

Abstract
Mitochondria are cellular energy powerhouses that play important roles in maintaining cell survival, cell death and cellular metabolic homeostasis. Timely removal of damaged mitochondria via autophagy (mitophagy) is thus critical for cellular homeostasis and function. Mitochondria are reticular organelles that have high plasticity for their dynamic structures and constantly undergo fission and fusion as well as movement through the cytoskeleton. In this review, we discuss the most recent progress on the molecular mechanisms and roles of mitochondrial fission/fusion and mitochondrial motility in mitophagy. We also discuss multiple pathways leading to the quality control of mitochondria in addition to the traditional mitophagy pathway under different conditions.
Abbreviations: APAP, acetaminophen; Bag4, Bcl2-associated athanogene 4; Bcl2L1, Bcl-2 like 1; BNIP3, Bcl-2/adenovirus E1B 19 kDa protein-interacting protein 3; CCCP, m-chloro phenyl hydrazine; Clec16a, C-type lectin domain family 16, member A; Drp1, dynamin-related protein 1; Fis1, mitochondrial fission 1; FUNDC1, Fun14 domain containing 1; Hif-1, hypoxia-inducing factor 1; HSPA1L, heat shock 70 kDa protein 1-like; LC3, microtubule-associated protein 1 light-chain 3; LIR, LC3-interacting region; MEFs, mouse embryonic fibroblasts; Mff, mitochondria fission factor; Mfn1, mitofusin 1; Mfn2, mitofusin 2; MDV, mitochondria-derived vesicles; MID49, mitochondrial dynamics protein of 49 kDa; Miro, mitochondrial Rho GTPase; MUL1, mitochondrial ubiquitin ligase 1; Nrdp1, neuregulin receptor degradation protein 1; OPA1, optic atrophy 1; PARL, presenilin-associatedrhomboid-like; PGAM5, phosphoglycerate mutase family member 5; PINK1, PTEN-induced putative kinase 1; ROS, reactive oxygen species; Smurf1, Smad-specific E3 ubiquitin protein ligase 1; SQSTM1, sequestosome 1; SNPH, syntaphilin; TOMM7, translocase of outer mitochondrial membrane 7; TOMM20, translocase of outer mitochondrial membrane 20; UBA, ubiquitin-associated; Usp30, ubiquitin-specific peptidase 30; VDAC, voltage-dependent anion channel
Keywords: Autophagy, Mitophagy, Parkin, Mitochondrial spheroids
Graphical abstract
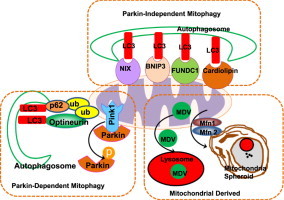
Highlights
-
•
Mitochondrial dynamics including mitochondrial fission/fusion and mitochondrial movement regulate mitochondrial homeostasis and quality.
-
•
Damaged mitochondria are removed via multiple mechanisms including mitophagy, mitochondrial derived vesicles and mitochondrial spheroids.
-
•
Mitophagy can occur in a Parkin-dependent or independent manner.
-
•
Parkin also regulates the formation of mitochondrial derived vesicles and mitochondrial spheroids.
Introduction
Mitochondria are the “power house” of the cell because they are the major site of ATP production for cell survival and many other vital cellular functions. It is well known that mitochondria act as central executioners of cell death including apoptotic and necrotic cell death. Therefore, mitochondrial quality must be well controlled to avoid cell death. Multiple mechanisms have been utilized by mitochondria to maintain their homeostasis. First, mitochondria have their own proteolytic system, allowing them to degrade misfolded proteins that could potentially disrupt mitochondrial function [1,2]. Second, damaged outer mitochondrial membrane proteins can be degraded by the proteasome [3]. Third, mitochondria can undergo constant fission and fusion to repair damaged components of the mitochondria, which allows for segregation of damaged mitochondria via the fission process and exchange of material between healthy mitochondria via the fusion process [4,5]. Fourth, a portion of mitochondria can bud off and form mitochondria-derived vesicles (MDV) under oxidative stress conditions, which further fuse with lysosomes to degrade oxidized mitochondrial proteins within MDV [6]. Fifth, damaged mitochondria can form mitochondrial spheroids and acquire lysosomal markers to possibly serve as an alternative pathway for removal of damaged mitochondria [7–9]. Finally, damaged mitochondria can be enveloped by autophagosomes to trigger their degradation in the lysosome via mitophagy [10–12]. This graphic review will focus on the current understanding of mitochondrial dynamics and the multiple mechanisms that regulate mitochondrial homeostasis.
Current mechanisms of mitochondrial quality control
Multiple mechanisms regulating mitochondrial quality control in yeast and mammals have been discovered recently. Below, we discuss regulation of mitochondrial quality control by various mechanisms including mitochondrial fission and fusion, Parkin-dependent and Parkin-independent mechanisms, MDV and mitochondrial spheroid formation.
Mitochondrial fission and fusion and motility in mitophagy
Mitochondria are dynamic organelles that continuously undergo fission and fusion, which are necessary for cell survival and adaptation to changing conditions needed for cell growth, division, and distribution of mitochondria during differentiation [4].
Mitochondrial fusion in mammals is mediated by the fusion proteins mitofusin 1 (Mfn1) and Mfn2 and optic atrophy 1 (OPA1). Mfn1 and Mfn2 are dynamin-related GTPases that are responsible for fusion of outer mitochondrial membranes. OPA1 is also a dynamin-related GTPase, which is responsible for fusion of inner mitochondrial membranes (Fig. 1A). Presenilin-associatedrhomboid-like (PARL) [13] and paraplegin (an AAA protease present in the mitochondrial matrix) [14] induce alternative splicing and alternative processing of OPA1 to generate eight OPA1 isoforms. However, OPA1 processing still occurs in PARL or paraplegin knockout MEF cells, suggesting that other factors may also be involved in OPA1 processing [15]. Yme can further cleave OPA1 under normal conditions to generate Short and Long forms of OPA1 (S-OPA1 and L-OPA1) [16], where L-OPA1 is integral in the inner membrane and S-OPA1 is located in the intermembrane space. L-OPA1 is further cleaved by the inducible protease OMA1 when mitochondria are depolarized by the mitochondrial uncoupler carbonyl cyanide m-chloro phenyl hydrazine (CCCP), resulting in mitochondrial fragmentation by preventing mitochondrial fusion [17,18]. The mitochondrial deacetylase SIRT3 is capable of deacetylating OPA1 and elevating its GTPase activity [19].
Fig. 1.
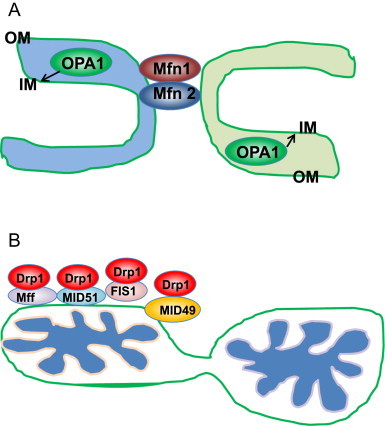
Mitochondrial fusion and fission in mammalian cells. (A) Mitochondrial fusion is mediated by large dynamin-related GTPase proteins Mfn1, Mfn2 and OPA1. Outer mitochondria membrane (OM) fusion is mediated by Mfn1 and Mfn2, whereas inner mitochondria membrane (IM) fusion is mediated by OPA1. (B) Mitochondria fission requires the recruitment of Drp1 from cytosol to mitochondria. Drp1 is also a dynamin-related GTPase protein that binds to four Drp1 receptor proteins Fis1, Mff, MID49 and MID51, which are mitochondria OM proteins.
Mitochondrial fission in mammals is mediated by dynamin-related protein 1 (Drp1), which is also a large GTPase. Drp1 is a cytosolic protein that can be recruited to the outer mitochondrial membrane to constrict mitochondria resulting in eventual division of a mitochondrion into two separate organelles. Drp1 interacts with four mitochondrial receptor proteins: fission 1 (Fis1), mitochondria fission factor (Mff), mitochondrial dynamics protein of 49 kDa (MID49) and MID51 (Fig. 1B). In mammalian cells, it seems that the interaction between Fis1 and Drp1 has a minor role in regulating mitochondrial fission whereas the interactions of Drp1 with the other three receptor proteins play prominent roles for fission [20–24]. In addition, Drp1 can also localize at the endoplasmic reticulum–mitochondria contact site, and the endoplasmic reticulum may play a role in the process of mitochondrial fission [25]. Accumulating evidence indicates that posttranslational modification of Drp1 is an important mechanism for regulating its function. During cell division, mitochondrial fission is essential for separating mitochondria into two daughter cells. At the onset of mitosis, Drp1 is phosphorylated by Cdk1/Cyclin B at Ser585, which increases Drp1 GTPase activity [26]. In contrast, reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase (PKA) and its dephosphorylation by phosphatase calcineurin at Ser656 leads to elongated mitochondria [27]. In addition, the mitochondrial phosphatase phosphoglycerate mutase family member 5 (PGAM5), dephosphorylates Drp1 at Ser637 and recruits Drp1 to mitochondria to induce mitochondrial fragmentation. This PGAM5-Drp1-mediated mitochondrial fragmentation was initially thought to play a critical role in programmed necrosis, but this notion has been challenged by recent findings that Drp1 knockout mouse embryonic fibroblasts (MEFs) were equally as sensitive to necrosis as wild type MEFs [28,29]. In addition to phosphorylation, Drp1 can also be ubiquitinated by MARCH V, a mitochondrial E3 ligase, or sumoylated by SUMO-1. The ubiquitination and sumoylation of Drp1 can either regulate the stability of Drp1 or recruit Drp1 to the actual mitochondrial dividing site and in turn regulate mitochondria fission [30–32].
In addition to fission and fusion, the movement of mitochondria through the cytoskeleton is also important for the cellular distribution and turnover of mitochondria [33]. Mitochondria in mammalian cells are mostly transported on microtubules using a kinesin motor towards the plus end and a dynein motor towards the minus end of microtubules [34] (Fig. 2). The attachment of mitochondria to the kinesin motor is regulated by a series of molecular adapters. The adapter protein Milton directly interacts with the outer mitochondrial membrane protein Mitochondrial Rho GTPase (Miro) and in turn links mitochondria to kinesin [34]. Interestingly, both Mfn1 and Mfn2 interact with Miro and Milton, and loss of Mfn2 alone is sufficient to cause a defect in mitochondrial transport in neurons [35]. In addition, the availability of extracellular glucose can also regulate mitochondrial motility in neurons. Most mitochondria in mouse axons are immobile, which is regulated by syntaphilin (SNPH). SNPH anchors axonal mitochondria to microtubules resulting in the local stalling of mitochondria. SNPH serves as a docking protein, and deletion of SNPH in mice increases axonal mitochondrial motility. In response to elevated Ca2+, Miro releases Kinesin-1, which then binds with SNPH to inhibit the mobilization of axonal mitochondria [36]. In addition, Milton is GlcNAcylated by O-GlcNac Transferase (OGT) in response to increased extracellular glucose levels, which diminishes mitochondrial mobility [37].
Fig. 2.
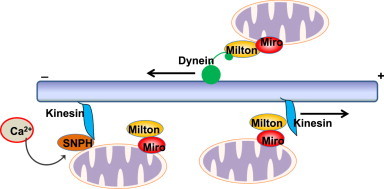
Regulation of mitochondrial motility. Mitochondria are transported on cytoskeleton microtubules by molecular motor proteins kinesin and dynein. Milton acts as an adapter molecule to link the motor proteins to the outer mitochondrial membrane protein miro, and kinesin transports mitochondria towards the plus end of microtubules whereas dynein transports mitochondria to the minus end of microtubules. When intracellular calcium levels rise, kinesin is dissociated from the Milton–miro complex and then binds to SNPH. SNPH inhibits the ATPase activity of kinesin to block the movement of mitochondria.
It has been suggested that mitochondria fission and fusion may serve as important quality control mechanisms for preserving mitochondria. For example, dysfunctional mitochondria may lose their fusion capacity by inactivating fusion or activating fission machineries to prevent the damaged mitochondria from incorporating back into the healthy mitochondrial network. After photo-labeling, mitochondria undergo continuous cycles of fission and fusion and show a striking heterogeneity with two sets of daughter mitochondria that either have increased or decreased membrane potential. Daughter mitochondria with higher membrane potential (presumably good quality mitochondria) proceed to fusion while depolarized daughter mitochondria (presumably bad quality mitochondria) are degraded by mitophagy [5]. Therefore, mitochondrial fission and fusion may serve as a quality control mechanism to preserve the better mitochondria via fusion and rid of the bad quality mitochondria via fission and subsequent mitophagy. When mitochondrial damage is considered mild, it will trigger the proteolytic mediated processing and inactivation of Opa1 resulting in the impairment of inner membrane fusion without affecting the outer membrane fusion (which is mediated by Mfn1 and Mfn2). Severe damage may activate Pink1–Parkin-mediated events resulting in the ubiquitination and proteasomal degradation of Mfn1 and Mfn2 and subsequent mitophagy (discussed in detail below). However, under nutrient replete conditions, Mfn1 and Mfn2 knockout MEFs do not show increased mitophagy, suggesting that mitochondrial fission alone is not enough to induce mitophagy. It seems that in addition to mitochondrial fragmentation, mitochondria must also be dysfunctional/depolarized and may also possibly need to recruit other autophagy receptor proteins for mitophagy induction to occur [10]. However, it is also likely that small mitochondria are relatively easier to be enwrapped by autophagosomes compared to larger elongated mitochondria. In addition to mitochondrial fission, the motility of mitochondria also seems to be important for mitophagy. Overexpression of Pink1 and Parkin can lead to Miro ubiquitination and degradation, suggesting a link between Pink1–Parkin and Miro in mitophagy [38]. Indeed, loss of Miro results in perinuclear clustering of mitochondria, which promotes mitophagy in HeLa cells. However, it remains unclear whether Miro would act as a mitophagy receptor or Miro–Milton–Kinesin-mediated mitochondria transport may normally prevent the mitophagy process. It will be interesting to determine whether SNPH-mediated mitochondrial motility would also be involved in mitophagy.
Parkin-dependent mitophagy
One of the best studied mechanisms for mitophagy in mammalian cells is the Pink1–Parkin-mediated mitophagy pathway [10,11]. Parkin is a cytosolic E3 ubiquitin ligase that translocates to depolarized mitochondria after treatment with the mitochondrial uncoupler CCCP and initiates their degradation via mitophagy [39]. The Parkin-dependent mitophagy pathway has been shown to require both Parkin and Pink1. Pink1 is a mitochondria serine/threonine kinase. Genetic studies from drosophila indicate that Pink1 and Parkin work on the same pathway, and Pink1 acts upstream of Parkin because overexpression of Parkin in Pink1-deficient drosophila partially rescued the Pink1 mutant phenotype while overexpression of Pink1 failed to do so in Parkin-deficient drosophila [40–42]. In addition, overexpression of Pink1 alone can initiate translocation of Parkin to mitochondria without mitochondrial damage [43]. In humans, mutations of both Pink1 and Parkin have been linked to familiar autosomal recessive Parkinsonism [44]. Taken together, all evidence supports the notion that Pink1 and Parkin work on the same pathway.
The level of Pink1 is normally undetectable in most cells because Pink1 is cleaved in the mitochondria by PARL and then degraded by mitochondrial peptidases [45]. However, Pink1 is no longer cleaved and becomes stabilized on the outer mitochondrial membrane when mitochondria are depolarized [45]. Pink1 then promotes Parkin-mediated mitophagy by recruiting Parkin to mitochondria and promoting Parkin E3 ligase activity. Pink1 directly phosphorylates Thr175 and Thr217 within Parkin's linker region, which promotes Parkin mitochondrial translocation [46]. In addition, phosphorylation of Parkin by Pink1 also activates Parkin's E3 ubiquitin ligase activity, enabling it to ubiquitinate mitochondrial proteins [47]. Pink1 also phosphorylates ubiquitin at Ser65, and the phospho-ubiquitin activates Parkin E3 ubiquitin ligase activity through a feed-forward mechanism [48,49]. Using a genome-wide small interfering RNA (siRNA) screening for genes that regulate Parkin mitochondrial translocation, the translocase of outer mitochondrial membrane 7 (TOMM7) was found to stabilize Pink1 on the outer mitochondrial membrane whereas SIAH3, a mitochondrial resident protein, de-stabilized Pink1 on mitochondria. Furthermore, two other proteins, HSPA1L (an HSP70 family protein) and Bcl2-associated athanogene 4 (BAG4, a nucleotide exchange factor for HSP70), positively and negatively regulate Parkin mitochondrial translocation, respectively [50]. In addition to Pink1, four of Parkin's different cognate E2 co-enzymes (UBE2D, UBE2L3, UBE2N and UBE2R1) also positively or negatively regulate Parkin’s activation, translocation and enzymatic functions during mitophagy [51,52]. Neuregulin receptor degradation protein 1 (Nrdp1), an E3 ubiquitin ligase, regulates the ubiquitination and proteasomal degradation of Parkin. Nrdp1 interacts with C-type lectin domain family 16, member A (Clec16a), a protein that is associated with type 1 diabetes mellitus, and deletion of Clec16a increases Parkin expression. Intriguingly, despite an increased expression of Parkin, pancreas-specific Clec16a knockout mice had abnormal mitochondria with reduced oxygen consumption and ATP concentration in pancreatic β cells due to defects of mitophagy. Subsequent studies revealed that Clec16a deficient fibroblasts had defective fusion of autophagosomes with lysosomes [53], suggesting that Clec16a also regulates autophagy in addition to Parkin-mediated mitophagy.
Once recruited to the mitochondria, Parkin ubiquitinates several mitochondrial outer membrane proteins including the mitochondrial fusion proteins Mfn1 and Mfn2, Miro, Translocase of outer mitochondrial membrane 20 (TOMM20), and vbltage-dependent anion channel (VDAC) to initiate mitophagy. Ubiquitination and proteasomal degradation of Mfn1 and Mfn2 results in mitochondrial fission and fragmentation [54–57]. Mitochondrial fission has been shown to be important for mitophagy induction in liver hepatocytes among other cell types [5,58]. Fragmented mitochondria can fuse together if they have normal membrane potential, but loss of membrane potential prevents fusion and leads to mitochondria segregation and subsequent degradation by mitophagy [5]. In contrast, excessive fusion of mitochondria has been shown to inhibit the mitophagy process [59]. These observations support the notion that mitochondrial fragmentation promotes mitophagy induction. In addition to regulating mitochondria fusion and fission, Parkin-induced ubiquitination of Miro induces mitochondrial arrest, which can segregate damaged mitochondria from healthy mitochondria prior to mitophagy [60]. Parkin-mediated mitophagy can be antagonized by Ubiquitin-specific peptidase 30 (USP30), a deubiquitinase localized to mitochondria, which removes ubiquitin from damaged mitochondria previously attached by Parkin. Defective mitophagy caused by pathogenic mutations in Parkin is rescued by knockdown of USP30. Moreover, knockdown of USP30 improves mitochondrial integrity in Parkin- or Pink1-deficient flies and protects flies against paraquat toxicity in vivo by ameliorating defects in dopamine levels, motor function and organismal survival [61].
Following Parkin-mediated ubiquitination of outer mitochondrial membrane proteins, the selective autophagy adapter protein p62/SQSTM1 (Sequestosome 1) is recruited to mitochondria and thought to play a role in mitophagy due to its capacity to directly interact with LC3 via its LC3 interacting region (LIR) [56,62–64]. However, it seems that the role of p62 in mitophagy is not essential [65,66], likely due to the presence of other compensatory or redundant similar autophagy receptor proteins. Interestingly, it was recently found that optineurin, another autophagy receptor protein, was also recruited to ubiquitinated mitochondria via its ubiquitin binding domain after Parkin activation on mitochondria. Optineurin induces autophagosome formation around the damaged mitochondria by recruiting double FYVE-containing protein 1 (DFCP1) and LC3 to damaged mitochondria [67]. In addition, Parkin also recruits Ambra1 (a Beclin-1 interacting protein) to depolarized mitochondria to initiate engulfment of damaged mitochondria by autophagosomes [68]. Taken together, it appears that Pink1 and Parkin regulate mitophagy at multiple levels including mitochondrial fragmentation (via Mfn1 and Mfn2), mitochondrial motility (via Miro), autophagy receptor protein (via p62 or optineurin), and autophagy machinery (via Ambra1). The molecular events of Pink1–Parkin-mediated mitophagy are summarized in Fig. 3.
Fig. 3.
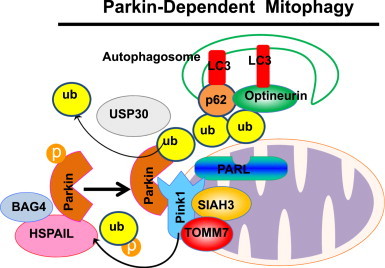
Molecular events for Parkin-dependent mitophagy. When mitochondria are depolarized in response to various insults, Parkin is translocated to the outer membrane of mitochondria. This process is regulated by PINK1, which is stabilized on depolarized mitochondria due to the inactivation of the mitochondrial protease PARL. Pink1 is also stabilized by the outer mitochondrial membrane protein TOMM7 but de-stabilized by SIAH3, a mitochondrial resident protein. PINK1 either directly phosphorylates Parkin or ubiquitin to promote Parkin translocation or its ligase activity. Cytosolic HSPA1L also promotes, whereas BAG4 inhibits, Parkin mitochondrial translocation. Once Parkin translocates to mitochondria, it promotes selective mitophagy through mitochondrial ubiquitination and recruitment of autophagy receptor proteins such as p62 and optineurin, which further recruit LC3 positive autophagosomes.
While there is a tremendous amount of evidence supporting the important role of Pink1–Parkin in regulating mitophagy, most of the studies so far used a cell culture model in which cells were ectopically overexpressed with Parkin and exposed to the mitochondrial uncoupler CCCP [10]. Studies on the role of Parkin in regulating mitophagy in vivo are scarce. Parkin knockout mice had decreased mitophagy and were more sensitive to myocardial infarction compared with wild type mice [69]. In aged mice or mice treated with doxorubicin, it was recently reported that cytosolic p53 bound to Parkin and inhibited Parkin mitochondrial translocation and mitophagy. Overexpression of Parkin attenuated functional decline of aged hearts [70]. However, a recent study showed that loss of Drp1 in mouse cardiomyocytes led to increased mitochondrial ubiquitination and p62 mitochondrial targeting, which was independent of Parkin [71]. We also found that Parkin is expressed in mouse livers, and Parkin translocated to hepatic mitochondria after mice were administrated with acetaminophen, which damages mitochondria in hepatocytes. Intriguingly, the levels of ubiquitination and p62 on mitochondria were not different between wild type and Parkin knockout mice after APAP treatment (Williams J. et al., manuscript under revision). These results suggest that there must be other E3 ligases compensating for the loss of Parkin to trigger Parkin-independent mitophagy. It is well known that Pink1 and Parkin knockout mice only bear subtle phenotypes related to dopaminergic neuronal degeneration. A recent elegant study discovered that mitochondrial ubiquitin ligase 1 (MUL1) compensates for the loss of Pink1 or Parkin, and MUL1 acts by increasing Mfn1 degradation. Overexpression of MUL1 in Drosophila almost completely rescued the phenotypes observed in Pink1/Parkin null Drosophila [72]. These results suggest that while Parkin plays an important role in mitophagy, Parkin-independent mechanisms may also be involved in regulating mitophagy (see below).
Parkin-independent mitophagy
Increasing evidence now supports that mitophagy can occur independent of Parkin. Several autophagy receptor proteins have been shown to localize on mitochondria and interact with LC3 to recruit autophagosomes to damaged mitochondria including Parkin-independent mediators such as Bcl2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), NIX (also called BNIP3L), and Fun14 Domain containing 1 (FUNDC1). Both BNIP3 and NIX are hypoxia-inducing factor-1 (HIF-1) target genes that contain an LIR to interact with LC3, which can promote mitophagy by recruiting autophagosomes to damaged mitochondria and protect against reactive oxygen species (ROS) accumulation [73]. BNIP3 and NIX activate autophagy by binding to Bcl-2, which dissociates the complex of Bcl-2 and Beclin-1, a protein necessary for initiation of autophagosome formation [74]. BNIP3 and NIX seem to have complementary roles in mitophagy because depletion of either BNIP3 or NIX did not affect mitophagy, but depletion of both BNIP3 and NIX inhibited the mitophagic response to hypoxic conditions [74]. In addition to its role in mitophagy induction during hypoxia, NIX is also required for removal of excess mitochondria during red blood cell maturation. The entry of mitochondria into autophagosomes for degradation was blocked in NIX-deficient mice, which led to life-span reduction of red blood cells and development of anemia [75,76].
In response to hypoxia, FUNDC1, an outer mitochondrial membrane protein, has also been shown to have a role in mitophagy [77]. During hypoxia, PGAM5 dephosphorylates FUNDC1 on Ser13 [78], which allows for it to interact with LC3 on autophagosome membranes via its LIR [77]. Moreover, Bcl2-like 1 (BCL2L1) prevents the dephosphorylation of FUNDC1 and mitophagy by inhibiting PGAM5 [79]. It seems that hypoxia induces FUNDC1-dependent mitophagy using a different mechanism. Unlike BNIP3 and NIX, the expression of FUNDC1 mRNA is decreased during hypoxia. It is currently unknown why several autophagy receptor proteins are needed during hypoxia-induced mitophagy, but dephosphorylated FUNDC1 has greater binding affinity for LC3 than NIX [80].
Cardiolipin is a phospholipid dimer synthesized in the inner mitochondrial membrane. Upon mitochondrial damage and depolarization, cardiolipin translocates to the outer mitochondrial membrane to initiate mitophagy [81,82]. In rotenone-treated SH-SY5Y cells and primary cortical neurons, increased LC3 co-localization with mitochondria was observed. Prevention of cardiolipin translocation to the outer mitochondrial surface inhibited GFP-LC3 co-localization with mitochondria and mitophagy [82]. Interestingly, cardiolipin regulates both apoptosis and mitophagy depending on its peroxidation status. Peroxidized cardiolipin that is present on the outer mitochondrial membrane initiates cell death through apoptosis, whereas non-peroxidized cardiolipin initiates mitophagy to protect the cell from apoptotic cell death [81].
In primary cultured mouse hepatocytes, two distinctive mitophagy termed Type I and Type II mitophagy have been defined by John Lemasters group. Type I mitophagy was induced by nutrient deprivation, whereas Type II mitophagy was induced by photodamage. Both types of mitophagy require the recruitment of GFP-LC3 positive autophagosomes to mitochondria, in which Type I but not type II mitophagy is blocked by PI3 kinase inhibitors [12]. Our laboratory recently found that Parkin is quickly degraded within 4–6 h culture in primary mouse hepatocytes (Ding et al., unpublished observations) thus it is likely that both Type I and Type II mitophagy are Parkin-independent.
As discussed above, in addition to Parkin, several other mitochondrial E3 ligases may also play a role in mitophagy. SMURF1 is an E3 ubiquitin ligase that was found to have a role in CCCP-induced mitophagy. Unlike Parkin, the ubiquitin ligase function of SMURF1 is not required for mitophagy induction. Instead, the C2 domain of SMURF1 is required for engulfment of damaged mitochondria by autophagosomes [83]. In addition to SMURF1, overexpression of MUL1 increases mitochondrial ubiquitination, mitochondrial fission and mitophagy in mouse skeletal muscle [84]. As mentioned above, AMBRA1 has been shown to interact with Parkin and regulates Parkin-mediated mitophagy [68]. However, AMBRA1 also binds directly with LC3 via its LIR and induces LC3-dependent but Parkin and p62-independent mitophagy [85]. Therefore, with the rapid progress on mitophagy research, it is anticipated that many other Parkin-independent mitophagy pathways will be revealed in the future. The possible molecular events of Parkin-independent mitophagy are summarized in Fig. 4.
Fig. 4.
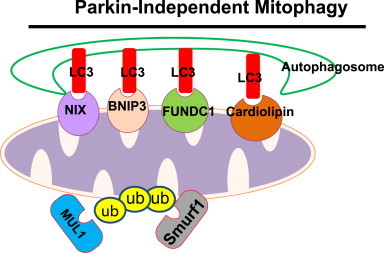
Molecular events for Parkin-independent mitophagy. In the absence of Parkin, BNIP3, NIX, FUNDC1 or cardiolipin directly interact with LC3 and recruit autophagosomes to damaged mitochondria. Moreover, other E3 ubiquitin ligases such as SMURF1 and MUL1 can also promote mitochondrial ubiquitination, p62 mitochondrial targeting and mitophagy.
Mitochondrial quality control regulation by vesicular trafficking to the lysosome and formation of mitochondrial spheroids
One intriguing puzzle in the mitophagy field is that not all mitochondrial proteins are degraded to the same extent during mitophagy. The current notion is that outer mitochondrial membrane proteins may be degraded via the proteasomal system whereas mitochondrial matrix proteins may be removed by autophagy [54,86]. However, it remains unknown how autophagy selectively removes some matrix proteins but not others. Abeliovich et al. elegantly showed that different mitochondrial matrix proteins were degraded at distinct different rates during yeast mitophagy [87]. The rates of mitophagic degradation of matrix proteins correlated with the degree of physical segregation of specific matrix proteins, which was regulated by the yeast fission molecule DNM1 [87]. Therefore, mitochondrial fission is important for mitochondrial matrix remodeling/segregation, which may result in selective degradation of mitochondrial matrix proteins by mitophagy.
Moreover, a novel pathway involving the formation of mitochondria-derived vesicles (MDVs) under conditions of mitochondrial oxidative stress that regulates mitochondrial protein turnover and quality has also been reported. Interestingly, this pathway also requires Parkin and Pink1 but it is different from canonical mitophagy because the formation of MDVs is stimulated by ROS production instead of mitochondrial depolarization. In addition, vesicles bud off of damaged mitochondria and are degraded in the lysosome independent of the canonical autophagy pathway [88]. MDVs contain oxidized proteins and may regulate mitochondrial quality faster than mitophagy to prevent complete mitochondrial depolarization while preserving mitochondrial function by selectively degrading damaged mitochondrial contents [6,88].
Our laboratory has also recently reported a novel mitochondrial remodeling and quality control mechanism, which we named mitochondrial spheroids [7–9]. Mitochondrial spheroids are structurally unique mitochondria that have a ring or cup-like morphology with squeezed mitochondrial matrix. Similar to autophagosomes, mitochondrial spheroids can enwrap contents of the cytosol such as endoplasmic reticulum, lipid droplets, or other mitochondria. They are also positive for lysosome proteins, but whether they actually degrade contents within their lumen remains to be determined. Formation of mitochondrial spheroids is independent of canonical autophagy pathways because it occurs in Atg5 or Atg7-deficient MEFs. The formation of mitochondrial spheroids requires the presence of ROS and either Mfn1 or Mfn2 [7]. Parkin negatively regulates the formation of mitochondrial spheroids by inducing proteasomal degradation of Mfn1 and Mfn2, suggesting that Parkin prevents mitochondrial spheroid formation in order for mitophagy to occur. Mitochondrial spheroids are detected in CCCP-treated MEFs and in mouse livers exposed to acetaminophen, acute alcohol or high fat diet [89]. These results suggest that formation of mitochondrial spheroids may represent general mitochondrial structural remodeling in response to various physiological and pathological stresses and could serve as an alternative mechanism for regulation of mitochondrial homeostasis. The molecular events for MDVs and mitochondrial spheroids formation are summarized in Fig. 5. Taken together, it seems that multiple pathways may serve as an alternative pathway for regulation of mitochondrial homeostasis independent of Pink1–Parkin or in the absence of canonical autophagy.
Fig. 5.
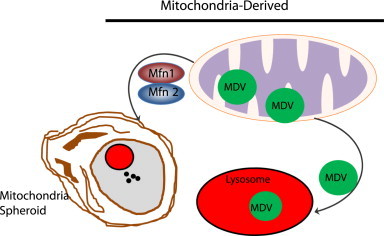
Mitochondria derived vesicles and mitochondrial spheroids. Under certain conditions, mitochondria can undergo direct remodeling to form mitochondrial spheroids, which are regulated by Mfn1 and Mfn2. Mitochondrial spheroids then further acquire lysosomal markers, possibly through fusion with lysosomes. Small vesicles that contain a subset group of mitochondrial proteins are generated from damaged mitochondria to form MDVs. The segregation of mitochondria to form MDVs is also regulated by Pink1 and Parkin. MDVs are fused with late endosomes and multivesicular bodies and then delivered to lysosomes where they are eventually degraded. Both the formation of mitochondrial spheroids and MDVs are independent of canonical autophagy machinery.
Concluding remarks and future perspectives
Mitochondria are dynamic organelles that play many important roles in maintaining cellular homeostasis and survival. Mitochondria are also the central executioners for cell death by releasing pro-cell death molecules from their inter membrane space or by generating toxic ROS. Therefore, it is vital to timely remove damaged mitochondria via mechanisms such as mitophagy. Mitochondrial fission and fusion as well as motility play an important role in mitophagy. Both Parkin-dependent and -independent mechanisms for induction of mitophagy help to maintain the quality control of mitochondria. In addition, damaged mitochondria may also undergo direct remodeling to form MDVs or mitochondrial spheroids. MDVs target to lysosomes for degradation of a specific subset of mitochondrial proteins, but whether mitochondrial spheroids can degrade their contents in the lysosome is currently unclear. Future work to determine the role of Parkin-dependent and -independent mitophagy in vivo models is needed to further elucidate the pathophysiological relevance of mitophagy.
Acknowledgments
The research work in Wen-Xing Ding's laboratory was supported in part by the NIAAA funds R01 AA020518, R01 DK102142, National Center for Research Resources (5P20RR021940), the National Institute of General Medical Sciences (8P20 GM103549 and T32 ES007079), and an Institutional Development award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20 GM103418). The authors want to thank Barbara Fegley from the electron microscopy core facility at the University of Kansas Medical Center for her excellent assistance for EM studies.
References
- 1.Baker, Haynes C.M. Mitochondrial protein quality control during biogenesis and aging. Trends in Biochemical Sciences. 2011;36(5):254–261. doi: 10.1016/j.tibs.2011.01.004. 21353780 [DOI] [PubMed] [Google Scholar]
- 2.Matsushima Y., Kaguni L.S. Matrix proteases in mitochondrial DNA function. Biochimica et Biophysica Acta. 2012;1819(9–10):1080–1087. doi: 10.1016/j.bbagrm.2011.11.008. 22172992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karbowski M., Youle R.J. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Current Opinion in Cell Biology. 2011;23(4):476–482. doi: 10.1016/j.ceb.2011.05.007. 21705204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van der Bliek A.M., Shen Q., Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harbor Perspectives in Biology. 2013;5(6):a011072. doi: 10.1101/cshperspect.a011072. 23732471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Twig G. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO Journal. 2008;27(2):433–446. doi: 10.1038/sj.emboj.7601963. 18200046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soubannier V. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLOS One. 2012;7(12):e52830. doi: 10.1371/journal.pone.0052830. 23300790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding W.X. Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. Journal of Biological Chemistry. 2012;287(50):42379–42388. doi: 10.1074/jbc.M112.413682. 23095748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding W.X. Electron microscopic analysis of a spherical mitochondrial structure. Journal of Biological Chemistry. 2012;287(50):42373–42378. doi: 10.1074/jbc.M112.413674. 23093403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin X.M., Ding W.X. The reciprocal roles of PARK2 and mitofusins in mitophagy and mitochondrial spheroid formation. Autophagy. 2013;9(11):1687–1692. doi: 10.4161/auto.24871. 24162069 [DOI] [PubMed] [Google Scholar]
- 10.Ding W.X., Yin X.M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biological Chemistry. 2012;393(7):547–564. doi: 10.1515/hsz-2012-0119. 22944659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Youle R.J., Narendra D.P. Mechanisms of mitophagy. Nature Reviews Molecular Cell Biology. 2011;12(1):9–14. doi: 10.1038/nrm3028. 21179058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemasters J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3) Redox Biology. 2014;2:749–754. doi: 10.1016/j.redox.2014.06.004. 25009776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cipolat S. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell. 2006;126(1):163–175. doi: 10.1016/j.cell.2006.06.021. 16839884 [DOI] [PubMed] [Google Scholar]
- 14.Ishihara N. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO Journal. 2006;25(13):2966–2977. doi: 10.1038/sj.emboj.7601184. 16778770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duvezin-Caubet S. OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria. Molecular Biology of the Cell. 2007;18(9):3582–3590. doi: 10.1091/mbc.E07-02-0164. 17615298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griparic L., Kanazawa T., van der Bliek A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. Journal of Cell Biology. 2007;178(5):757–764. doi: 10.1083/jcb.200704112. 17709430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ehses S. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. Journal of Cell Biology. 2009;187(7):1023–1036. doi: 10.1083/jcb.200906084. 20038678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Head B. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. Journal of Cell Biology. 2009;187(7):959–966. doi: 10.1083/jcb.200906083. 20038677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samant S.A. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Molecular and Cellular Biology. 2014;34(5):807–819. doi: 10.1128/MCB.01483-13. 24344202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westermann B. Mitochondrial fusion and fission in cell life and death. Nature Reviews Molecular Cell Biology. 2010;11(12):872–884. doi: 10.1038/nrm3013. 21102612 [DOI] [PubMed] [Google Scholar]
- 21.Gandre-Babbe S., van der Bliek A.M. The novel tail-anchored membrane protein mff controls mitochondrial and peroxisomal fission in mammalian cells. Molecular and Cellular Biology. 2008;19(6):2402–2412. doi: 10.1091/mbc.E07-12-1287. 18353969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otera H. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. Journal of Cell Biology. 2010;191(6):1141–1158. doi: 10.1083/jcb.201007152. 21149567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmer C.S. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Reports. 2011;12(6):565–573. doi: 10.1038/embor.2011.54. 21508961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Losón O.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Molecular and Cellular Biology. 2013;24(5):659–667. doi: 10.1091/mbc.E12-10-0721. 23283981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedman J.R. ER tubules mark sites of mitochondrial division. Science. 2011;334(6054):358–362. doi: 10.1126/science.1207385. 21885730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taguchi N. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. Journal of Biological Chemistry. 2007;282(15):11521–11529. doi: 10.1074/jbc.M607279200. 17301055 [DOI] [PubMed] [Google Scholar]
- 27.Cribbs J.T., Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Reports. 2007;8(10):939–944. doi: 10.1038/sj.embor.7401062. 17721437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148(1–2):228–243. doi: 10.1016/j.cell.2011.11.030. 22265414 [DOI] [PubMed] [Google Scholar]
- 29.Moujalled D.M. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death & Disease. 2014;5:e1086. doi: 10.1038/cddis.2014.18. 24577084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karbowski M., Neutzner A., Youle R.J. The mitochondrial E3 ubiquitin ligase March5 is required for Drp1 dependent mitochondrial division. Journal of Cell Biology. 2007;178(1):71–84. doi: 10.1083/jcb.200611064. 17606867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura N. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Reports. 2006;7(10):1019–1022. doi: 10.1038/sj.embor.7400790. 16936636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harder Z., Zunino R., McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Current Biology. 2004;14(4):340–345. doi: 10.1016/j.cub.2004.02.004. 14972687 [DOI] [PubMed] [Google Scholar]
- 33.Mishra P., Chan D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nature Reviews Molecular Cell Biology. 2014;15(10):634–646. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saxton W.M., Hollenbeck P.J. The axonal transport of mitochondria. Journal of Cell Science. 2012;125(9):2095–2104. doi: 10.1242/jcs.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misko A. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the miro/Milton complex. Journal of Neuroscience. 2010;30(12):4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010. 20335458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y., Sheng Z.H. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. Journal of Cell Biology. 2013;202(2):351–364. doi: 10.1083/jcb.201302040. 23857772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pekkurnaz G. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell. 2014;158(1):54–68. doi: 10.1016/j.cell.2014.06.007. 24995978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu S. Parkinson's disease-associated kinase PINK1 regulates miro protein level and axonal transport of mitochondria. PLOS Genetics. 2012;8(3) doi: 10.1371/journal.pgen.1002537. 22396657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Narendra D. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. Journal of Cell Biology. 2008;183(5):795–803. doi: 10.1083/jcb.200809125. 19029340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark I.E. Drosophila pink1 is required for mitochondrial function and interacts genetically with Parkin. Nature. 2006;441(7097):1162–1166. doi: 10.1038/nature04779. 16672981 [DOI] [PubMed] [Google Scholar]
- 41.Park J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by Parkin. Nature. 2006;441(7097):1157–1161. doi: 10.1038/nature04788. 16672980 [DOI] [PubMed] [Google Scholar]
- 42.Yang Y. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(28):10793–10798. doi: 10.1073/pnas.0602493103. 16818890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawajiri S. PINK1 is recruited to mitochondria with Parkin and associates with LC3 in mitophagy. FEBS Letters. 2010;584(6):1073–1079. doi: 10.1016/j.febslet.2010.02.016. 20153330 [DOI] [PubMed] [Google Scholar]
- 44.Kitada T. Mutations in the Parkin gene cause autosomal recessive juvenile Parkinsonism. Nature. 1998;392(6676):605–608. doi: 10.1038/33416. 9560156 [DOI] [PubMed] [Google Scholar]
- 45.Jin S.M. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. Journal of Cell Biology. 2010;191(5):933–942. doi: 10.1083/jcb.201008084. 21115803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim Y. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochemical and Biophysical Research Communications. 2008;377(3):975–980. doi: 10.1016/j.bbrc.2008.10.104. 18957282 [DOI] [PubMed] [Google Scholar]
- 47.Sha D., Chin L.S., Li L. Phosphorylation of Parkin by Parkinson disease-linked kinase PINK1 activates Parkin E3 ligase function and NF-kappaB signaling. Human Molecular Genetics. 2010;19(2):352–363. doi: 10.1093/hmg/ddp501. 19880420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kane L.A. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. Journal of Cell Biology. 2014;205(2):143–153. doi: 10.1083/jcb.201402104. 24751536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koyano F. Ubiquitin is phosphorylated by PINK1 to activate Parkin. Nature. 2014;510(7503):162–166. doi: 10.1038/nature13392. 24784582 [DOI] [PubMed] [Google Scholar]
- 50.Hasson S.A. High-content genome-wide RNAi screens identify regulators of Parkin upstream of mitophagy. Nature. 2013;504(7479):291–295. doi: 10.1038/nature12748. 24270810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fiesel F.C. A specific subset of E2 ubiquitin-conjugating enzymes regulate Parkin activation and mitophagy differently. Journal of Cell Science. 2014;127:3488–3504. doi: 10.1242/jcs.147520. 24928900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Geisler S. The ubiquitin-conjugating enzymes UBE2N, UBE2L3 and UBE2D2/3 are essential for Parkin-dependent mitophagy. Journal of Cell Science. 2014;127(15):3280–3293. doi: 10.1242/jcs.146035. 24906799 [DOI] [PubMed] [Google Scholar]
- 53.Soleimanpour S.A. The diabetes susceptibility gene Clec16a regulates mitophagy. Cell. 2014;157(7):1577–1590. doi: 10.1016/j.cell.2014.05.016. 24949970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chan N.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Human Molecular Genetics. 2011;20(9):1726–1737. doi: 10.1093/hmg/ddr048. 21296869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gegg M.E. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Human Molecular Genetics. 2010;19(24):4861–4870. doi: 10.1093/hmg/ddq419. 20871098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geisler S. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature Cell Biology. 2010;12(2):119–131. doi: 10.1038/ncb2012. 20098416 [DOI] [PubMed] [Google Scholar]
- 57.Poole A.C. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLOS One. 2010;5(4):e10054. doi: 10.1371/journal.pone.0010054. 20383334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim I., Lemasters J.J. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. American Journal of Physiology—Cell Physiology. 2011;300(2):C308–C317. doi: 10.1152/ajpcell.00056.2010. 21106691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Twig G., Shirihai O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxidants and Redox Signaling. 2011;14(10):1939–1951. doi: 10.1089/ars.2010.3779. 21128700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X. PINK1 and Parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147(4):893–906. doi: 10.1016/j.cell.2011.10.018. 22078885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bingol B. The mitochondrial deubiquitinase USP30 opposes Parkin-mediated mitophagy. Nature. 2014;510(7505):370–375. doi: 10.1038/nature13418. 24896179 [DOI] [PubMed] [Google Scholar]
- 62.Ding W.X. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. Journal of Biological Chemistry. 2010;285(36):27879–27890. doi: 10.1074/jbc.M110.119537. 20573959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang C. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLOS One. 2011;6(6):e20975. doi: 10.1371/journal.pone.0020975. 21687634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manley S., Williams J.A., Ding W.X. Role of p62/SQSTM1 in liver physiology and pathogenesis. Experimental Biology and Medicine (Maywood) 2013;238(5):525–538. doi: 10.1177/1535370213489446. 23856904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Narendra D. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6(8):1090–1106. doi: 10.4161/auto.6.8.13426. 20890124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Okatsu K. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells. 2010;15(8):887–900. doi: 10.1111/j.1365-2443.2010.01426.x. 20604804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wong Y.C., Holzbaur E.L. Optineurin is an autophagy receptor for damaged mitochondria in Parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(42):E4439–E4448. doi: 10.1073/pnas.1405752111. 25294927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van Humbeeck C. Parkin interacts with Ambra1 to induce mitophagy. Journal of Neuroscience. 2011;31(28):10249–10261. doi: 10.1523/JNEUROSCI.1917-11.2011. 21753002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kubli D.A. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. Journal of Biological Chemistry. 2013;288(2):915–926. doi: 10.1074/jbc.M112.411363. 23152496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoshino A. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nature Communications. 2013;4:2308. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- 71.Kageyama Y. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO Journal. 2014 doi: 10.15252/embj.201488658. 25349190 (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yun J. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife. 2014;3:e01958. doi: 10.7554/eLife.01958. 24898855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang H. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. Journal of Biological Chemistry. 2008;283(16):10892–10903. doi: 10.1074/jbc.M800102200. 18281291 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74.Bellot G. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Molecular and Cellular Biology. 2009;29(10):2570–2581. doi: 10.1128/MCB.00166-09. 19273585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schweers R.L. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(49):19500–19505. doi: 10.1073/pnas.0708818104. 18048346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sandoval H. Essential role for nix in autophagic maturation of erythroid cells. Nature. 2008;454(7201):232–235. doi: 10.1038/nature07006. 18454133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu L. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature Cell Biology. 2012;14(2):177–185. doi: 10.1038/ncb2422. 22267086 [DOI] [PubMed] [Google Scholar]
- 78.Chen G. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Molecular Cell. 2014;54(3):362–377. doi: 10.1016/j.molcel.2014.02.034. 24746696 [DOI] [PubMed] [Google Scholar]
- 79.Wu H. The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy. 2014;10(10):1712–1725. doi: 10.4161/auto.29568. 25126723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Novak I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Reports. 2010;11(1):45–51. doi: 10.1038/embor.2009.256. 20010802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ren M., Phoon C.K.L., Schlame M. Metabolism and function of mitochondrial cardiolipin. Progress in Lipid Research. 2014;55:1–16. doi: 10.1016/j.plipres.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 82.Chu C.T. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nature Cell Biology. 2013;15(10):1197–1205. doi: 10.1038/ncb2837. 24036476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Orvedahl A. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480(7375):113–117. doi: 10.1038/nature10546. 22020285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lokireddy S. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metabolism. 2012;16(5):613–624. doi: 10.1016/j.cmet.2012.10.005. 23140641 [DOI] [PubMed] [Google Scholar]
- 85.Strappazzon F. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of Parkin and p62/SQSTM1. Cell Death and Differentiation. 2014 doi: 10.1038/cdd.2014.190. 25215947 (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yoshii S.R. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. Journal of Biological Chemistry. 2011;286(22):19630–19640. doi: 10.1074/jbc.M110.209338. 21454557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Abeliovich H. Involvement of mitochondrial dynamics in the segregation of mitochondrial matrix proteins during stationary phase mitophagy. Nature Communications. 2013;4:2789. doi: 10.1038/ncomms3789. 24240771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McLelland G. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO Journal. 2014;33(4):282–295. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ni H.M. Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox Biology. 2013;1(1):427–432. doi: 10.1016/j.redox.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]