULK1 (Atg1), which then coordinates interactions of other critical proteins in the autophagy cascade [5], leading to encapsulation of cellular constituents in a double-membrane vesicle called the autophagosome. The autophagosome then fuses with the lysosome, leading to degradation of the compartmentalized contents and release of essential building blocks such as amino acids for reutilization (Fig. 2). This form of autophagy is commonly activated as a survival mechanism to degrade damaged cellular components and to maintain sufficient nutrient and biosynthetic stores under conditions of bioenergetic distress [6]. Autophagy is also important in regulating the life-cycle of numerous cellular organelles, such as mitochondria, endoplasmic reticulum, and peroxisomes, and it has other specialized roles in the cell that utilize different strategies to target cargo for degradation (for review, see [7]). In this graphical review, we discuss primarily the major form of autophagy, i.e., macroautophagy, and its role in the biology of the VSMC.
Fig. 2.
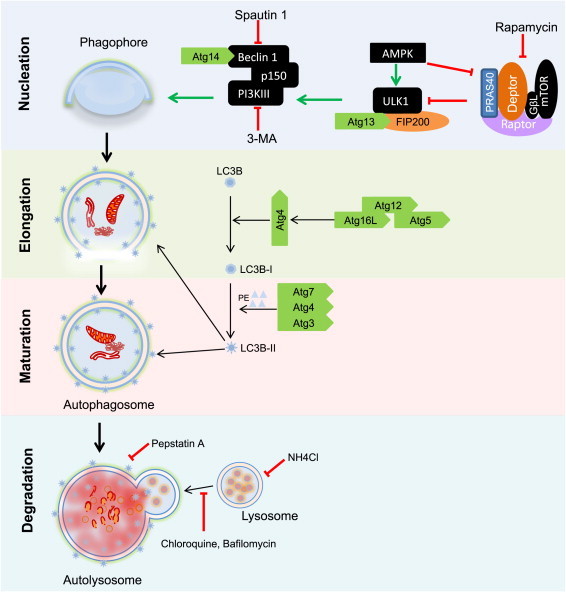
Regulation of autophagy. The activation of autophagy commences with the activation of Atg1 (ULK1) by upstream signaling molecules, e.g., adenosine 5′ monophosphate kinase (AMPK), which causes its dissociation from raptor. Activated Atg1 signals through other downstream mediators such as PI3KIII leading to the recruitment of scaffolding molecules important for the nucleation of the phagophore. The maturation of the phagophore to a double membrane bound vesicle (the autophagosome) depends on two ubiquitin-like conjugation systems. In the first system, Atg7 and Atg10 catalyze the conjugation of Atg12 to Atg5 to form an Atg12–Atg5 complex, which is stabilized by Atg16L. The second conjugation reaction involves 2 steps: first, Atg4 activates Atg8 (LC3); the activated LC3, or LC3-I, is then conjugated to phosphatidylethanolamine (PE) to form LC3-PE or LC3-II, a reaction catalyzed by Atg7 and Atg3. LC3-II is recruited to both the outer and inner faces of the growing autophagosome and it is required for autophagosome maturation. The final step of autophagy involves the fusion of the autophagosome with the lysosome leading to degradation of intra-autophagosomal contents by lysosomal enzymes.
Importance of autophagy in VSMC phenotype
The fact that VSMCs can display at least three different phenotypic identities suggests an ability to respond effectively to numerous extracellular and intracellular stimuli. Indeed, numerous growth factors appear to be important regulators of VSMC phenotype, in part by modulating autophagic activity [4]. Of pathological importance, platelet-derived growth factor (PDGF)-BB – which is secreted by numerous cell types following vascular injury and is known to promote rapid contractile-to-synthetic phenotype transition of VSMCs – was recently shown to activate autophagy. Autophagy initiated by PDGF-BB helps to remove contractile proteins as well as proteins damaged by lipid electrophiles [8,9]; thus, this form of autophagy appears to hasten transition to the synthetic VSMC phenotype and increase cell survival under conditions associated with increased oxidative stress (Fig. 3). The mechanism by which PDGF-BB activates autophagy remains to be identified.
Fig. 3.
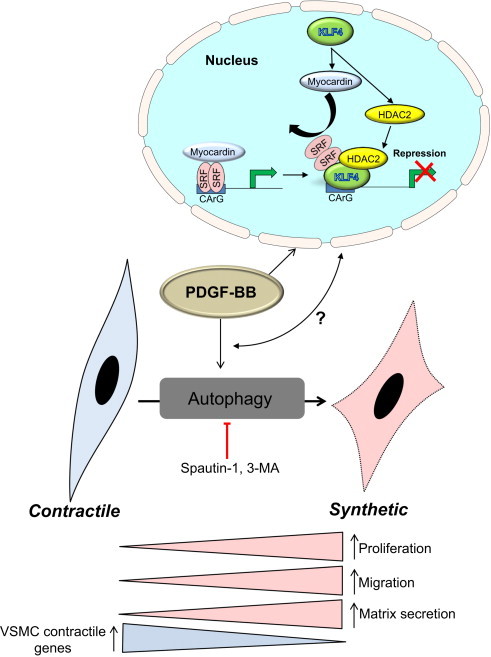
Regulation of autophagy and phenotype switching by PDGF-BB. A characteristic feature of smooth muscle cells in the media of the healthy artery is the expression of contractile proteins such as α-smooth muscle actin, calponin, and smooth muscle cell myosin heavy chain-II (MHC-II). However, diseased vessels commonly present a de-differentiated smooth muscle cell phenotype having lower contractile protein abundance and higher levels of proteins such as collagen I, osteopontin, and vimentin, indicative of the synthetic phenotype. A robust inducer of this phenotype is PDGF-BB, which is elevated after vascular injury or during disease. In cultured cells, PDGF-BB regulates the expression of numerous genes that underlie the synthetic phenotype program. Additionally, PDGF-BB activates autophagy, which hastens degradation of the contractile apparatus to hasten the emergence of the synthetic phenotype. Inhibition of autophagy in vitro using 3-methlyadenine (3-MA) or spautin-1 stabilizes the contractile machinery, thereby preserving the contractile phenotype, and it prevents excessive migration, proliferation and extracellular matrix production commonly evoked by exposure to PDGF-BB. How the autophagic program converges and collaborates with the metabolic and transcriptional machinery to control VSMC phenotype requires additional elucidation.
Autophagy is activated in VSMCs by multiple other conditions and signaling agents including starvation (particularly lack of amino acids), metabolic stress, hypoxia, reactive species, drugs, growth factors, and cytokines (Fig. 4; reviewed in [4]). The form(s) of autophagy elicited by these species are not equal and have different consequences. For example, PDGF-BB elicits a cytoprotective form of autophagy, while other growth factors, cytokines, and secreted factors, such as osteopontin [10], Sonic hedgehog (Shh) [11], tumor necrosis factor-α [12], and angiotensin II [13], activate forms of VSMC autophagy that could, under some conditions, promote cell death. Moreover, drugs such as telmisartan and atorvastatin induce forms of autophagy associated with decreased lipid droplet accumulation and diminished calcification, respectively; these forms of autophagy appear to play a causal role in the VSMC phenotypic changes, as inhibition of autophagy prevented phenotypic changes [14,15]. Rapamycin-based drugs commonly found in drug-eluting stents (e.g., everolimus and sirolimus) activate autophagy; yet unlike PDGF, which increases cell proliferation and potentiates the synthetic phenotype, these drugs promote a contractile phenotype. Similarly, verapamil and emodin induce autophagy and prevent cell proliferation [16,35].
Fig. 4.
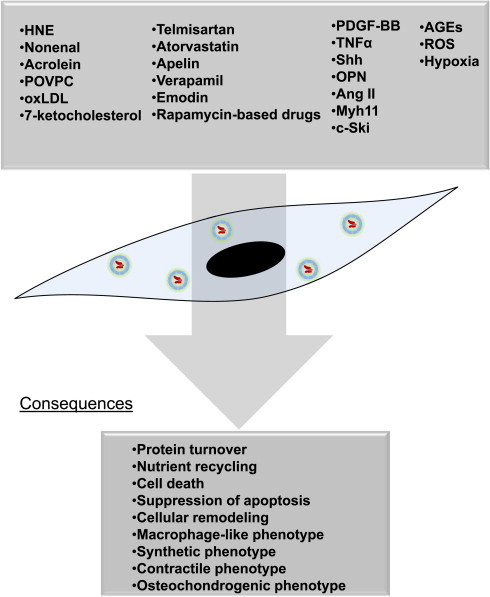
Inducers and inhibitors of autophagy in VSMCs. Autophagy in VSMCs can be activated by metabolic stressors, inflammatory signals, mitogens, cytokines, specific pathways regulating cellular homeostasis (e.g., ER stress), and reactive species resulting from oxidative stress. The following stimuli have been shown to regulate autophagy in VSMCs: ROS from multiple sources including the mitochondrial electron transport chain, NADPH oxidases, peroxisomes and the cytochrome P450 system; reactive lipid species such as free aldehydes (e.g., 4-hydroxynonenal; HNE) and 7-ketocholesterol; bioactive core aldehydes such as 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (POVPC); both oxLDL and non-oxidized cholesterol; advanced glycation end products (AGEs); inflammatory cytokines, growth factors and other proteins such as: tumor necrosis factor-α (TNF-α), osteopontin (OPN), PDGF-BB, sonic hedgehog (shh), insulin and insulin-like growth factor-1 (IGF-1), angiotensin II, apelin; Myh11; c-Ski; metabolic stressors such as hypoxia and nutrient deprivation or excess; pharmacological agents and drugs such as telmisartan, atorvastatin, everolimus, sirolimus, rapamycin; and inorganic phosphate (Pi). These agents may either increase or decrease autophagic flux, leading to several potential outcomes, including, but not limited to, protein and organelle turnover, changes in cell viability and responses to stress, and changes in VSMC phenotype.
Although several studies illustrate that autophagy occurs in VSMCs of restenotic, atherosclerotic, and hypertensive vessels [4], few studies to date have tested the direct role of VSMC autophagy in vascular health and disease. The use of Cre-lox technology and smooth muscle-targeted transgenic models could help to address the role of autophagy directly in VSMCs. Such studies have been performed in other cell types pertinent to vascular pathology such as macrophages and endothelial cells [17,18]. Two fundamental questions remain to be addressed: (1) How does VSMC autophagy affect vascular health, disease etiology, and the progression of pathologies such as restenosis and atherosclerosis and (2) How do different autophagy-inducing stimuli coordinate apparently disparate forms of autophagy that elicit specific VSMC phenotypes?
Role of redox state and reactive species in VSMC autophagy
Importantly, many autophagic stimuli have a redox component, which suggests that changes in the reduced and oxidized forms of pyridine nucleotides, the relative abundance of antioxidants and oxidants, and the generation of electrophiles may be initiating factors in the autophagic program. For example, growth factors and cytokines such as PDGF-BB and TNF-α involve initial bursts of free radicals or sustained elevations in oxidative stress [19–21]. Hence, the mechanisms underlying activation of autophagy by these and other autophagic stimuli likely involve the redox state of the cell. This is supported by the fact that reactive lipids, advanced glycation end products, and free radical species regulate autophagy activity in VSMCs (recently reviewed in [4]). Oxidized LDL is a potent inducer of autophagy that contains reactive aldehydic and ketone species, which educe a cytoprotected phenotype or cell death [22–26], depending on the concentration. Moreover, mitochondrial-derived superoxide has been shown to be an important instigator of inorganic phosphate (Pi)-induced autophagy, which prevents calcification. Interestingly, reactive lipids such as 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (POVPC) and 4-hydroxynonenal (HNE) both induce autophagy, yet POVPC promotes the synthetic phenotype [27,28] while HNE does not appear to do so ([4,8,22]; and unpublished observations).
Controlled modulation of autophagy for treatment of vascular disease
The wealth of evidence showing that autophagy controls VSMC function and responses to stress supports the idea that modulating its activity in vivo may be a viable therapeutic option for preventing or mitigating vascular disease; however, this approach has several difficult challenges to overcome. The different “forms” of autophagy elicited by various stimuli and its two-edged nature – which promotes either survival or death, or differentially modulate VSMC phenotypic identity – suggest that bluntly regulating its activity with inhibitors that overtly diminish or activate autophagy could have deleterious effects. Rather, a controlled stimulation or inhibition of autophagy may be effective in modulating some facets of vascular disease. It is unlikely that inhibitors of autophagy such as 3-methyladenine (3-MA) would be effective because of the likely off-target effects of inhibiting the PI3K/Akt pathway. Numerous studies suggest that modest stimulation of autophagy could be beneficial, as rapamycin-eluting stents both increase autophagy and prevent VSMC hyperproliferation in vivo; however, the rapamycin-based drugs used in stents appear to prevent proliferation via inhibiting mTORC1 activity and regulating expression of the cell cycle machinery [29]. Hence, it is unclear what the role of autophagy may be in the therapeutic effects of these agents.
Drugs that currently appear to be more specific for regulating autophagy, such as spautin-1, are promising candidates for therapy (Fig. 5). In cell culture studies, spautin-1 was shown to inhibit autophagy in an Akt-independent manner and to prevent completely the VSMC hyperproliferation caused by PDGF [8]. Nevertheless, to our knowledge, this drug has not been tested in vivo. However, other inhibitors of autophagy have been shown to have beneficial effects in vascular disease. For example, chloroquine was shown to prevent hypertension in a monocrotaline mouse model [30].
Fig. 5.
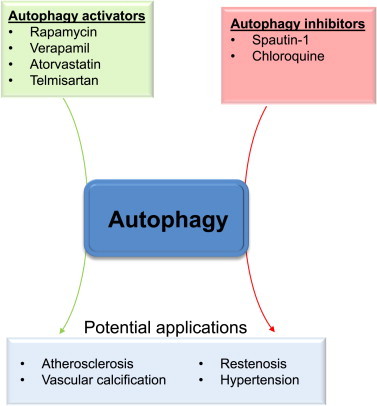
Pharmacological regulation of autophagy to treat vascular diseases. Pharmacological activators and inhibitors of autophagy could be used to mitigate vascular disease. It is likely that controlled activation of autophagy could have beneficial actions in atherosclerosis, whereas its modest inhibition could diminish restenosis and perhaps hypertension. Promising small molecules include spautin-1 (to inhibit autophagy) and certain clinically available drugs, the latter of which may impart some of their effects through stimulating autophagic flux.
It is likely that activation of autophagy may be a viable approach for diseases such as atherosclerosis. Pharmacological agents that simulate caloric restriction or activate sirtuins, both of which increase autophagy [31], could promote modest increases in autophagy and mitigate disease progression. Indeed, caloric restriction and sirtuin 1 have been shown to have beneficial effects in the context of atherosclerosis by diminishing oxidative stress or protecting against DNA damage [32–34]. Moreover, clinically approved drugs such as verapamil and atorvastatin, which are known to induce autophagy in VSMCs, may represent promising candidates to modulate autophagy and VSMC phenotype in specific vascular disease states. As reviewed elsewhere [35,36], numerous other drugs have been shown to modulate autophagy in the cardiovascular system, but whether these drugs regulate autophagy in VSMCs in vivo remains unanswered. A more thorough understanding of how different stimuli coordinate phenotypic changes via autophagy would undoubtedly help in developing targeted therapies. An ‘ideal’ autophagic regulator would likely be capable of discriminating basal from pathological autophagy or modulate only those metabolic pathways that are deregulated in disease. Future studies will hopefully shed light on the role of autophagy in maintaining VSMC health and identify the mechanisms by which autophagy is affected in cardiovascular diseases, the knowledge of which could be used to derive new and more efficacious therapies.
Conflicts of interest
None.
Acknowledgment
The authors acknowledge funding from the National Institute of Health (GM103492 and HL78825).
References
- 1.Webb R.C. Smooth muscle contraction and relaxation. Advances in Physiology Education. 2003;27(1–4):201–206. doi: 10.1152/advan.00025.2003. 14627618 [DOI] [PubMed] [Google Scholar]
- 2.Owens G.K., Kumar M.S., Wamhoff B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiological Reviews. 2004;84(3):767–801. doi: 10.1152/physrev.00041.2003. 15269336 [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nature Medicine. 2000;6(4):389–395. doi: 10.1038/74651. 10742145 [DOI] [PubMed] [Google Scholar]
- 4.Salabei J.K., Hill B.G. Implications of autophagy for vascular smooth muscle cell function and plasticity. Free Radical Biology and Medicine. 2013;65:693–703. doi: 10.1016/j.freeradbiomed.2013.08.003. 23938401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim J. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology. 2011;13(2):132–141. doi: 10.1038/ncb2152. 21258367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh R., Cuervo A.M. Autophagy in the cellular energetic balance. Cell Metabolism. 2011;13(5):495–504. doi: 10.1016/j.cmet.2011.04.004. 21531332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ravikumar B. Regulation of mammalian autophagy in physiology and pathophysiology. Physiological Reviews. 2010;90(4):1383–1435. doi: 10.1152/physrev.00030.2009. 20959619 [DOI] [PubMed] [Google Scholar]
- 8.Salabei J.K. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochemical Journal. 2013;451(3):375–388. doi: 10.1042/BJ20121344. 23421427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J. c-Ski inhibits autophagy of vascular smooth muscle cells induced by oxLDL and PDGF. PLoS ONE. 2014;9(6):e98902. doi: 10.1371/journal.pone.0098902. 24887307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng Y.H. Osteopontin stimulates autophagy via integrin/CD44 and p38 MAPK signaling pathways in vascular smooth muscle cells. Journal of Cellular Physiology. 2012;227(1):127–135. doi: 10.1002/jcp.22709. 21374592 [DOI] [PubMed] [Google Scholar]
- 11.Li H. Sonic hedgehog promotes autophagy of vascular smooth muscle cells. American Journal of Physiology—Heart and Circulatory Physiology. 2012;303(11):H1319–H1331. doi: 10.1152/ajpheart.00160.2012. 23023870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia G. Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol. Cell Biol. 2006;84(5):448–454. doi: 10.1111/j.1440-1711.2006.01454.x. 16942488 [DOI] [PubMed] [Google Scholar]
- 13.Yu K.Y. Mitochondrial KATP channel involvement in angiotensin II-induced autophagy in vascular smooth muscle cells. Basic Research in Cardiology. 2014;109(4):416. doi: 10.1007/s00395-014-0416-y. 24847907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li B.H. Telmisartan-induced PPARgamma activity attenuates lipid accumulation in VSMCs via induction of autophagy. Molecular Biology Reports. 2014 doi: 10.1007/s11033-014-3757-6. 25249228 [DOI] [PubMed] [Google Scholar]
- 15.Liu D. Atorvastatin protects vascular smooth muscle cells from TGF-beta 1-stimulated calcification by inducing autophagy via suppression of the beta-catenin pathway. Cellular Physiology and Biochemistry. 2014;33(1):129–141. doi: 10.1159/000356656. 24481040 [DOI] [PubMed] [Google Scholar]
- 16.Salabei J.K. Verapamil stereoisomers induce antiproliferative effects in vascular smooth muscle cells via autophagy. Toxicology and Applied Pharmacology. 2012;262(3):265–272. doi: 10.1016/j.taap.2012.04.036. 22627060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng N. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E−/− mice. Scientific Reports. 2014;4(5519) doi: 10.1038/srep05519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao X. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metabolism. 2012;15(4):545–553. doi: 10.1016/j.cmet.2012.01.022. 22445600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sundaresan M. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270(5234):296–299. doi: 10.1126/science.270.5234.296. 7569979 [DOI] [PubMed] [Google Scholar]
- 20.Chen X. Role of reactive oxygen species in tumor necrosis factor-alpha induced endothelial dysfunction. Current Hypertension Reviews. 2008;4(4):245–255. doi: 10.2174/157340208786241336. 20559453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abe J., Berk B.C. Reactive oxygen species as mediators of signal transduction in cardiovascular disease. Trends in Cardiovascular Medicine. 1998;8(2):59–64. doi: 10.1016/S1050-1738(97)00133-3. 21235913 [DOI] [PubMed] [Google Scholar]
- 22.Hill B.G. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochemical Journal. 2008;410(3):525–534. doi: 10.1042/BJ20071063. 18052926 [DOI] [PubMed] [Google Scholar]
- 23.Haberzettl P., Hill B.G. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biology. 2013;1(1):56–64. doi: 10.1016/j.redox.2012.10.003. 24024137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu K.D. Autophagy plays a protective role in free cholesterol overload-induced death of smooth muscle cells. Journal of Lipid Research. 2010;51(9):2581–2590. doi: 10.1194/jlr.M005702. 20484746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinet W. Interactions between cell death induced by statins and 7-ketocholesterol in rabbit aorta smooth muscle cells. British Journal of Pharmacology. 2008;154(6):1236–1246. doi: 10.1038/bjp.2008.181. 18469840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding Z.F. Regulation of autophagy and apoptosis in response to ox-LDL in vascular smooth muscle cells, and the modulatory effects of the microRNA hsa-let-7g. International Journal of Cardiology. 2013;168(2):1378–1385. doi: 10.1016/j.ijcard.2012.12.045. 23305858 [DOI] [PubMed] [Google Scholar]
- 27.Cherepanova O.A. Oxidized phospholipids induce type VIII collagen expression and vascular smooth muscle cell migration. Circulation Research. 2009;104(5):609–618. doi: 10.1161/CIRCRESAHA.108.186064. 19168440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pidkovka N.A. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circulation Research. 2007;101(8):792–801. doi: 10.1161/CIRCRESAHA.107.152736. 17704209 [DOI] [PubMed] [Google Scholar]
- 29.Rosner D., McCarthy N., Bennett M. Rapamycin inhibits human in stent restenosis vascular smooth muscle cells independently of pRB phosphorylation and p53. Cardiovascular Research. 2005;66(3):601–610. doi: 10.1016/j.cardiores.2005.01.006. 15914125 [DOI] [PubMed] [Google Scholar]
- 30.Long L. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal Bone morphogenetic protein Type II receptor degradation. Circulation Research. 2013;112(8):1159–1170. doi: 10.1161/CIRCRESAHA.111.300483. 23446737 [DOI] [PubMed] [Google Scholar]
- 31.Morselli E. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Disease. 2010;1:e10. doi: 10.1038/cddis.2009.8. 21364612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorenne I. Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation. 2013;127(3):386–396. doi: 10.1161/CIRCULATIONAHA.112.124404. 23224247 [DOI] [PubMed] [Google Scholar]
- 33.Dolinsky V.W., Dyck J.R. Calorie restriction and resveratrol in cardiovascular health and disease. Biochimica et Biophysica Acta. 2011;1812(11):1477–1489. doi: 10.1016/j.bbadis.2011.06.010. 21749920 [DOI] [PubMed] [Google Scholar]
- 34.Guo Z. Dietary restriction reduces atherosclerosis and oxidative stress in the aorta of apolipoprotein E-deficient mice. Mechanisms of Ageing and Development. 2002;123(8):1121–1131. doi: 10.1016/s0047-6374(02)00008-8. 12044962 [DOI] [PubMed] [Google Scholar]
- 35.Salabei J.K., Conklin D.J. Cardiovascular autophagy: crossroads of pathology, pharmacology and toxicology. Cardiovascular Toxicology. 2013;13(3):220–229. doi: 10.1007/s12012-013-9200-8. 23408289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nemchenko A. Autophagy as a therapeutic target in cardiovascular disease. Journal of Molecular and Cellular Cardiology. 2011;51(4):584–593. doi: 10.1016/j.yjmcc.2011.06.010. 21723289 [DOI] [PMC free article] [PubMed] [Google Scholar]