

Abstract
4-Hydroxynonenal (HNE) is a cytotoxic and genotoxic lipid oxidation secondary product which is formed endogenously upon peroxidation of cellular n-6 fatty acids.
However, it can also be formed in food or during digestion, upon peroxidation of dietary lipids. Several studies have evidenced that we are exposed through food to significant concentrations of HNE that could pose a toxicological concern. It is then of importance to known how HNE is metabolized after oral administration. Although its metabolism has been studied after intravenous administration in order to mimick endogenous formation, its in vivo fate after oral administration had never been studied.
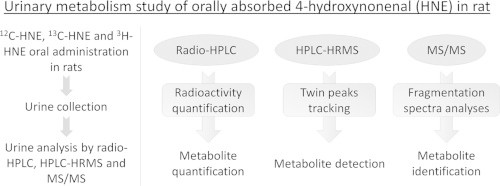
In order to identify and quantify urinary HNE metabolites after oral administration in rats, radioactive and stable isotopes of HNE were used and urine was analyzed by radio-chromatography (radio-HPLC) and chromatography coupled with High Resolution Mass Spectrometry (HPLC–HRMS).
Radioactivity distribution revealed that 48% of the administered radioactivity was excreted into urine and 15% into feces after 24 h, while 3% were measured in intestinal contents and 2% in major organs, mostly in the liver. Urinary radio-HPLC profiles revealed 22 major peaks accounting for 88% of the urinary radioactivity. For identification purpose, HNE and its stable isotope [1,2-13C]-HNE were given at equimolar dose to be able to univocally identify HNE metabolites by tracking twin peaks on HPLC–HRMS spectra. The major peak was identified as 9-hydroxy-nonenoic acid (27% of the urinary radioactivity) followed by classical HNE mercapturic acid derivatives (the mercapturic acid conjugate of di-hydroxynonane (DHN-MA), the mercapturic acid conjugate of 4-hydroxynonenoic acid (HNA-MA) in its opened and lactone form) and by metabolites that are oxidized in the terminal position. New urinary metabolites as thiomethyl and glucuronide conjugates were also evidenced. Some analyses were also performed on feces and gastro-intestinal contents, revealing the presence of tritiated water that could originate from beta-oxidation reactions.
Keywords: 4-Hydroxynonenal, Lipoperoxidation, Metabolism, HRMS, Isotope tracking
Abbreviations: a.m.u, atomic mass unit; DHN, 1,4-dihydroxy-2-nonene; DHN-MA, 1,4-dihydroxy-2-nonane mercapturic acid; dpm, desintegration per minute; HNA, 4-hydroxy-2-nonenoic acid; HNA-MA, 4-hydroxy-2-nonanoic mercapturic acid; HNE, 4-hydroxynon-2-enal; 3HNE, [4-3H]-4-hydroxynon-2-enal; 12C-HNE, 4-hydroxynon-2-enal; 13C-HNE, [1,2-13C2]-4-hydroxynon-2-enal; HPLC–HRMS, High Pressure Liquid Chomatography–High Resolution Mass Spectrometry; MS/MS, tandem mass spectrometry
Graphical abstract
Highlights
-
•
Stable isotope tracking was used to study HNE urinary fate after oral administration.
-
•
Twenty-two HNE metabolites were identified and quantified in urine.
-
•
The major urinary metabolite was 9-hydroxy-nonenoic acid.
-
•
Oxidation in terminal position was a prominent biotransformation pathway.
-
•
New urinary metabolites as thiomethyl and glucuronide conjugates were evidenced.
Introduction
4-Hydroxynonenal (HNE) is an α,β-unsaturated aldehydic secondary lipid peroxidation product, arising from omega-6 polyunsaturated fatty acid oxidation. It has been extensively studied in numerous pathological states in which its generation is a consequence of oxidative stress accompanying inflammatory events. However, this product is not only a mere product of lipid peroxidation: it is a reactive compound that can play a biological role or have cytotoxic and genotoxic properties, depending on its concentration, through covalent attachment to macromolecules.
In addition to its endogenous formation, HNE can be found in various foodstuffs, upon non enzymatic oxidation of dietary PUFAs due to an imbalance between pro- and anti-oxidant compounds, or due to food-processing oxidation during cooking or storage. Seppanen and Csallany have reported HNE formation in thermally oxidized oils, and its presence in food fried in those oils to a concentration reaching 600 mg/kg of oil [1,2]. HNE can be found in meat products, or in fish in concentration reaching 5 mg/kg in case of prolonged storage [3,4] and in PUFA fortified infant formula and baby food (up to 0.8 mg/kg, 10 days after package opening) [5]. Our group has shown that heme iron concentration may have a prominent role [6].
Several studies have evidenced the oral toxicity of HNE. Nishikawa et al. reported an acute toxicity of HNE after a single dose given by gavage (from 10 to 1000 mg/kg BW). They observed a diffuse liver cell necrosis 14 days after the treatment in all treated animals, while rats that died few hours after the 1000 mg/kg dosing presented extensive kidney tubular necrosis [7]. They concluded that HNE administered at high dose can induce lethal renal damage before severe hepatic damage occurs. Such hepatotoxicity was reported after injection in the portal vein of 4-hydroxy-hexenal (HHE), an analog of HNE formed upon omega-3 PUFAs [8]. Chung et al. reported a liver tumorigenic activity of crotonaldehyde, another α, β-unsaturated aldehyde, after oral exposure in drinking water for 113 weeks. Wacker et al. [9] reported a slight increase of HNE specific propano-adducts in forestomach DNA in rats treated with single oral dose of HNE (500 mg/kg BW). Kang et al. reported nephro-and hepatotoxicity of HNE after 4 weeks of daily dose ranging from 0.5 to 12.5 mg/kg BW [10]. From this study, these authors have calculated a no-adverse-effect-level (NOAEL) below 0.5 mg/kg/day for HNE.
Taken together, those studies show that we are exposed through food to significant concentrations of HNE that could pose a toxicological concern. It is then of importance to known how HNE is metabolized after oral administration. If some studies have been done, by our group [11,12] and others [13,14], on the in vivo metabolism of HNE or HHE after parenteral administration, none has been achieved to elucidate oral fate of HNE.
In the present work, we used a strategy based on both radioactive (i.e. [4-3H]-HNE) and stable (i.e. [1,2-13C]-HNE) isotope labeling to elucidate its oral metabolic fate, that is to say to identify and quantify the major HNE metabolites in urine.
Material and methods
Chemicals
Reagents were purchased from Sigma-Aldrich (France). Solvents for HPLC were of analytical grade and purchased from Fisher, France. Ultrapure water from MilliQ system (gradient A10, Millipore) was used for HPLC.
[4-3H]-4-Hydroxynon-2(E)-enal (3H-HNE) was obtained by acid hydrolysis of aldehyde protected diethyl acetal precursor, synthesized as previously described [15], and purified by thin layer chromatography (Fig. 1). The synthesis was carried out at CEA, Service des Molecules Marquees, CEN, Saclay, France. Radiochemical purity (>95%) was checked by HPLC before administration to rats. Specific activity was 222 GBq/mol [1,2-13C2]-4-Hydroxynon-2(E)-enal (13C-HNE) (Fig. 1): 13C-HNE was prepared from ethyl [1,2-13C2]-2-bromoacetate according to our published method [16], with modifications as previously described [17]. 4-Hydroxynon-2(E)-enal (HNE): HNE was prepared from ethyl 2-bromoacetate by the same method as for 13C-HNE. 4,9-dihydroxy-2-nonenoic acid (9-hydroxy-HNA) standard was obtained as previously described [12]. 1,4-dihydroxynonane mercapturic acid (DHN-MA) and 4-hydroxynonanoic mercapturic acid (HNA-MA) standards were synthesized as previously described [11].
Fig. 1.
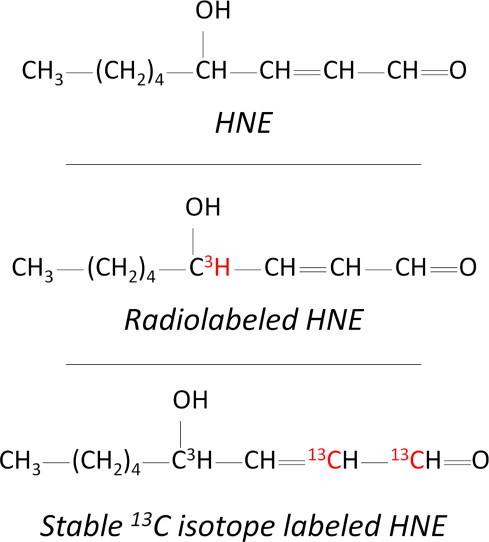
Labeling position of the HNE isotopes used for metabolite tracking and quantification.
Animal treatments
Male Wistar rats (200 g) were purchased from Charles River. Animal care was in accordance with our local ethic committee (number TOXCOM/0008/FG). 3 Rats were treated by gavage with 0.9 ml of an equimolar mixture of HNE and 13C-HNE and of 3H-HNE diluted in water. 3 Other rats were treated by gavage with 1 ml of the same mixture of 12C-HNE and 13C-HNE diluted in water, without radiolabeled HNE. These treatments contained 1 mg/ml of HNE, 1 mg/ml of 13C-HNE and, when appropriate, 0.97 MBq/ml of 3H-HNE. HNE concentration was checked spectrophotometrically (λ, 223 nm; ε, 13,750 l/mol/cm).
The rats were housed in individual metabolism cages and food (A03 breeding diet, SAFE, France) and tap water were provided ad libitum. 24 h Feces and 0–6 h and 6–24 h urine were collected. Rats were sacrificed by CO2 inhalation. Several organs and tissues were collected and stored at − 80 °C until analysis. Organs and tissues coming from rats treated with radiolabeled HNE were used for distribution of radioactivity and radio-HPLC analyses. Urine of rats treated with HNE and 13C-HNE mixture only was used for HPLC/HRMS analyses (1st set of experiments, Fig. 2). For radio-HPLC analyses followed by MS/MS analyses (2nd set of experiments, Fig. 2), a mixture of radioactive (1/3) and non radioactive (2/3) urine was used, in order to be able to detect radioactive peaks but to avoid an important radioactive contamination of mass spectrometry equipment.
Fig. 2.
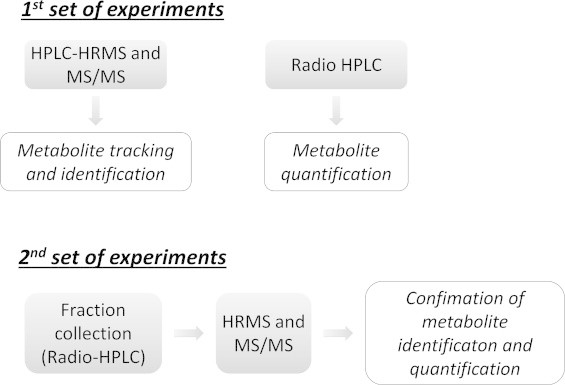
Workflow for metabolites identification and quantification.
Radioactivity determination
Radioactivity was counted PerkinElmer Tri-carb 2910 TR liquid scintillation analyzer (PerkinElmer, France), using Ultima Gold (PerkinElmer) as scintillation cocktail. Urine was analyzed directly, while intestinal contents and 0–24 h homogenized feces were first dissolved in 1 M NaOH before being counted for radioactivity. Rat tissues and organs were homogenized and sampled (n = 3, 100–200 mg) before combustion by a Packard Sample Oxidizer (Model 307, PerkinElmer) and then sample radioactivity was counted using Monophase S (PerkinElmer) as scintillation cocktail.
“Metabolite tracking” strategy (Figs. 2 and 3)
Fig. 3.
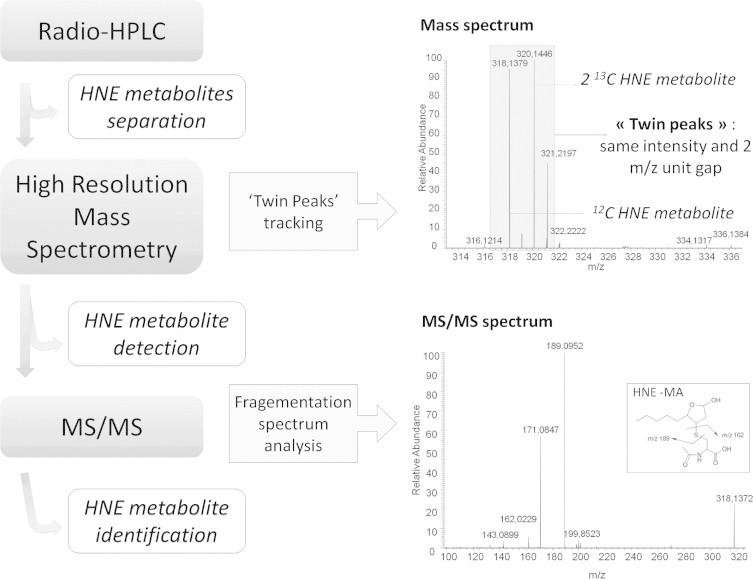
Work flow of the 2nd set of experiments. Metabolites were identified using radio-HPLC separation, ‘twin peaks’ tracking by High Resolution Mass Spectrometry and further ion fragmentation by MS/MS.
Metabolite tracking strategy has been previously used by our group to study in vitro HNE metabolism in colon epithelial cells [17,18]. In the present study, urine and feces coming from rats treated with HNE were analyzed by HPLC–HRMS for twin peaks tracking and identification purpose and by radio-HPLC for quantification purpose. Urine was first fully analyzed by HPLC–HRMS and radio HPLC (Fig. 2, 1st set of experiment) but quantification and identification results were difficult to match on the basis of retention time, due to slight differences between the two techniques (even if the same column was used for chromatography). To clarify and to confirm our results, we undertook a second set of experiments (Fig. 2), analyzing radio-HPLC collected fractions by HRMS and MS/MS (Fig. 3). For feces samples, we only realized the 2nd set of experiments as metabolites concentrations were too low to achieve a full analysis.
Radio-HPLC system
The system consisted of two HPLC Kontron 420 pumps (Kontron, Selabo, France), managed with the HPLC software Diamir (Varian Medical Systems, USA). The HPLC system was equipped with a 500 µl loop and an Interchrom Strategy KR100 column C18 (5 µm, 4.6×250 mm) (Interchim, France) kept in an oven thermostated at 35 °C and connected to an on-line radioactivity analyzer (Packard Flo-one, Flow scintillation analyzer) using Flo-scint as the scintillation cocktail or connected to a Gilson FC 204 fraction collector (Gilson, France) for 4 tubes/min collection, using Packard Ultima Gold as scintillation cocktail, when appropriate. Two mobile phases were used. Mobile phase A containing: water/acetonitrile/acetic acid: 97.5/2.5/0.1; and mobile phase B containing: water/acetonitrile/acetic acid: 40/60/0.1. Elution gradient was as follows: 100% A from 0 to 4 min, a linear gradient from 4 to 20 min from 0% to 6% B, a linear gradient from 20 to 25 min from 6% to 25% B, a plateau at 25% B from 25 to 35 min, a linear gradient from 35 to 50 min from 25% to 90% B, a plateau at 90% B from 50 to 60 min.
Urine was injected directly whereas intestinal contents and 0–24 h homogenized feces were extracted by 2 volumes of 0.5 M NaCl.
HPLC–HRMS analysis (1st set of experiments)
Urine sample analysis was achieved using HPLC coupled to an LTQ Orbitrap XL high-resolution mass spectrometer (Thermo, Fisher Scientific, Les Ulis, France). Briefly, 25 µl of urine was injected into an HPLC system using the same column and the same mobile phases and gradients as for radio-HPLC profiling, and a 1/4 post-column splitting HRMS acquisitions were performed using electrospray ionization (ESI) in the negative mode with the following operating parameters: spray voltage (−3.5 kV), heated transfer capillary temperature (300 C), heated transfer capillary voltage (−1 V), tube lens voltage (−50 V), sheath gas (N2) flow rate 40 au, auxiliary gas (N2) flow rate 5 au. Full MS spectra were recorded at a resolution of 30,000 from m/z 120–750.
Chromatograms for 0–6 h and 6–24 h urine and mass spectra were analyzed for 0–6 h and 6–24 h urine and feces samples using Xcalibur software (Thermo Scientific). Twin peaks were detected on the basis of their m/z difference of 2.0067 a.m.u. corresponding to similar chemical formula for 12Cn and 12C(n−2)13C2 compounds using the MetWorks software package (Thermo Scientific). The search criteria for chemical formula determination were restricted to a mass error below 5 ppm and C, H, O, N, S, P, K and Na were the only accepted elements. Further MS/MS fragmentation was achieved on twin peaks, for HNE metabolite identification. For MS/MS experiments, excitation parameters (isolation width (1.5 a.m.u.), normalized collision energy (35%), excitation time (30 ms)) were adjusted in order to get maximum structural information for the compound of interest. When possible, high resolution was used (R=7500) for fragment ion composition assignment. All analyses were achieved under automatic gain control conditions using helium as damping as well as collision gas for MS/MS experiments.
HRMS and MS/MS infusion experiments (2nd set of experiments) (Fig. 3)
Fractions of interest collected from urine and feces samples were passed through a SPE cartridge. Obtained purified fractions were evaporated under a nitrogen stream, and taken up by the appropriate volume of methanol–water (50/50, v/v) to get a concentration of 1–5 ng/µl. Samples were infused at a flow rate of 8 µl/min into the LTQ Orbitrap XL high-resolution mass spectrometer. The same mass spectrometry parameters as described above were used for HRMS acquisitions and MS/MS experiments.
Results and discussion
Radioactivity distribution
Radioactivity distribution was analyzed in the group treated with 3H-HNE in urine, feces, intestinal contents and major organs (Fig. 4). 24 h After radiolabeled HNE oral administration, most radioactivity was found in urine (48%). 15% of the orally given dose was found in feces. Around 3% was measured in the intestinal contents (including 1.7% in cecum content) and around 2% in major organs (including 1.5% in liver). Total radioactivity recovery was 68% knowing that this result is non-exhaustive given that only the radioactivity in major organs was measured. In a previous study done by our group on HNE metabolism following intra-venous administration, 67% and 3% of the administered radioactivity was found in urine and feces, respectively, 48 h after treatment [11].
Fig. 4.
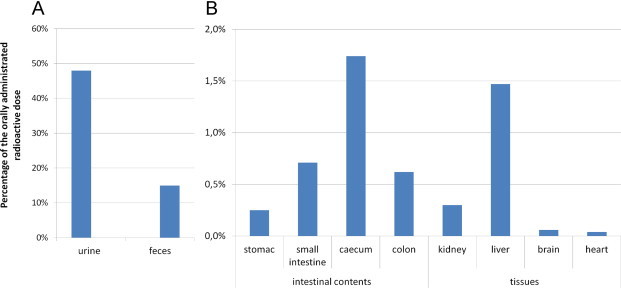
Radioactivity distribution after 3H-HNE oral administration in rats (n=3). Percentage of the given radioactivity found in 0–24 h urine and feces (A), intestinal contents and tissues after 24 h (B) (volume or weight related).
Urinary HNE metabolites identification and quantification
First, we performed radio-HPLC analyses on urine collected within 24 h after oral administration of 3H-HNE (Fig. 5A). Then, we performed radio-HPLC analyses on urine collected over the first 6 h after treatment and on urine collected between 6 and 24 h after treatment (Fig. 5B and C).
Fig. 5.
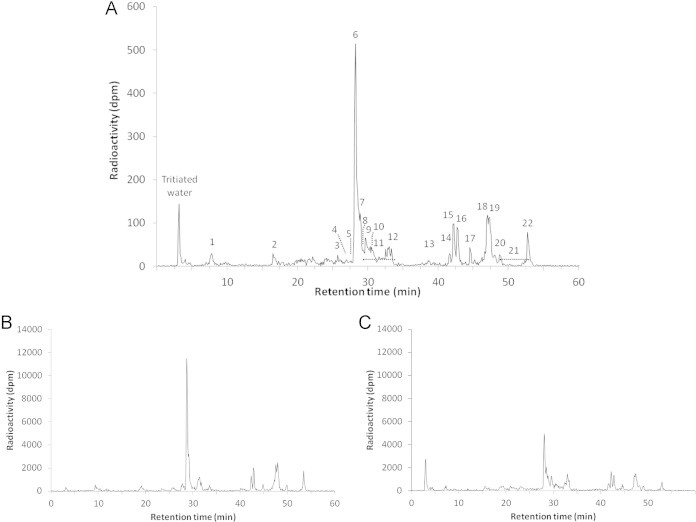
Radio-HPLC profiles of 3H-HNE urinary metabolites after oral administration in rats. A: 0–24 h urine. B: 0–6 h urine. C: 6–24 h urine.
On 0–24 h urine, a first peak eluted at 3.1 min, then the major peak eluted at 28.2 min, followed by a group of peaks eluting up to 33 min. Finally, a second group of peaks eluted between 41 and 52.5 min. The analyzed peaks represent 88% of the urinary radioactivity.
When 0–6 h urine was analyzed by radio-HPLC, the relative abundance of the first peak was very reduced compared to the major peak. In contrast, in 6–24 h urine analysis, the first peak became one of the most important, whereas the relative abundance of the major peak was greatly reduced.
Tritiated water
In radio-HPLC profiles we obtained from 0–24 h and 6–24 h rat urine, a peak which retention time corresponded to the column void volume was observed. This peak was not present when samples were passed through a SPE cartridge. To tentatively identify this peak, we have tested different methods, using evaporation or activated charcoal treatment of the SPE cartridge unretained fraction of urine. Activated charcoal added to the unretained fraction for 24 h, followed by filtration on a Whatman GF/A filter, did not decrease the radioactivity of the unretained fraction. This result allowed us to identify this peak as tritiated water. However, evaporation of this fraction led to a minor concentration of radioactivity in the unretained fraction. This could be explained by a weak enrichment of tritium, due to isotope discrimination during water phase change [19].
Moreover, when organ samples were left for several hours for drying purpose, an important loss of radioactivity (55%) was observed, probably indicating tritiated water evaporation.
Tritiated water has been observed previously by our group during radio-HPLC analysis of urine after intra-venous HNE treatment [12] or of liver slices incubated with HNE [20]. It represented 3.5% of 0–24 h urinary radioactivity and was completely unretained by all kinds of SPE cartridge. Moreover, it was recovered in the condensate after evaporation. Other authors have reported the presence of tritiated water in several in vitro and in vivo 3H-HNE metabolic studies [21,22] but using tritiated HNE with a different labeling position: [2-3H]-HNE compared to the [4-3H]-HNE used in our group. These authors attributed the formation of tritiated water to the first step of β-oxidation of HNA. Indeed, they reported a decrease of tritiated water formation by the use of 4-pentaenoic acid, an inhibitor of β-oxidation [21]. Interestingly, a decrease in tritiated water following 4-pentaenoic acid treatment was also observed in a study done in our group with the tritium located on carbon 4, probably indicating further β-oxidation steps [20]. The fact that in the present study, but also in the previous studies done by our group [12,20], the formation of tritiated water increased with time after dosing or incubation time, indicates probably a multi-step process. Indeed, in the present study, tritiated water accounted for 1.1% of urinary radioactivity for 0–6 h urine, while it represented 15.9% of the urinary radioactivity for the 6–24 h urine. Alary et al. [12] found no tritiated water in the 0–2 h urine, 3.5% in the 2–24 h urine and 28% in the 24–48 h urine, following intra-venous HNE treatment. Moreover, in the present study, tritiated water represented the major radioactive peak in the intestinal contents 24 h following exposure (Fig. 9). All together these results indicate an important catabolism of HNE, given by oral or parenteral route, through β-oxidation of HNA and possibly also through ω-oxidation of HNA and further β-oxidation steps originating from the carbon 9 of HNA as recently reported by some authors [23].
Fig. 9.
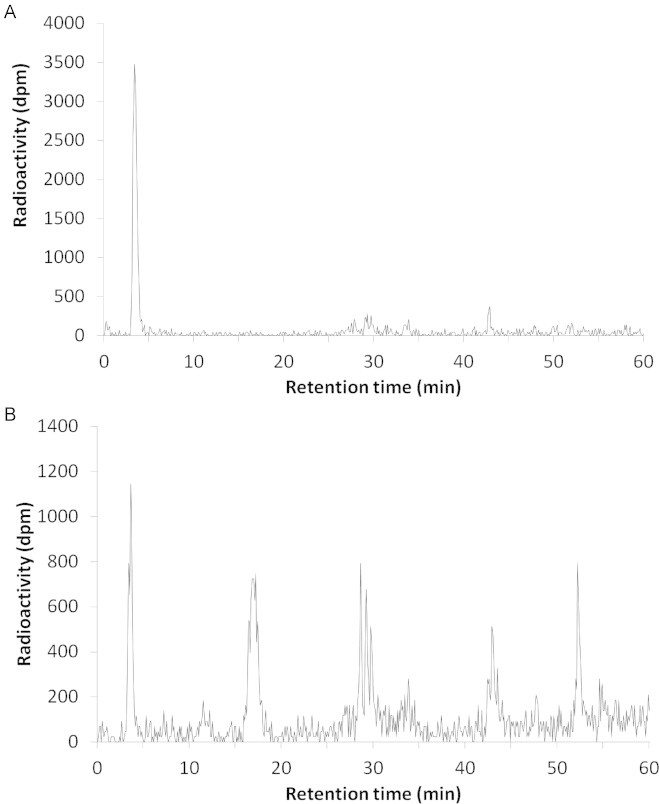
Cecal radio-HPLC profile at 24 h (A) and fecal 0–24 h radio-HPLC profile (B) of rats treated with HNE.
Other HNE metabolites identification
Peak 1 eluted at 7.8 min (Fig. 5A). Using our metabolite tracking technique with high resolution mass spectrometry for highlighting HNE metabolites on the basis of their 12C/13C2 characteristic isotopic pattern, twin peaks were evidenced at m/z 173.0456 (12C7H9O5, calc. 173.0444, Δ=0.5 ppm) and 175.0523 (12C513C2H9O5, calc. 175.0512, Δ=0.5 ppm) (Table 1). High resolution accurate mass measurements allowed accessing elemental composition of these ions, which were both in agreement with the molecular formula C7H9O5. The MS/MS spectrum of the m/z 173 precursor ion exhibited 2 fragment ions corresponding to two subsequent decarboxylations (m/z 129 and m/z 85 ions) that indicated the presence of a dicarboxylic acid. On the basis of the molecular formula and of this fragmentation pattern, peak 1 can correspond to 7-carboxy-4-hydroxy-heptenoic acid. Such metabolite has not been observed in our previous studies. This metabolite could originate from a β-oxidation step originating from the carbon 9 of HNA, as we observed the “twin compound” still retaining the stable isotope labeling on carbon 1 and 2 (ion at m/z 175), precluding any possibility of β-oxidation step on the carbons 1 and 2 of HNA. The presence of this metabolite accounting for 2% of urinary radioactivity reinforces the HNE disposition pathway evidenced recently by Jin et al. [23].
Table 1.
Metabolites identified in 0–24 h urine of rats treated with HNE.
| Peak (% of urinary radioactivity) |
Ion m/z |
Fragments |
Proposed structure | |||
|---|---|---|---|---|---|---|
| From HNE from [13C]-HNE | Chemical formula | 12C fragment m/z ions | 13C fragment m/z ions | Fragment interpretation | ||
| 1 (2.07) | 173.0456 175.0523 | C7H9O5 | 155 | [M–H–H2O]− | 7-Carboxy-4-hydroxy-heptenoic acid | |
| 137 | [M–H–2H2O]− | |||||
| 129 | [M–H–COO]− | |||||
| 111 | [M–H–COO–H2O]− | |||||
| 85 | [M–H–COO–COO]− | |||||
| 2 (2.20) | 205.1082 207.1148 | C9H17O5 | 187 | [M–H–H2O]− | 4,8,9-Trihydroxy-nonanoic acid | |
| 169 | [M–H–2H2O]− | |||||
| 161 | [M–H–COO]− | |||||
| 145 | [M–H–CHOH–CHOH]− | |||||
| 3 (1.85) | 185. 0821 187.0888 | C9H13O4 | 167 | 169 | [M–H–H2O]− | 9-Carboxy-HNE |
| 141 | 143 | [M–H–COO]− | ||||
| 123 | 125 | [M–H–COO–H2O]− | ||||
| 4 (1.29) | 201.0769 203.0836 | C9H13O5 | 183 | [M–H–H2O]− | 9-Carboxy-HNA | |
| 157 | [M–H–COO]− | |||||
| 139 | [M–H–COO-H2O]− | |||||
| 5 (0.23) | 203.0926 205.0992 | C9H15O5 | 185 | [M–H–H2O]− | 9-Carboxy-4-Hydroxy-nonanoic acid | |
| 159 | [M–H–COO]− | |||||
| 141 | [M–H–COO–H2O]− | |||||
| 6 (26.72) | 187.0977 189.1044 | C9H15O4 | 169 | 171 | [M–H–H2O]− | 9-Hydroxy-HNA |
| 143 | 144 | [M–H–COO]− | ||||
| 113 | 114 | [M–H–CHOH]− | ||||
| 7 (3.74) | 350.1281 352.1348 | C14H24NO7S | 332 | 334 | [M–H–H2O]− | 9-Carboxy-DHN-MA |
| 306 | 308 | [M–H–COO]− | ||||
| 221 | 223 | [A+S]− | ||||
| 162 | 162 | [B]− | ||||
| 128 | 128 | [B–S]− | ||||
| 364.1064 366.1136 | C14H22NO8S | 346 | [M–H–H2O]− | 9-Carboxy-HNA-MA | ||
| 320 | [M–H–COO]− | |||||
| 235 | [A+S]− | |||||
| 162 | [B]− | |||||
| 8 (1.25) | 203.0020 205.0091 | C9H15O5 | 185 | [M–H–H2O]− | 8,9-Dihydroxy-HNA | |
| 172 | [M–H–CHOH]− | |||||
| 141 | [M–H–COO–H2O]− | |||||
| 121 | [M–H–COO–2H2O]− | |||||
| 9 (5.22) | 185.0818 187.0882 | C9H13O4 | 167 | 169 | [M–H–H2O]− | 9-Oxo-HNA or 9-Hydroxy-4-oxo-nonenoic acid |
| 141 | 142 | [M–H–COO]− | ||||
| 123 | 124 | [M–H–COO–H2O]− | ||||
| 348.1109 350.1177 | C14H22NO7S | 304 | 306 | [M–H–COO]− | 9-Carboxy-HNE-MA | |
| 219 | 221 | [A+S]− | ||||
| 162 | 162 | [B]− | ||||
| 10 (2.14) | 336.1486 338.1552 | C14H26NO6S | 207 | 209 | [A+S]− | 9-Hydroxy-DHN-MA |
| 189 | 191 | [A+S–H2O]− | ||||
| 159 | 160 | [A+S–H2O–CHOH]− | ||||
| 128 | 128 | [B–S]− | ||||
| 11 (2.35) | 235.1035 237.1077 | C10H19O4S | 191 | [M–H–COO]− | 3-Thiomethyl-9-Hydroxy-HNA | |
| 187 | [M–H–S–CH3]− | |||||
| 249.0803 251.0867 | C10H17O5S | 205 | [M–H–COO]− | 3-Thiomethyl-9-Carboxy-HNA | ||
| 201 | [M–H–SCH3]− | |||||
| 12 | No twin peak | |||||
| 13 (1.46) | 332.1172 334.1239 | C14H22NO6S | 203 | 205 | [A+S]− | 9-Hydroxy-HNA-lactone-MA |
| 162 | 162 | [B]− | ||||
| 346.0964 348.1031 | C14H20NO7S | 301 | 304 | [M–H–COO]− | 9-Carboxy-HNA-lactone-MA | |
| 217 | 219 | [A+S]− | ||||
| 162 | 162 | [B]− | ||||
| 14 (1.10) | 199.0598 201.0667 | C9H11O5 | 181 | 183 | [M–H–H2O]− | 9-Carboxy-4-oxo-nonenoique acid |
| 155 | 157 | [M–H–COO]− | ||||
| 137 | 139 | [M–H–COO-H2O]− | ||||
| 15 (4.38) | 189.1132 191.1198 | C9H17O4 | 171 | [M–H–H2O]− | 4, (8 or 9)-Dihydroxy-nonanoic acid | |
| 129 | [M–H–CH2COOH]− | |||||
| 99 | ||||||
| 59 | [CH2COOH] | |||||
| 16 (3.92) | 189.1133 191.1199 | C9H17O4 | 171 | 173 | [M–H–H2O]− | 4, (8 or 9)-Dihydroxy-nonanoic acid |
| 129 | 129 | [M–H–CH2COOH]− | ||||
| 59 | 61 | [CH2COOH] | ||||
| 17 | No twin peak | |||||
| 333.1554 335.1621 | C15H25O8 | 193 | 193 | Glucuronide | 4-Glucuronide-DHN | |
| 157 | 159 | [M–H-glucuronide]− | ||||
| 347.1346 349.1416 | C15H23O9 | 193 | 193 | Glucuronide | 4-Glucuronide-HNA | |
| 18 (6.53) | 320.1539 322.1603 | C14H26NO5S | 191 | 193 | [A+S]− | DHN-MA |
| 128 | 128 | [B–S]- | ||||
| 19 (6.98) | 334.1331 336.1395 | C14H24NO6S | 290 | 292 | [B–COO]− | HNA-MA |
| 205 | 207 | [A+S]− | ||||
| 162 | 162 | [B]− | ||||
| 128 | 128 | [B–S]− | ||||
| 320.1539 322.1603 | C14H26NO5S | 191 | 193 | [A+S]− | DHN-MA | |
| 128 | 128 | [B–S]− | ||||
| 20 (1.35) | 318.1379 320.1445 | C14H24NO5S | 274 | 276 | [B–COO]− | HNE-MA |
| 189 | 191 | [A+S]− | ||||
| 171 | 173 | [A+S–H2O]− | ||||
| 162 | 162 | [B]− | ||||
| 334.1328 336.1396 | C14H24NO6S | 205 | 207 | [A+S]− | HNA-MA | |
| 162 | 162 | [B]− | ||||
| 128 | 128 | [B–S]− | ||||
| 219.1064 221.1130 | C10H19O3S | 175 | [M–H–COO]− | 3-Thiomethyl-HNA | ||
| 171 | [M–H–SCH3]− | |||||
| 21 (4.31) | 316.1225 318.1285 | C14H22NO5S | 162 | 162 | [B]− | HNA–lactone-MA |
Peak 2 eluted at 16.5 min and accounted for 2.2% of urinary radioactivity. Twin peaks at m/z 205.1086/207.1148, corresponding to the C9H17O5 molecular formula were obtained. The MS/MS spectrum of the m/z 205 ion exhibited a fragment ion corresponding to a decarboxylation (m/z 161 ion) and to the loss of a terminal CH2OH−CH2OH (m/z 145 ion). Based on these information, the structure of 4,8,9-tri-hydroxy-nonanoic acid was tentatively assigned to peak 2. Such chemical structure has not been evidenced in our previous studies. However, ω−1 oxidation has been reported by Jin et al. [23].
Peak 3 eluted at 25.7 min and accounted for 1.85% of urinary radioactivity. The C9H13O4 molecular formula could be deduced from the twin peaks observed at m/z 185.0821/187.0888. MS/MS of the m/z 185 ion exhibited a fragment ion corresponding to a decarboxylation (m/z 141 ion). As the m/z 143 ion (and not m/z 142) was observed following fragmentation of the m/z 187 twin precursor ion, it can be concluded that the decarboxylation did not concern the labeled C1 carbon atom and that consequently the carboxylic acid function is located at the carbon 9 position. This peak should then correspond to 9-carboxy-DHN.
The HNE metabolite tracking allowed us to tentatively identify those minor peaks as new HNE metabolites.
Peak 4 eluted at 26.9 min and represented 1.3% of urinary radioactivity. Twin peaks corresponding to the C9H13O5 molecular formula were measured at m/z 201.0769/203.0836. The MS/MS spectrum of the m/z 201 precursor ion exhibited major fragment ions corresponding to dehydration and decarboxylation processes (m/z 183, 157 and 139 fragment ions). This peak should then correspond to 9-carboxy-HNA. Such a metabolite has been previously observed in the rat, after intravenous administration [12] and in mice after intraperitoneal treatment [24].
Peak 5 eluted at 27.6 min and represented 0.23 % of urinary radioactivity and displayed twin peaks at m/z 203.0926/205.0992 corresponding to the C9H15O5 molecular formula. Similarly as for peak 4, MS/MS carried out on the m/z 203 ion yielded a fragment ions corresponding to dehydration and decarboxylation (m/z 185, 159 and 141 ions). This peak was then tentatively identified as 9-carboxy-4-hydroxy-nonanoic acid. This minor peak has not been evidenced before.
Peak 6 was the major peak obtained on the radio-HPLC analysis; it eluted at 28.2 min and represented 27% of the urinary radioactivity. Twin peaks were measured at m/z 187.0977 (12C9H15O4, calc. 187.0965, Δ=0.7 ppm)/189.1044 (12C713C2H15O4, calc. 189.1032, Δ=0.6 ppm), allowing to propose C9H15O4 as molecular formula. The MS/MS spectrum of the m/z 187 ion exhibited a fragment ion corresponding to a decarboxylation (m/z 143 ion, Fig. 7). Opposite to what has been observed for Peak 3, an m/z 144 ion was observed following fragmentation of the m/z 189 twin peak (corresponding to a 45 a.m.u. loss). It can thus be concluded that the decarboxylation involved the labeled C1 carbon atom of the metabolite. Consecutive losses of 30 a.m.u. (m/z 113 and 114 fragment ions) from both m/z 143 and 144 ions indicated the presence of a CH2OH moiety in the terminal position. Moreover, this peak coeluted with 9-hydroxy-HNA standard. This allowed us to identify this metabolite as 9-hydroxy-HNA. As for 9-carboxy-HNA, 9-hydroxy-HNA has been observed in urine of rats and mice after parenteral HNE administration. Such terminal oxidation process could involve cytochrome P450 4A family because fibrate treated mice excreted more of those compounds than untreated ones, or than PPARα-deficient and fibrate treated ones [12,24]. Cytochrome P450 4A enzymes specifically catalyze the ω- and ω−1-hydroxylation of medium and long chain fatty acids [25]. The presence of 9-hydroxy-HNA as a major metabolite in the present study indicated a prominent role of cytochrome P450 4A family in HNE metabolization after oral administration.
Fig. 7.
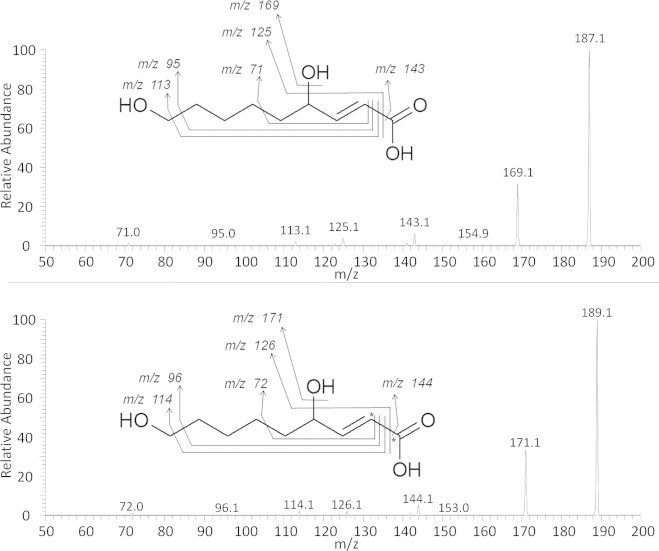
Fragmentation mass spectra and interpretation for ions 187.0977 (12C-HNE metabolite) and 189.1044 (13C-HNE metabolite).
Peak 7 was not fully resolved from Peak 6. It eluted at 28.9 min and represented approximately 4% of urinary radioactivity. However, this peak revealed the occurrence of two pairs of twin peaks. The first pair of ions at m/z 350.1281/352.1348 (molecular formula C14H24NO7S) gave upon fragmentation the characteristic fragment ions of a mercapturic acid conjugate at m/z 162 [B]− and of and m/z 128 [B−S]− (see Fig. 6 for fragment notation of mercapturic acid derivatives). MS/MS fragmentation of the pair of m/z 350/352 ions also yielded fragment ions corresponding to a decarboxylation (m/z 306/308 ions), indicating that this decarboxylation should be on the carbon 9 as it was the case for Peak 3. Intense fragment ions of were observed at m/z 221/223 and corresponded to [A+S]− fragments. Taken together, those results indicated that this metabolite should be identified as 9-carboxy-DHN-MA. The second pair of twin ions was detected at m/z 364.1069/366.1136, and gave upon fragmentation almost the same fragment ions as the previous pair, with a mass difference of 14 a.m.u., which could be attributed to the occurrence of a COOH group instead of a CH2OH group. This metabolite could then be identified as 9-carboxy-HNA-MA. Mercapturic acids derivatives of ω-oxidized HNE metabolites have been identified previously in rat urine, after intravenous administration [12]. Mercapturic acid metabolites derive from conjugation of the parent compound to glutathione, catalyzed by glutathione S-transferases, and the subsequent metabolization of the glutathione moiety into mercapturic acid [26].
Fig. 6.
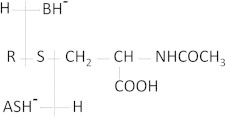
General fragmentation pattern and fragment ion notation of mercapturic acid derivatives.
Peak 8 eluted at 29.3 min and represented 1.25% of urinary radioactivity. Twin peaks were detected at m/z 203.0020/205.0091, in agreement with a C9H15O5 molecular formula, as observed for Peak 5. However, the MS/MS fragmentation of this metabolite was quite different to that of Peak 5. Opposite to Peak 5, this peak gave an m/z 173 fragment ion characteristic of the elimination of CH2O from a CH2OH group present in the terminal position. Accordingly, this peak should then correspond to 8,9-dihydroxy-HNA.
Peak 9 eluted at 29.6 min and represented 5.2% of urinary radioactivity. Exact mass measurements revealed the occurrence of two pairs of twin peaks. The first pair was observed at m/z 185.0818/187.0882 (C9H13O4), as for Peak 3. However, as for Peak 6, the MS/MS spectra of this compound showed a loss of 44 a.m.u. (yielding the m/z 141 fragment ion) from the m/z 185 precursor ion whereas a loss of 45 a.m.u. (yielding the m/z 142 fragment ion) occurred from the m/z 187 precursor ion, indicating that these fragment ions originated in a decarboxylation occurring on carbon 1. This metabolite should then correspond to 9-oxo-HNA. However, a structure corresponding to 9-hydroxy-4-oxo-HNA could also be in agreement with the mass spectrometric data. The second pair of twin peaks was observed at m/z 348.1109/350.1177 (C14H22NO7S). As for Peak 7, this metabolite gave upon fragmentation the characteristic m/z 162 [B]− ion of a mercapturic acid conjugate. MS/MS fragmentation of the m/z 348/350 ions exhibited fragment ions corresponding to a decarboxylation (m/z 304/306 ions) indicating that this decarboxylation should take place on the C9 carbon atom (loss of 44 a.m.u. for both m/z 348 and 350 precursor ions). These results allowed the identification of this metabolite as 9-carboxy-HNE-MA.
Peak 10 eluted at 30.5 min and represented 2.1% of urinary radioactivity, and presented twin peaks at m/z 336.1486/338.1552 (C14H26NO6S). Fragment ions corresponding to [B−S]− (m/z 128) and to [A+S]− (m/z 207/209) were observed in MS/MS, allowing to identify peak 10 as a mercapturic acid conjugate. Consecutive water and alcohol group losses from the 207/209 ions, respectively, led to the 189/191 and 159/160 fragment ions, in accordance with a loss of the labeled alcohol group on the C1 carbon atom. This metabolite was thus identified as hydroxy-DHN-MA, the other alcohol group likely being located on the C9 carbon atom.
Radioactivity between Peak 10 and 12 (named 11 in Fig. 5A) corresponded to 2.3% of urinary radioactivity. Two pairs of twin peaks could be detected at m/z 235.1011/237.1077 and 249.0803/251.0867 giving both, upon fragmentation, fragment ions corresponding respectively to a CO2 loss (−44 a.m.u.) and to a methyl-thiol group loss (−48 a.m.u.). Both elemental compositions deduced from exact mass measurements and MS/MS fragmentation data allowed us to identify these metabolites as 9-hydroxy-3-thiomethyl-HNA and 3-thiomethyl-carboxy-HNA. Those metabolites were seen only in the 6–24 h urine, indicating either several steps in their formation or the intervention of microbial flora in the distal part of the intestine. This is, to our knowledge, the first report of thiomethyl metabolites of HNE. Their formation could arise from two putative ways. The first one could involve the action of hydrogen sulfide (H2S) which is known to be a metabolic product of intestinal flora and is present in its free form in the distal parts of the intestine (for a review, see [27]). Hydrogen sulfide has been reported to scavenge HNE in vitro in neuronal cell culture and to prevent HNE adduction to cellular proteins [28]. This thiol compound could then be methylated by thiol S-methyltransferases in the intestine, as colon and cecal mucosa are believed to be major sites of thiol methylation [29]. The second one could involve the so-called mercapturic acid shunt. Most of the time, glutathione conjugates are transformed into mercapturic acids by two subsequent reactions, namely the cleavage of the γ-glutamyl bond of the glutathione moiety, leaving a thioether of cysteine, which is N-acetylated, forming a mercapturic acid derivative readily excreted into urine. In the mercapturic acid shunt, the cysteine conjugate is subjected to β-lyase action, giving a thiol compound, which can be methylated by thiol S-methyltransferases. β-Lyases from microbiota present in the distal parts of the intestine may play an important role in this process [30].
Peak 12 revealed no interpretable mass spectrometry signal, despite the presence of radioactivity detected at this retention time.
Peak 13 eluted at 38.5 min and represented 1.5% of urinary radioactivity. This peak was also a mixture of two pairs of twin peaks at m/z 332.1172/334.1239 and 346.0964/348.1031, respectively. Both pairs presented, upon fragmentation, the [B]− (m/z 162) and [A+S]− (m/z 203/205 and 217/219) ions characteristic of mercapturic acid conjugates. Under MS/MS, the m/z 346/348 pair also gave the m/z 302/304 fragment ions, corresponding to the elimination of CO2 from the C9 carbon atom. Those compounds were thus respectively identified as 9-hydroxy-HNA–lactone-MA (although the position of the hydroxyl group on carbon 9 cannot be confirmed), and 9-carboxy-HNA–lactone-MA.
Peak 14 eluted at 41.1 min and represented 1.1% of urinary radioactivity. High resolution MS analysis revealed only one pair of twin peaks at m/z 199.0598/201.0667 with a molecular formula of C9H11O5. Fragment ions corresponding to a decarboxylation on the C9 carbon atom (m/z 155/157 ions) were observed, enabling to hypothesize the presence of 9-carboxy-4-oxo-nonenoic acid. However, as this metabolite should have lost the tritium on carbon 4, the radioactive peak should correspond to another metabolite that could not be characterized by LCMS.
Peak 15 and 16 eluted at 42.1 and 42.7 min and represented 4.38% and 3.9% of urinary radioactivity, respectively. For both of them, MS analysis gave a pair of twin peaks at m/z 189.1132(3)/191.1198(9) (Table 1) in agreement with a C9H17O4 chemical formula. The MS/MS analysis revealed the presence of a fragment ion at m/z 129 for both metabolites and both twin peaks, together with the complementary twin fragment ions at m/z 59/61, indicating the loss of a CH3COOH group on carbon 1 (Fig. 8). Taken together, these results indicate the presence of hydroxylated 4-hydroxy-nonanoic acid. The presence of two radioactive peaks suggests the occurrence of two region-isomers bearing the hydroxyl group on two different positions (likely on C9 for one metabolite and on C8 for the other). It is noteworthy that the fragmentation pattern is quite different from the one of hydroxylated-HNA (Peak 6) for which a loss of 44 a.m.u. (CO2) instead of 60 a.m.u. (CH3COOH) was observed, probably due to the presence of the double bond between C2 and C3, precluding any fragmentation at this location. Those peaks were thus tentatively identified as 4,8-hydroxy-nonanoic acid and 4,9-hydroxy-nonanoic acid. Those metabolites are the saturated counterparts of hydroxy-HNA. Reduction of the HNE double bond has previously been described by our group in in vitro studies performed on colonocytes. It was followed by an oxidation of the aldehyde function [17]. This saturation pathway could involve NADPH-dependent alkenal/one oxidoreductase as reported by some authors [31]. 4,8-Hydroxy-nonanoic acid and 4,9-hydroxy-nonanoic acid have been reported by Jin et al. [23] and by Laurent et al. [20], as metabolites of HNE.
Fig. 8.
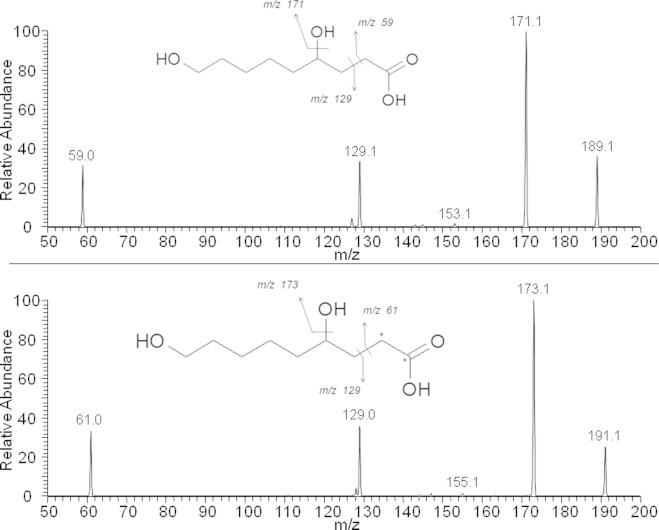
Fragmentation mass spectra and interpretation for ions 189.1132 (12C-HNE metabolite) and 191.1198 (13C-HNE metabolite).
Peak 17 revealed no interpretable mass spectrometric signal. Radioactivity between peak 17 and peak 18 gave two pairs of twin peaks, at m/z 333.1554/335.1621, 347.1346/349.1416 giving either a fragment ion at m/z 193 or fragment ions corresponding to a loss of 176 a.m.u. All these features can be considered as characteristic of glucuronidated metabolites [32]. Twin peaks at m/z 333/335 and at 347/349 should then correspond to DHN–glucuronide and HNA–glucuronide, respectively, with a respective chemical formula of C15H25O8 and C15H23O9. To our knowledge, this is the first report of glucuronide conjugates as metabolites of HNE.
Peak 18 and 19 eluted at 47.0 and 47.3 min and represented respectively 6.5% and 7% of urinary radioactivity. They both revealed twin peaks at m/z 320.1539/322.1603 (C14H26NO5S) upon high resolution MS analysis, with a common fragment ion at m/z 128 and twin fragments at 191/193, characteristic of the mercapturic acid of DHN. DHN-MA has been reported by our group to be the major HNE metabolite after intravenous administration [11]. The present study showed that DHN-MA, although being quantitatively an important HNE metabolite, was not the major one after oral administration. This can be attributed to the important oxidative metabolism observed by our group in vitro in intestinal epithelial cells [17]. In Peak 19, we observed the concomitant presence of a twin peak at m/z 334.1331/336.1395 (C14H24NO6S) giving in MS/MS a twin fragment of m/z 290/292 characteristic of the loss of a carboxylate group (not located on the C1 carbon atom), together with the presence of characteristic fragment ions of a mercapturic acid conjugate, indicating the presence of HNA-MA in this radioactive peak.
Peak 20 eluted at 48.7 min and represented 1.35% of urinary radioactivity. This peak exhibited two pairs of twin peaks at m/z 318.1379/320.1445 (C14H24NO5S) and 334.1328/336.1396 (C14H24NO6S). MS/MS analysis yielded fragment ions at m/z 162 and 128 that are characteristic of mercapturic acid conjugates and twin fragment ions at m/z 189/191 and m/z 205/207, respectively corresponding to the complementary ions of [HNE+S]− and [HNA+S]−, confirming the presence of HNE-MA and HNA-MA. This peak co-eluted with HNA-MA standard.
Analyses carried out between Peak 20 and 21 revealed the presence of twin peaks at m/z 219.1064/221.1130 (C10H19O3S) indicating the presence of a sulfur atom in the molecule. Fragmentation of the m/z 219 ion gave daughter ions at m/z 175 and m/z 171 that were respectively attributed to the loss of a carboxylic group and of a thiomethyl group respectively, indicating that this compound could correspond to a thiomethyl derivative of HNA.
Peak 21 eluted at 52.7 min and represented 4.31% of urinary radioactivity. MS analysis revealed twin peaks at m/z 316.1225/318.1285 (C14H22NO5S) both giving upon fragmentation an intense peak at m/z 162, characteristic of a mercapturic acid. This fragmentation is in accordance with the presence of HNA–lactone-MA.
DHN-MA, HNE-MA, HNA-MA, HNA–lactone-MA are expected urinary metabolites that have been observed in most in vivo studies done on HNE metabolism [11,13]. Some of them, such as DHN-MA, are used as lipid peroxidation biomarkers [33,34].
Intestinal contents and feces analyses
We also analyzed by radio-HPLC intestinal contents (stomach, small intestine, cecum and colon) and feces.
As well as in 0–24 h and 6–24 h urine analyses, intestinal contents and feces radio-HPLC profiles displayed an important peak with an early retention time. As demonstrated earlier, this peak was identified as tritiated water.
In intestinal contents, it was clearly the major peak, representing between 50% and 71% of the radioactivity in the sample (Fig. 9A).
In contrast, tritiated water represented only 16% of the total radioactivity in feces (Fig. 9B). It could be explained by the fact that an important part of the water present in intestinal contents is reabsorbed in the colon.
Four other groups of peaks could be determined at 17.2 min, 28.6 min, 42.9 min and 52.2 min.
In order to attempt to identify those fecal metabolites, we performed HRMS and MS/MS analyses of radio-HPLC collected fractions. Unfortunately, no twin peaks were identified in those samples, probably because of the necessary concentration/evaporation steps before MS/MS analyses, due to low concentration of metabolites in the samples studied.
Conclusion
The metabolite tracking methodology allowed the specific identification of HNE urinary metabolites and gave valuable insights into chemical function positions. All together these results indicate a prominent role of oxidation pathways, on carbon 1 bearing the aldehyde function and also on the terminal carbon, in the metabolization of HNE after oral administration. These first oxidation steps are most probably followed by β-oxidation steps originating from the carbon 1 and/or 9 of HNA and finally the production of water. Those pathways are quantitatively more important than the mercapturic acid pathway with more than 50% of total urinary radioactivity, while mercapturic conjugates represent around 30% of urinary radioactivity. Those oxidation pathways underline the importance of aldehyde dehydrogenase (ALDH) and of cytochrome P450 4A enzyme families in the respective oxidation of the HNE aldehyde function on carbon 1 and the terminal oxidation on carbon 9. As a consequence of those prominent oxidative pathways involved in HNE metabolization after oral administration, DHN-MA did not represent the major urinary metabolite as reported after intravenous administration [11]. The huge excretion of this metabolite after a heme-iron rich diet in rats, almost 10 µg/24 h, shows that important quantities of HNE can be absorbed and be metabolized, and could also be related to promotion of colon carcinogenesis [33,35].
The metabolite tracking methodology allowed also to specifically detect quantitatively novel minor HNE urinary metabolites such as thiomethyl and glucuronide derivatives. Studies are under progress to determine if those metabolites are specific of an oral administration, and in the case of the HNA-thiomethyl metabolites, if their parent thiol compounds could bear some toxicity.
Conflict of interest
The authors report no potential conflicts of interest.
Acknowledgments
We thank ITMO Cancer (Plan cancer 2009–2013)/INCa/INSERM for financial support for the “NeoMeaTox” project. This work was also supported by COST CM1001.
We also want to thank Florence Blas-y-Estrada for animal care.
References
- 1.Seppanen C.M., Saari Csallany A. Formation of 4-hydroxynonenal, a toxic aldehyde, in soybean oil at frying temperature. Journal of American Oil Chemists' Society. 2002;79(10):1033–1038. [Google Scholar]
- 2.Seppanen C.M., Csallany A.S. Incorporation of the toxic aldehyde 4-hydroxy-2-trans-nonenal into food fried in thermally oxidized soybean oil. Journal of American Oil Chemists' Society. 2004;81(12):1137–1141. [Google Scholar]
- 3.Sakai T., Kuwazuru S. A lipid peroxidation-derived aldehyde, 4-hydroxy-2-nonenal, contents in several fish meats. Fisheries Sciences. 1995;61(3):527–528. doi: 10.1271/bbb.59.1379. http:dx.doi.org/10.2331/fishsci.61.527. [DOI] [PubMed] [Google Scholar]
- 4.Sakai T., Shimizu Y., Kawahara S. Effect of NaCl on the lipid peroxidation-derived aldehyde, 4-hydroxy-2-nonenal, formation in boiled pork. Bioscience, Biotechnology and Biochemistry. 2006;70(4):815–820. doi: 10.1271/bbb.70.815. 16636446 [DOI] [PubMed] [Google Scholar]
- 5.Surh J., Lee S., Kwon H. 4-Hydroxy-2-alkenals in polyunsaturated fatty acids-fortified infant formulas and other commercial food products. Food Additives and Contaminants. 2007;24(11):1209–1218. doi: 10.1080/02652030701422465. 17852396 [DOI] [PubMed] [Google Scholar]
- 6.Gasc N., Taché S., Rathahao E., Bertrand-Michel J., Roques V., Guéraud F. 4-Hydroxynonenal in foodstuffs: heme concentration, fatty acid composition and freeze-drying are determining factors. Redox Report. 2007;12(1):40–44. doi: 10.1179/135100007X162257. 17263907 [DOI] [PubMed] [Google Scholar]
- 7.Nishikawa A., Sodum R., Chung F.L. Acute toxicity of trans-4-hydroxy-2-nonenal in Fisher 344 rats [corrected] Lipids. 1992;27(1):54–58. doi: 10.1007/BF02537060. 1608305 [DOI] [PubMed] [Google Scholar]
- 8.Segall H.J., Wilson D.W., Dallas J.L., Haddon W.F. Trans-4-hydroxy-2-hexenal: a reactive metabolite from the macrocyclic pyrrolizidine alkaloid senecionine. Science. 1985;229(4712):472–475. doi: 10.1126/science.4012327. 4012327 [DOI] [PubMed] [Google Scholar]
- 9.Wacker M., Wanek P., Eder E. Detection of 1,N2-propanodeoxyguanosine adducts of trans-4-hydroxy-2-nonenal after gavage of trans-4-hydroxy-2-nonenal or induction of lipid peroxidation with carbon tetrachloride in F344 rats. Chemico-Biological Interactions. 2001;137(3):269–283. doi: 10.1016/s0009-2797(01)00259-9. 11566294 [DOI] [PubMed] [Google Scholar]
- 10.Kang S.C., Kim H.-W., Kim K.B., Kwack S.J., Ahn I.Y., Bae J.Y. Hepatotoxicity and nephrotoxicity produced by 4-hydroxy-2-nonenal (4-HNE) following 4-week oral administration to Sprague-Dawley rats. Journal of Toxicology and Environmental Health A. 2011;74(12):779–789. doi: 10.1080/15287394.2011.567952. 21541880 [DOI] [PubMed] [Google Scholar]
- 11.Alary J., Bravais F., Cravedi J.P., Debrauwer L., Rao D., Bories G. Mercapturic acid conjugates as urinary end metabolites of the lipid peroxidation product 4-hydroxy-2-nonenal in the rat. Chemical Research in Toxicology. 1995;8(1):34–39. doi: 10.1021/tx00043a004. 7703364 [DOI] [PubMed] [Google Scholar]
- 12.Alary J., Debrauwer L., Fernandez Y., Paris A., Cravedi J.P., Dolo L. Identification of novel urinary metabolites of the lipid peroxidation product 4-hydroxy-2-nonenal in rats. Chemical Research in Toxicology. 1998;11(11):1368–1376. doi: 10.1021/tx980068g. 9815199 [DOI] [PubMed] [Google Scholar]
- 13.De Zwart L.L., Hermanns R.C.A., Meerman J.H.N., Commandeur J.N.M., Vermeulen N.P.E. Disposition in rat of [2–3H]-trans-4-hydroxy-2,3-nonenal, a product of lipid peroxidation. Xenobiotica. 1996;26(10):1087–1100. doi: 10.3109/00498259609167424. [DOI] [PubMed] [Google Scholar]
- 14.Winter C.K., Segall H.J., Jones A.D. Distribution of trans-4-hydroxy-2-hexenal and tandem mass spectrometric detection of its urinary mercapturic acid in the rat. Drug Metabolism and Disposition. 1987;15(5):608–612. 2891475 [PubMed] [Google Scholar]
- 15.Bravais F., Rao D., Alary J., Rao R.C., Debrauwer L., Bories G. Synthesis of 4-hydroxy[4-3H]-2(E)-nonen-1-al-diethylacetal. Journal of Labelled Compounds and Radiopharmaceuticals. 1995;36(5):471–477. [Google Scholar]
- 16.Jouanin I., Sreevani V., Rathahao E., Guéraud F., Paris A. Synthesis of the lipid peroxidation product 4-hydroxy-2(E)-nonenal with13C stable isotope incorporation. Journal of Labelled Compounds and Radiopharmaceuticals. 2008;51(2):87–92. [Google Scholar]
- 17.Baradat M., Jouanin I., Dalleau S., Taché S., Gieules M., Debrauwer L. 4-hydroxy-2(E)-nonenal metabolism differs in Apc(+/+) cells and in Apc(Min/+) cells: it may explain colon cancer promotion by heme iron. Chemical Research in Toxicology. 2011;24(11):1984–1993. doi: 10.1021/tx2003036. 21967605 [DOI] [PubMed] [Google Scholar]
- 18.Jouanin I., Baradat M., Gieules M., Taché S., Pierre F.H.F., Guéraud F. Liquid chromatography/electrospray ionisation mass spectrometric tracking of 4-hydroxy-2(E)-nonenal biotransformations by mouse colon epithelial cells using [1,2-13C2]-4-hydroxy-2(E)-nonenal as stable isotope tracer. Rapid Communications in Mass Spectrometry. 2011;25(19):2675–2681. doi: 10.1002/rcm.5033. [DOI] [PubMed] [Google Scholar]
- 19.IRSN . 2012. Tritium and the Environment. Radionuclide Sheet. http://www.irsn.fr/EN/Research/publications-documentation/radionuclides-sheets/environment/Pages/Tritium-environment.aspx. [Google Scholar]
- 20.Laurent A., Perdu-Durand E., Alary J., Debrauwer L., Cravedi J.P. Metabolism of 4-hydroxynonenal, a cytotoxic product of lipid peroxidation, in rat precision-cut liver slices. Toxicology Letters. 2000;114(1–3):203–214. doi: 10.1016/s0378-4274(99)00301-x. 10713486 [DOI] [PubMed] [Google Scholar]
- 21.Siems W.G., Zollner H., Grune T., Esterbauer H. Metabolic fate of 4-hydroxynonenal in hepatocytes: 1,4-dihydroxynonene is not the main product. Journal of Lipid Research. 1997;38(3):612–622. 9101442 [PubMed] [Google Scholar]
- 22.Grune T., Siems W.G., Petras T. Identification of metabolic pathways of the lipid peroxidation product 4-hydroxynonenal in situ perfused rat kidney. Journal of Lipid Research. 1997;38(8):1660–1665. 9300788 [PubMed] [Google Scholar]
- 23.Jin Z., Berthiaume J.M., Li Q., Henry F., Huang Z., Sadhukhan S. Catabolism of (2E)-4-hydroxy-2-nonenal via ω- and ω-1-Oxidation stimulated by ketogenic diet. Journal of Biological Chemistry. 2014;289(46):32327–32338. doi: 10.1074/jbc.M114.602458. 25274632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guéraud F., Alary J., Costet P., Debrauwer L., Dolo L., Pineau T. In vivo involvement of cytochrome P450 4A family in the oxidative metabolism of the lipid peroxidation product trans-4-hydroxy-2-nonenal, using PPARalpha-deficient mice. Journal of Lipid Research. 1999;40(1):152–159. 9869661 [PubMed] [Google Scholar]
- 25.Aoyama T., Hardwick J.P., Imaoka S., Funae Y., Gelboin H.V., Gonzalez F.J. Clofibrate-inducible rat hepatic P450s IVA1 and IVA3 catalyze the Omega- and (omega-1)-hydroxylation of fatty acids and the Omega-hydroxylation of prostaglandins E1 and F2 alpha. Journal of Lipid Research. 1990;31(8):1477–1482. 2280187 [PubMed] [Google Scholar]
- 26.Mulder G.J. Conjugation Reactions in Drug Metabolism: An Integrated Approach. CRC Press, Taylor and Francis; London: 1990. [Google Scholar]
- 27.Blachier F., Davila A.-M., Mimoun S., Benetti P.-H., Atanasiu C., Andriamihaja M. Luminal sulfide and large intestine mucosa: friend or foe? Amino Acids. 2010;39(2):335–347. doi: 10.1007/s00726-009-0445-2. 20020161 [DOI] [PubMed] [Google Scholar]
- 28.Schreier S.M., Muellner M.K., Steinkellner H., Hermann M., Esterbauer H., Exner M. Hydrogen sulfide scavenges the cytotoxic lipid oxidation product 4-HNE. Neurotoxocity Research. 2010;17(3):249–256. doi: 10.1007/s12640-009-9099-9. 19680736 [DOI] [PubMed] [Google Scholar]
- 29.Weisiger R.A., Pinkus L.M., Jakoby W.B. Thiol S-methyltransferase: suggested role in detoxication of intestinal hydrogen sulfide. Biochemical Pharmacology. 1980;29(20):2885–2887. doi: 10.1016/0006-2952(80)90029-5. 7437088 [DOI] [PubMed] [Google Scholar]
- 30.Arias I.M., Boyer J.L., Fausto N. Liver: Biology and Pathobiology. 3. Raven Pr; New York: 1994. [Google Scholar]
- 31.Dick R.A., Kwak M.K., Sutter T.R., Kensler T.W. Antioxidative function and substrate specificity of NAD(P)H-dependent alkenal/one oxidoreductase. A new role for leukotriene B4 12-hydroxydehydrogenase/15-oxoprostaglandin 13-reductase. Journal of Biological Chemistry. 2001;276(44):40803–40810. doi: 10.1074/jbc.M105487200. 11524419 [DOI] [PubMed] [Google Scholar]
- 32.Levsen K., Schiebel H.-M., Behnke B., Dötzer R., Dreher W., Elend M. Structure elucidation of phase II metabolites by tandem mass spectrometry: an overview. Journal of Chromatography A. 2005;1067(1–2):55–72. doi: 10.1016/j.chroma.2004.08.165. 15844510 [DOI] [PubMed] [Google Scholar]
- 33.Pierre F., Peiro G., Taché S., Cross A.J., Bingham S.A., Gasc N. New marker of colon cancer risk associated with heme intake: 1,4-dihydroxynonane mercapturic acid. Cancer Epidemiology, Biomarkers and Prevention. 2006;15(11):2274–2279. doi: 10.1158/1055-9965.EPI-06-0085. 17119057 [DOI] [PubMed] [Google Scholar]
- 34.Peiro G., Alary J., Cravedi J.-P., Rathahao E., Steghens J.-P., Guéraud F. Dihydroxynonene mercapturic acid, a urinary metabolite of 4-hydroxynonenal, as a biomarker of lipid peroxidation. Biofactors (Oxford England) 2005;24(1–4):89–96. doi: 10.1002/biof.5520240110. 16403967 [DOI] [PubMed] [Google Scholar]
- 35.Pierre F., Tache S., Guéraud F., Rerole A.L., Jourdan M.-L., Petit C. Apc mutation induces resistance of colonic cells to lipoperoxide-triggered apoptosis induced by faecal water from haem-fed rats. Carcinogenesis. 2007;28(2):321–327. doi: 10.1093/carcin/bgl127. 16885197 [DOI] [PubMed] [Google Scholar]