

Abstract
The past decade has seen an unprecedented increase in our understanding of the biology and etiology of head and neck squamous cell carcinomas (HNSCC). Genome-wide sequencing projects have identified a number of recurrently mutated genes, including unexpected alterations in the NOTCH pathway and chromatin related genes. Gene-expression profiling has identified 4 distinct genetic subtypes which show some parallels to lung squamous cell carcinoma biology. The identification of the human papilloma virus as one causative agent in a subset of oropharyngeal cancers and their association with a favorable prognosis has opened up avenues for new therapeutic strategies. The expanding knowledge of the underlying molecular abnormalities in this once very poorly understood cancer should allow for increasingly rational clinical trial design and improved patient outcomes.
Keywords: Genomics, Head and neck cancer, Mutation, Gene expression, Copy number
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common malignancy in the world, with an annual world-wide incidence of over 600,000 cases per year and 350,000 deaths per year.1 Cancers in the head and neck can originate from a variety of subsites including the lip, oral cavity, nasopharynx, oropharynx, and larynx. The most common causes of head and neck cancers are tobacco and alcohol, which work synergistically, and are responsible for 70–75% of cases.2, 3 In parts of Asia, betel-quid chewing also plays a significant role in the development of malignancy.4 On a molecular level, these cancers often have p53 mutations and many display chromosomal instability.5
More recently, the human papilloma virus (HPV) has been shown to promote HNSCC and primarily cause oropharyngeal cancers (a sub-site in the head and neck).6 Researchers have found significant differences in the pathogenesis and prognosis of HPV-related malignancies and HPV-negative malignancies and have tended to classify HPV associated malignances as a separate biologic entity. Unlike HPV-negative cancers, HPV-related oropharyngeal malignancies are not as strongly associated with tobacco and alcohol exposure. They are, instead, related to sexual behavior, which is a conduit to transmit HPV.7, 8 Relative to HPV-negative malignancies, HPV-positive cancers are associated with a more favorable prognosis.7, 9 The incidence of HPV-positive cancer is rising, while incidence of HPV-negative cancers is decreasing.
Treatment of HNSCC is made challenging due to the diversity of the anatomic sites in the head and neck and the critical normal structures that may be near a particular tumor site. Often, the care of a patient requires a multi-disciplinary team of surgeons, radiation oncologists, medical oncologists, nutritionists, gastroenterologists, and speech and swallowing therapists, amongst others. Treatment options for HNSCC can consist of definitive surgical resection, definitive chemo-radiation, or a combination of surgery and chemo-radiation. Despite the availability of aggressive treatments, the 5-year survival rate for head and neck malignancies remains relatively poor at 65%, with only modest gains in the past few years.10
An understanding of the molecular and genetic abnormalities leading to oncogenesis in head and neck malignancies has dramatically increased in the past decade. Initial attempts to understand the genetic etiology of head and neck cancers focused on cytogenetic studies. The development of microarray technology enabled the classification of HNSCC into distinct types based on gene expression. More recently, next-generation sequencing technologies have allowed multiple groups of researchers to sequence a large number of tumors to identify novel mutated tumor suppressor genes and oncogenes. An underlying rationale for these researchers' genomic profiling studies was to gain a more thorough understanding of the molecular abnormalities in head and neck cancer to help guide the development of new therapeutics. Before reviewing the major discoveries of each of these profiling techniques, we will briefly discuss genetic factors that may pre-dispose individuals to develop HNSCC.
Genetic predisposition to HNSCC
Although most cases of HNSCC are induced by carcinogens or viral infection, a small fraction of cases are familial in nature. The syndromic etiology with the clearest link of an increased risk of HNSCC is Fanconi anemia, an autosomal recessive genomic-instability syndrome associated with bone marrow failure, leukemia, congenital defects, and sensitivity to cross-linking agents.11 Cohort studies of Fanconi anemia patients have demonstrated a highly elevated risk of HNSCC with the ratio of the observed number of HNSCC cases to expected number of cases ranging between 240 and 706.12, 13, 14 Treatment of these patients requires care as they are sensitive to common chemotherapeutics used in HNSCC and may be sensitive to radiotherapy as well, although this latter point is controversial.15 Rare clusters of HNSCC have also been reported in families with germ-line mutations in CDKN2A and ATR.16, 17
In non-syndromic families, initial case–control studies demonstrated a genetic predisposition, with first-degree relatives having a 3.5 fold increased risk of HNSCC. Subsequent pooled analysis, however, led to an odds ratio of 1.7.18, 19, 20, 21 It has been hypothesized that genetic differences in pathways such as DNA repair, carcinogen metabolism, and cell cycle control may increase the risk of carcinogenesis from exposure to tobacco or alcohol.22 Phenotypic differences in these processes in lymphocytes from HNSCC patients and controls have been identified.23, 24 Candidate gene-based studies have generated mixed results, with the notable exception of a nearly 9000 patient series that identified SNPs in multiple alcohol dehydrogenase (ADH) genes associated with a decreased risk of an upper aero-digestive malignancy.25, 26, 27, 28 Subsequently, a genome-wide association study in upper aero-digestive malignancies validated the ADH SNPs and also identified a SNP in HELQ (a DNA repair gene) as being associated with risk of malignancy. Cumulatively, these SNPs only accounted for 4% of the familial risk.29
Expression profiling
One of the first attempts to genomically profile HNSCC was with mRNA profiling. Unsupervised hierarchal clustering of the transcriptome of 60 HNSCC patients30 identified 4 subtypes – basal, atypical, mesenchymal type, and classical subtype – which have subsequently been verified by other investigators.31 Classical tumors exhibit alterations in expression of genes involved in oxidative stress, such as KEAP/NFEL2. The atypical cluster is enriched with HPV-positive tumors (discussed further below). Mesenchymal tumors demonstrate an elevation in expression of genes associated with epithelial-to-mesenchymal transition (EMT). The basal subtype has an expression pattern similar to basal epithelial cells in airways and is named for its similarity to the basal type in squamous cell carcinoma of the lung.
The four subtypes do not show a significant correlation with age or smoking status, but do appear to be related to site of origin.31 Still, each anatomic subsite, except for hypopharyngeal cancer, is present to some extent in each cluster, suggesting that expression-based subtypes reflect a biology that, at least in part, transcends anatomic sub-site. Whether these subtypes provide prognostic information in and of themselves is unclear at present, given the small size of the studies to date examining the issue. Initial reports indicated these expression-based groups may predict for recurrence-free survival, however, those findings have not been subsequently replicated.31 Of significant interest is that there is a strong correlation between each of these subtypes and their corresponding expression-based subtypes in squamous cell carcinoma of the lung.31 That is, the basal, mesenchymal, and classical subtypes in HNSCC strongly correlate with the basal, secretory, and classical subtypes in lung cancer. In other malignancies such as breast cancer and glioblastoma, expression-based subtypes have helped guide translational research as well as therapeutic development; their utility in HNSCC remains to be determined but they potentially could guide research efforts.32, 33
Multiple investigators have developed expression-based signatures to predict clinical behavior of HNSCC, such as lymph node metastasis, hypoxia, or radiosensitivity.34, 35, 36, 37, 38 Roepman developed a 102 gene signature that is predictive of the propensity for lymph node metastasis which theoretically could be used to guide decisions regarding lymph node dissections.34 The predictor, however, was only able to correctly determine lymph node status in 61 of 82 patients, and the authors noted that this was of only incremental improvement to clinical decision-making. Onken et al developed an expression signature for nodal metastasis from a mouse model of oral cavity cancer.39 Interestingly, this signature could predict nodal metastasis development for human oral cavity cancers in a training set and a small validation set. However, additional validation is still needed.
Several groups have looked into expression signatures to identify tumors that are hypoxic, and therefore, likely to be resistant to radiotherapy. Some groups have looked for hypoxic gene signatures in head and neck cancers whereas others have performed combined analysis over multiple cancers. In general, these signatures have demonstrated prognostic value in small cohorts.35, 36, 37 The hypoxic signature by the Danish group was used to retrospectively analyze data from the DANHCA 5 trial, which was a randomized trial of radiotherapy ± nimorazole (a hypoxic radiosensitizer) in HNSCC.40 This group found that the benefit of nimorazole on local control and DFS was limited to patients whose tumors were deemed to be “more hypoxic” by their expression signature.41 This suggests that their signature may be predictive and could help select patients for future clinical trials using hypoxic sensitizers.
Mutational analysis and alterations in specific genes and pathways in HNSCC
The first two large-scale sequencing efforts in HNSCC identified a number of recurrently mutated genes. The majority of these were in tumor suppressor genes rather than oncogenes.42, 43 Stransky et al performed whole exome-sequencing on 74 tumor/normal pairs, with 150-fold mean coverage, while Agrawal et al sequenced 32 tumor/normal pairs, with a mean coverage of 77-fold and performed targeted follow-up sequencing in an additional 88 patients. For HPV-negative tumors, Stransky et al found a mutation rate of 4.83 per Mb while Agrawal et al found a mutation rate of 20.6 per Mb. The large discrepancy between the two groups may have arisen from differences in bioinformatics techniques of mutation calling or differences in coverage of sequencing. Since these initial studies, two other groups have looked at mutations specifically in oral cavity cancers44, 45 (Table 1 for study details).
Table 1.
Major sequencing studies.
| Stransky | Agrawal | Pickering | India ICGC |
|
|---|---|---|---|---|
| N | 74 | 32 | 40 | 50 |
| Disease sitesa | OC, OP, H, L, SC | OC, OP, L, H | OC | OC |
| Smokers | 89% | 75% | 78.6% | 96% |
| HPV positivity | 14% | 25% | 2.5% | 26% |
| Mean coverage | 150 | 77d | 119 | 38d |
| Whole exome | 72 | 32 | 40 | 50 |
| Whole genome | 2 | 0 | 0 | 0 |
| Additional tumors with limited sequencing | NA | 88 | NA | 60 |
| Significantly mutated genesb | 39 | 6 | 53 | 10 |
| Mutations per MB (HPV+; HPV−) | 2.28; 4.83 | 4.8; 20.6 | NRc | 4.07; 3.36 |
Sites: OC: Oral Cavity, OP: Oropharynx, H: Hypopharynx, L: Larynx; SC sinonasal cavity.
Significantly mutated here is as determined by authors of paper (some studies included formal statistical testing, while others did not).
Not reported.
Group used multiple sequencing platforms, highest mean coverage is reported.
In a pan-cancer analysis, Stratton and colleagues identified HNSCC as possessing the 9th highest average mutational load out of 30 tumor types studied, with patients having a range of mutations from less than 1 mutation per Mb to over 100 mutations per Mb.46 They identified several active mutational processes in HNSCC, including mutations due to tobacco exposure, aging, UV light, and alterations in the apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) enzymes. Multiple groups have found signatures of tobacco exposure in HNSCC, which often include guanosine-to-thymidine transversions.43, 46 Although alterations in APOBEC have been shown to cause mutations in other malignancies, most prominently breast cancer, it is not clear these enzymes are active in HNSCC as of yet.47 Instead of looking at the type of mutations, Mroz et al used sequencing data to estimate tumor heterogeneity, which they called the mutant-allele tumor heterogeneity (MATH) score. This score was prognostic in both HPV-positive and HPV-negative patients.48
In HPV-negative tumors, the predominant finding from these sequencing efforts was that a large number of tumor suppressor genes were mutated. The Broad group identified 39 genes with recurrent mutations (q ≤ 0.1), whereas the JHU group identified 6 genes with mutations in more than 4% of their study (Table 2). Mutations and other genomic alterations in individual genes and key pathways are reviewed individually below. Alterations in selected pathways are summarized in Fig. 1. Of the non-coding mutations, an interesting one occurs in the promoter of TERT in nearly 20% of HNSCC, resulting in increased expression of telomerase.49
Table 2.
Frequency of selected genes recurrently mutated in 4 major sequencing studies to date.
| Gene | Stransky | Agrawala | Pickering | India ICGC |
|---|---|---|---|---|
| p53 | 62% | 47% | 66% | 62% |
| CDKN2A | 12% | 9.2% |  |
 |
| CASP8 | 8% |  |
10% | 34% |
| FAT1 | 12% | |
28% | 40% |
| NOTCH1 | 14% | 15% | 9% | 16% |
| HRAS | 5% | 4% | 9% | 12% |
| PIK3CA | 8% | 6% | 11% | 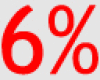 |
| MLL2 |  |
|
 |
 |
| FBXW7 |  |
5% |  |
|
Red indicates genes were not identified as significant in that study.
Did not use formal statistical testing.
Figure 1.

Alterations in selected pathways in head and neck squamous cell cancers. Green: Frequency of mutations. Red: frequency of amplification. Blue: Frequency of deletion. (A) Mitogenic Pathway Alterations. (B) Cell Cycle Alterations. (C) NOTCH Signaling. (D) Oxidative Stress Response. See text for more details of alterations in each pathway (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
TP53
Long before the era of genome sequencing, TP53 had been recognized as a frequently mutated gene in HNSCC, with modern series estimating 46–73% of HNSCC cases having a mutation.42, 43, 44, 50, 51, 52, 53 TP53 is a tumor suppressor gene that functions primarily as a transcription factor and regulates the expression of hundreds of downstream target genes in response to a variety of cellular stresses.54, 55 It is composed of 393 amino acids and contains 4 domains, including a highly conserved DNA binding domain (DBD). It plays a key role in a number critical cellular functions, including the response to DNA damage and oncogenic stress. DNA damage leads to activation of p53, triggering cell cycle arrest and an attempt to repair the damage, which if not completed, can trigger apoptosis or senescence. Mutations in TP53 appear to be acquired early in the pathogenesis of HNSCC and are present in pre-malignant lesions, suggesting an important role in early oncogenesis.5, 51, 56
Most of the missense mutations in TP53 cluster in the DNA binding domain (DBD) whereas frame-shift and nonsense mutations are more equally distributed throughout the gene. One third of the mutations in the DBD occurs within 6 hotspot residues and result in preventing binding to DNA.55 Since p53 forms a tetramer, many of these missense mutations are expected to have a dominant negative effect.57 Work has indicated that some of these mutations may also result in a “gain-of-function” phenotype, perhaps resulting from expression of other off-target genes. However, the significance of this remains unclear.57, 58 Besides mutation, p53 may be inactivated in other ways such as via MDM2 over-expression or amplification (MDM2 is a negative regulator of p53) or deletion of CDKN2A (negatively regulates MDM2).59 Inactivation of p53 also occurs in HPV-positive tumors and is discussed in more detail below.
Whether alterations in TP53 are prognostic for overall survival has been somewhat controversial, with early studies showing conflicting results.54, 60 Limitations in these earlier studies include the reliance on immunostaining, which is often discordant with mutational status, and a failure to sequence the entire length of the TP53 gene.61, 62 More recent and thorough sequencing-based analysis shows that functional mutations appear to result in significantly worse outcomes.50, 63 For instance, Poeta and colleagues examined a cohort of 420 patients treated with surgery ± radiotherapy and found that mutations in TP53 were associated with worse overall survival (HR = 1.4, p = 0.009). Patients with truncating mutations or disrupting mutations in the DNA binding domain had significantly worse overall survival (HR = 1.7, p < 0.001), whereas non-functional mutations alone were not statistically associated with worse outcomes (HR = 1.2, p = 0.16).
As p53 plays an integral role in handling genotoxic stress, multiple groups found that TP53 mutation results in worse outcomes in patients uniformly treated with either definitive radiotherapy or post-operative radiotherapy.63, 64, 65 However, as TP53 mutations also are prognostic in patients treated with surgery alone, it is unclear if mutational status is a true predictive marker. Further, the mechanism by which p53 promotes radio-resistance remains unclear.66 In cell types susceptible to p53-mediated apoptosis, loss-of-function would theoretically induce radio-resistance. p53-mediated apoptosis, however, does not appear to be the dominant mode of cell death in epithelial tumors.66 Recent in-vitro work with HNSCC cell lines has suggested that a subset of p53 mutations may have a gain-of-function phenotype that promotes senescence and subsequent radio-resistance.65 Future investigation and more accurate models will be needed to sort out this controversy.
Cell cycle alterations: CDKN2A, CCND1, and RB1
There is homozygous deletion of CDKN2A in nearly 30% of HNSCC and the gene is mutated in another 10–20% of samples, and is frequently subject to LOH.67, 68, 69 The majority of mutations are inactivating and includes frame shifts, nonsense mutations, and splice site changes. Additional epigenetic alterations may lead to this gene being inactivated in up to 80% of tumors.70 One possible strategy to target this alteration would be to inhibit CDK4/CDK6. Recently, Palbociclib, a CDK4/6 inhibitor, increased progression free survival in a cohort of breast cancer patients, suggesting such an approach may be efficacious.71
The next most commonly altered cell cycle gene in HNSCC is CCND1, which is amplified in over 20% of tumors.68 CCND1 encodes cyclin D1, which serves as a cofactor for CDK4 and CDK6 to phosphorylate Rb.72 Again, CDK4/6 inhibitor could serve as a method to target these tumors, and both p16 loss and CCND1 amplification are being investigated as biomarkers in the Palbociclib trial. In the TCGA data, genetic alteration of one of either CCND1 or CDKN2A occurs in nearly 60% of cases and in the study by Pickering et al, it was 94%.45 RB1, the retinoblastoma gene, also appears to be either mutated or deleted in approximately 5% of patients.
EGFR
Epidermal growth factor receptor (EGFR) is a member of the HER/erbB family of receptor tyrosine-kinases and is one of the primary targetable alterations in cancers.73, 74 When activated by binding its ligand, EGFR homodimerizes or heterodimerizes with other members of the ErbB family, stimulating its tyrosine kinase activity and leading to auto-phosphorylation of tyrosine residues in its cytoplasmic tail.75 This can lead to activation of downstream signaling cascades including RAS/RAF/MAPK, PI3K/AKT/mTOR (discussed further below), and/or JAK/STAT. These pathways are important regulators of proliferation, invasion, angiogenesis and metastasis.
Therapeutics targeting the EGFR pathway have been introduced into the clinical setting for a number of malignancies, including HNSCC.76, 77 Cetuximab, a chimeric murine/human monoclonal antibody against EGFR, has improved overall survival compared to standard treatment in the locally advanced and metastatic settings, although adoption in the locally advanced setting remains controversial.78, 79, 80, 81 Two humanized antibodies against EGFR, however, have failed to improve overall survival in either the recurrent/metastatic setting or the locally advanced setting.82, 83, 84 Similarly two first-generation, small molecule, tyrosine kinase inhibitors against EGFR (gefitinib and erlotinib) have been disappointing.85, 86, 87
Although EGFR is expressed on the surface of the majority of HNSCC88 tumors and the degree of overexpression is associated with a worse prognosis, the muddled picture from therapeutic interventions in the pathway has led to uncertainty on exactly how this pathway is activated in HNSCC.89, 90, 91, 92 Unlike adenocarcinoma of the lung where mutations in the kinase domain result in constitutive activation of EGFR, such mutations are rare in HNSCC.69, 93 Amplification of EGFR occurs in a minority of patients and the presence of amplification or increased IHC expression has not correlated with efficacy of EGFR inhibition.94, 95 Of note, amplifications in EGFR appear to occur less frequently in HPV-positive patients, although the therapeutic implications of this are unclear.
The molecular and clinical trial results suggest that perhaps cetuximab's efficacy is mediated more by its antibody-dependent cell-mediated cytotoxicity then blockade of the EGFR pathway.76, 96, 97 Alternatively, resistance to EGFR inhibition may occur quickly by up-regulation of other erbB receptors, such as ERBB2 (HER2/NEU).98 Strategies addressing both these possibilities are being actively pursued clinically with the development of pan-erbB inhibitors and monoclonal antibodies as well as the pairing of cetuximab with various immune check-point inhibitors in development.
PI3K/AKT/mTOR
PIK3CA is the 2nd most commonly mutated gene across human cancers and alterations in this pathway are thought to play an important role in cancer cell growth, survival, and metabolism.99, 100 The PI3K family of kinases consists of three classes, with class IA being most frequently involved in cancer. Activation of PI3K leads to phosphorylation of phosphatidylinositols (PIP2, a second messenger) and subsequent activation of AKT, one of the main effectors of PI3K signaling.100 Activated forms of AKT have been found in the majority of HNSCC tumors analyzed by tissue microarrays.88 AKT further activates mTOR, which is also responsible for integrating cellular signals regarding nutrient levels, cellular energy stores, and stress. One mechanism of PI3K activation in HNSCC is by receptor-tyrosine kinases, such as EGFR. Although EGFR cannot directly active PI3K, if EGFR heterodimerizes with ERBB3 it can subsequently activate PI3K signaling.101
Another mechanism of activation of PI3K pathway in HNSCC involves mutation, with mutations occurring in PI3K catalytic subunit, p110α (encoded by PIK3CA gene). Approximately 20% of tumors have mutations in PIK3CA, however these mutations occur much more commonly in HPV-positive tumors compared to HPV-negative tumors.68 Mutations often occur in one of two hotspot locations in the kinase or helical domain and promote constitutive signaling through the pathway.102 Further, PIK3CA is amplified in 20% of HNSCC overall. PTEN loss or mutation occurs in 3% of HNSCC. PTEN encodes a phosphatase that shuts off PI3K signaling by dephosphorylating PIP3 to PIP2. Genomic alterations in other members of the PI3K pathway typically occur at frequencies of less than 5% in HNSCC.
Multiple therapeutic agents that target this pathway are under investigation in both the upfront and metastatic/recurrent setting. In general, agents targeting this pathway include sole PI3K inhibitors (isoform selective and pan-inhibitors), dual PI3K-mTOR inhibitors, AKT inhibitors, and mTOR inhibitors. In other malignancies, responses to these drugs as single agents have been disappointing and perhaps suggest the development of feedback loops counteracting inhibition.103 Further, initial experience with dual PI3K-mTOR agents appear to be limited by toxicity. To-date, however, experience in HNSCC remains limited and trials are presently on-going.77
NOTCH1/p63/FBXW7
The discovery of recurrent mutations in NOTCH1 was one of the key novel findings of the initial HNSCC exome-sequencing projects.42, 43 Notch1 is a transmembrane receptor that plays a role in cell differentiation and embryonic development.104, 105 Signaling is thought to be mediated by cell-to-cell contact, enabling binding of Notch to one of five known ligands. After receptor activation by ligand binding, a cascade of proteolytic cleavages take place that result in the release of Notch1's intracellular domain (NICD). NICD subsequently translocates to the nucleus where it interacts with CBF1 (CSL) and mastermind-like (MAML1) leading to transcriptional activation of multiple genes. Well characterized genes under control of this pathway include the HES and HRT family of genes, notch ligands, CCND1, MYC, and others.
Notch appears to be able to function as either a tumor suppressor or an oncogene depending on cell context. In TCGA data, NOTCH1 is mutated in 18.6% of cases, with over 35% of mutations leading to a frameshift or nonsense codon.68 The majority of missense mutations appear in EGF-like domain repeats, which are important for ligand binding. This pattern of mutations has suggested that Notch's role in HNSCC may be as a tumor suppressor gene, although the exact mechanism of tumor suppression remains unclear. In a murine model, deletion of Notch1 results in hyperplasia and carcinogen-induced skin cancer, supporting a tumor suppressor function.106 Similar loss-of-function mutations have been seen in cutaneous SCC and lung SCC and cell-based testing of mutations in the EGF-like domain have demonstrated loss of signaling.107 In contrast, Notch1 mutations are oncogenic in T-cell acute lymphoblastic leukemia and cause ligand-independent cleavage of Notch1, ultimately leading to constitutive activation.108 Notch's oncogenic functions are thought to be mediated by prompting cell cycle progression and inhibition of apoptosis.105
The other Notch receptors are mutated in 2–4% of cases with a roughly similar mutational spectrum. In keratinocytes of both human and mouse origin, Notch1 signaling interacts in a negative feedback loop with TP63. TP63 is a p53 family member gene, which has a role in keratinocyte cell fate determination. TP63 is found amplified in 20% of tumors in the TCGA set and interestingly, amplifications appear to be mutually exclusive with NOTCH1 mutation (p = 0.053).68 FBXW7 is ubiquitin ligase that targets Notch for degradation and is found mutated in 4.7% of cancers of HNSCC.109 FBXW7 may have a role outside of Notch signaling, as it targets many other known oncogenes including cyclin E, MYC, and JUN.
Oxidative stress response: NRF2/KEAP1/CUL3
Genes involved in oxidative stress are altered by mutation or copy number change in nearly 20% of HNSCC, and by IHC, NRF2 is overexpressed in 90% of tumors.68, 110 NRF2 (encoded by the NFE2L2 locus) is a transcription factor that activates the cellular antioxidant response.111 It is typically constitutively expressed under basal conditions and kept in the cytoplasm and degraded by KEAP1 and CUL3. CUL3 is a core component of an E3 ubiquitin ligase complex, which relies on KEAP1, a substrate specific adaptor, to successfully ubiquinate NRF2 and promote its degradation. The interaction between NRF2 and KEAP1 is mediated by NRF2's Neh2 domain and KEAP1's Kelch/DGR domain. In the presence of oxidative stress, the interaction between KEAP1 and NRF2 is inhibited, leading to the accumulation of NRF2 in the cytoplasm, subsequent translocation to the nucleus, and thereafter transcriptional activation of its target. Increased activation of NRF2 may promote cancer cell growth and protect against cytotoxic therapies.111
In HNSCC, NRF2 is mutated in 6% of patients and amplified in another 6%. Mutations are clustered in the Neh2 domain, specifically in two motifs important for binding with Keap1. The mutations appear to be gain-of-function mutations which inhibit interaction with KEAP1, and subsequently lead to an up-regulation of NRF2.112 KEAP1 is mutated in 4% of tumors and rarely homozygously deleted. The majority of mutations are missense mutations and they occur both in domains involved with binding to NRF2 and those responsible for binding to CUL3. These also presumably lead to increased cytoplasmic NRF2, and subsequent activation of the pathway. Lastly, CUL3 is mutated or deleted in approximately 6% of patients.
In the TCGA data set, genomic alterations in the NRF2 pathway have correlated with worse overall survival.113 Although not yet validated in another HNSCC cohort, alterations in the NRF2 pathway may be prognostic in other malignancies such as NSCLC.114 One reason for worse outcomes would be resistance to cytotoxic treatments. Multiple studies have shown that elevated NRF2 levels lead to chemoresistance in a variety of cancer cell lines that appears to be reversible with siRNA inhibition of NRF2.111, 115, 116 Similarly, NRF2 alterations as determined by expression signature also correlate with resistance to radiotherapy in cell-line models.117
Chromatin related genes: MLL2, NSD1, EP300, and other epigenetic changes
The discovery of recurrent mutations in chromatin modifying genes was one of the major discoveries of cancer sequencing projects in general. In HNSCC, several chromatin related genes are recurrently mutated, and at least one of these genes is mutated in 32% of cases. Chromatin consists of proteins and DNA and facilitates the packaging of genomic DNA within the nucleus. The basic functional unit is a nucleosome, which consists of a histone octamer and 147 bp of DNA. Chromatin modifying genes can covalently modify histone proteins, which alters the structure of the nucleosome or alternatively alter DNA.118 These modifications play a critical role in regulating DNA-based processes, such as transcription and DNA repair.
MLL2 (aka KMTD2) is a histone methyltransferase that is mutated in 19% of HNSCC. Over 50% of the mutations lead to a frame-shift, truncation or splice site alteration, suggesting a tumor suppressor function. MLL2 can methylate several lysine residues on histones, including H3K4, which is often associated with positive gene regulation. In leukemia's, MLL translocations are quiet common and their role in carcinogenesis is much better understood than the role of the more recently discovered MLL2 point mutations.119, 120 NSD1 is another histone methyltransferase that is mutated in 10% of patients, with, again, a strong bias towards truncating and nonsense mutations.
EP300 is histone acetyltransferase that is mutated in 7% of HNSCC. EP300 and its paralog CBP (CREB-binding protein) are transcriptional co-activators, which share 75% sequence similarity and although they have some distinct roles, they share many functions.121 Acetylation of histones by EP300 leads to relaxing the superstructure of chromatin and enables the transcription of proximal genes. EP300 can also directly interact with a wide range of transcription factors, pro-proliferation proteins, and tumor suppressors directly, including c-MYC, c-MYB, CREB, c-JUN, c-FOS, p53, STAT1, BRCA1, and SMAD homologous proteins. Given its cooperation with such a wide variety of proteins, it can promote opposing cellular processes depending on cell type and context.
FAT1/CASP8
Clustering analysis of mutations reported by the International Cancer Genome Consortium (ICGC) demonstrated that FAT1 and CAPS8 mutations may define a distinct subtype of HNSCC.44 FAT1 is mutated in 23% of tumors, with a predominance of nonsense and frame-shift mutations, suggesting a tumor suppressor function. FAT1 encodes a large transmembrane protein, a protocadherin, which contains 30+ cadherin repeats as well as 4–5 EGF-like domains.122 FAT1 is thought to suppress cancer cell growth by playing a role in Wnt signaling by binding to β-catenin and limiting its ability to translocate to the nucleus.123 Truncating mutations would inhibit this functionality and promote tumor cell growth. Of note, the FAT family of genes (FAT1-4) are also mutated in glioblastoma, colorectal cancer, gastric, ovarian, and pancreatic cancers. CASP8 is mutated in 9% of HNSCC and encodes a caspase that plays an important role in programmed cell death (apoptosis). CASP8 is specifically involved in the extrinsic pathway of apoptosis signaling and is activated by membrane bound receptors such as Fas and TNF.124 Upon activation, Casp-8 triggers the execution phase of apoptosis.
Chromosomal instability and copy number alterations
Among HPV-negative HNSCC, 80% display significant chromosomal instability and over 40% of tumors have undergone whole genome duplication.5, 45, 125 The mechanisms underlying this instability are unclear, but include both alterations at the chromosomal arm-level and smaller focal alterations (megabase-pair in size).126 The amount of chromosomal instability is known to vary between gene-expression defined molecular subtypes.31 On average, an individual tumor will have 7.3 arm-level alterations and 16 focal alterations.45 TCGA and others have identified a large number of recurrent alterations, including 12 arm-level amplification events, 14 arm-level deletions, 26 focal amplifications, and 49 focal deletions.69 Some of the most commonly lost regions include 3p, 4p, and 9p, whereas those commonly gained include 3q, 7p, and 8q.127, 128
Califano and colleagues studied 87 lesions – including hyperplasia, dysplasia, carcinoma in situ, and invasive cancer – using microsatellite analysis to determine which chromosomal losses occurred early in carcinogenesis.129 These investigators identified losses on 3p and 9p as early events that often occur in premalignant lesions. Chromosome 9p contains several tumor suppressor genes, the most notable being CDNK2A, which is involved in cell cycle regulation as discussed previously. The tumor suppressor(s) on 3p responsible for oncogenesis in HNSCC remains unclear.130 Recent genomic analysis of squamous cell carcinoma of the esophagus also identified common deletions in 3p and 9p, and overall demonstrated a very similar copy number landscape to HNSCC, suggesting a close molecular relationship.131
Increasing levels of chromosomal instability in pre-malignant lesions have been associated with an increased risk of progression to malignant disease in multiple retrospective studies.132, 133, 134 Across a variety of malignancies, once cancer has already developed, increasing chromosomal instability also appears to be associated with worse outcome.135 Similarly, in head and neck cancer, increasing chromosomal instability has been associated with a worse outcome and a more severe malignant phenotype in multiple studies, including worse overall survival and higher risk of lymph node metastasis.136, 137, 138 In terms of predicting response to treatment, one small case control study compared the copy number profile of tumors sensitive to chemoradiotherapy vs. those that were resistant and found a difference in the patterns of gains and losses.139
HPV
Human Papillomavirus (HPV) family of viruses are double-stranded DNA viruses with a propensity to infect squamous epithelium.140, 141 The HPV family is composed of both low and high-risk strains, which is determined by a strain's ability to lead to malignant progression. HPV has long been known to induce malignancies such as cervical, anal, and vulvar cancers. Definitive evidence linking HPV as a causative agent in oropharyngeal cancer didn't emerge until the turn of the century.6 Since then, it has become clear that HPV-related oropharyngeal cancers are a distinct entity with a better prognosis compared to traditional smoking and alcohol related head and neck cancers.9 Further, epidemiologic trends have suggested a dramatic increase of HPV-related malignancies over the past two decades, with the primary risk factor relating to sexual behavior.7, 142 HPV16 is the main subtype associated with HNSCC, although a number of other subtypes have also been reported.7, 142, 143 The role of HPV in the pathogenesis of non-oropharyngeal head and neck cancers remains unclear at present and is an area of active investigation.
The HPV genome consist of 8000 basepairs and is divided into 3 segments, an early coding region (E), a late coding region (L), and a long control region (LCR).141 The early and late regions contain genes important for different parts of the viral life cycle whereas the LCR contains regulatory elements important for replication and transcription. In cancer, the HPV genome can exist either in an episomal form or integrated into the host genome. The viral load and transcription of HPV-related genes in tumors can vary on the order of several magnitudes between different tumors.143, 144 Integration into the host genome was thought to favor common fragile sites, but can occur randomly. Recent work has suggested recurrent insertions in RAD51 and ERBB2.143, 145 Integration is also thought to promote oncogenesis.
HPV contains two oncogenes, E6 and E7, that inactivate p53 and Rb respectively and are thought to be important mediators in producing a malignant phenotype. E6 reduces p53 activity by binding to E6-AP (UBE3A), a ubiquitin-protein ligase, and targeting p53 for ubiquitination and degradation.146 E6 can also promote transcription of hTERT, facilitating immortalization of cells. Inhibition of Rb by E7 leads to cell cycle progression as discussed above (see Cell Cycle alterations). E7 inhibits Rb by facilitating ubiquitination and degradation of the protein. Although the E7 protein from low-risk HPV serotypes is also able to bind to Rb, E7 from high-risk HPV serotypes is much more efficient. Both E6 and E7 also interact with a variety of other host proteins that may also play a role in oncogenesis.141
Given their distinct etiology, the mutational, expression, and copy number profiles of HPV-related cancers are distinct from their HPV-negative counterparts. For instance, both of the initial sequencing projects demonstrated 2–4 fold less mutations per megabase for HPV-positive tumors compared to their HPV-negative counterparts.42, 43 The Indian ICGC sequencing project did not identify a difference in mutation rate between HPV-positive and negative tumors. However their samples consisted entirely of oral cavity tumors, where the role of HPV in the underlying pathogenesis of malignancy remains unclear.44 Similarly, which genes are mutated is strongly influence by whether or not the malignancy is HPV-related. For example, p53 is almost never mutated in HPV-related malignancies, as p53 is inhibited by E6. In addition, mutations in PIK3CA appear to be more common in HPV-positive malignancies compared to HPV-negative malignancies (see above). In terms of expression profiling, HPV-related malignancies cluster with atypical tumors; however, not all atypical tumors are HPV-related.31
As HPV-positive tumors have a better prognosis, a number of clinical trials are underway investigating various ways of de-escalating treatment to reduce morbidity of treatment. The molecular differences between HPV-positive and negative tumors clearly play a role in their prognosis and will also help guide de-escalation trial design. For instance, some have found an inverse relationship between HPV and the presence of EGFR by IHC staining.147, 148, 149 However, at present there is no definitive clinical data to suggest a difference in efficacy for anti-EGFR therapy in HPV-positive compared to HPV-negative patients.150
Conclusion and future directions
Sequencing efforts and integrative genomics have revealed a great deal about the molecular abnormalities that underlie HNSCC. Unfortunately, they have also demonstrated marked heterogeneity, which will make optimizing therapeutic intervention difficult. In HNSCC, oncogenic mutations are rare and mutations in tumor suppressor genes dominate. Restoring function of a tumor suppressor pharmacologically is difficult. Targeting these alterations may require exploring synthetic lethal approaches.43 Although any individual tumors may still have some targetable alterations (i.e. amplification or mutation in a known oncogene), it is unclear in many cases if these are passengers or driving events.45 Furthermore, oncogenic pathways can be activated by non-genetic mechanisms, which would not have been detected by genomic efforts. Now that a genetic blueprint for HNSCC has been completed, the challenge moving forward will be to identify ways to use this information to develop improved diagnostic and therapeutic modalities.
Conflict of interest
The authors have no conflict of interest to disclose.
Acknowledgments
We thank Dr. Nancy Lee for thoughtful discussions. T.A.C. was partially supported by the Adenoid Cystic Carcinoma Research Foundation (ACCRF) and a grant from the National Institutes of Health (R21 DE023229).
Footnotes
Peer review under responsibility of Chongqing Medical University.
References
- 1.Ferlay J., Shin H.R., Bray F., Forman D., Mathers C., Parkin D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. Dec 15 2010;127(12):2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Hashibe M., Brennan P., Chuang S.C. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol Biomarkers Prev. Feb 2009;18(2):541–550. doi: 10.1158/1055-9965.EPI-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blot W.J., McLaughlin J.K., Winn D.M. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. Jun 1 1988;48(11):3282–3287. [PubMed] [Google Scholar]
- 4.Chen Y.J., Chang J.T., Liao C.T. Head and neck cancer in the betel quid chewing area: recent advances in molecular carcinogenesis. Cancer Sci. Aug 2008;99(8):1507–1514. doi: 10.1111/j.1349-7006.2008.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leemans C.R., Braakhuis B.J., Brakenhoff R.H. The molecular biology of head and neck cancer. Nat Rev Cancer. Jan 2011;11(1):9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 6.Gillison M.L., Koch W.M., Capone R.B. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. May 3 2000;92(9):709–720. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 7.Chaturvedi A.K., Engels E.A., Pfeiffer R.M. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. Nov 10 2011;29(32):4294–4301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillison M.L., Broutian T., Pickard R.K. Prevalence of oral HPV infection in the United States, 2009-2010. JAMA. Feb 15 2012;307(7):693–703. doi: 10.1001/jama.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ang K.K., Harris J., Wheeler R. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. Jul 1 2010;363(1):24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howlader N., Noone A., Krapcho M. 2013. SEER Cancer Statistics Review; pp. 1975–2011. Accessed April 2014. [Google Scholar]
- 11.Moldovan G.L., D'Andrea A.D. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenberg P.S., Alter B.P., Ebell W. Cancer risks in fanconi anemia: findings from the German Fanconi Anemia Registry. Haematologica. Apr 2008;93(4):511–517. doi: 10.3324/haematol.12234. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg P.S., Greene M.H., Alter B.P. Cancer incidence in persons with Fanconi anemia. Blood. Feb 1 2003;101(3):822–826. doi: 10.1182/blood-2002-05-1498. [DOI] [PubMed] [Google Scholar]
- 14.Kutler D.I., Singh B., Satagopan J. A 20-year perspective on the International Fanconi Anemia Registry (IFAR) Blood. Feb 15 2003;101(4):1249–1256. doi: 10.1182/blood-2002-07-2170. [DOI] [PubMed] [Google Scholar]
- 15.Alter B.P. Radiosensitivity in Fanconi's anemia patients. Radiother Oncol. Mar 2002;62(3):345–347. doi: 10.1016/s0167-8140(01)00474-1. [DOI] [PubMed] [Google Scholar]
- 16.Schneider-Stock R., Giers A., Motsch C. Hereditary p16-Leiden mutation in a patient with multiple head and neck tumors. Am J Hum Genet. Jan 2003;72(1):216–218. doi: 10.1086/345397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka A., Weinel S., Nagy N. Germline mutation in ATR in autosomal- dominant oropharyngeal cancer syndrome. Am J Hum Genet. Mar 9 2012;90(3):511–517. doi: 10.1016/j.ajhg.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Negri E., Boffetta P., Berthiller J. Family history of cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Int J Cancer. Jan 15 2009;124(2):394–401. doi: 10.1002/ijc.23848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Copper M.P., Jovanovic A., Nauta J.J. Role of genetic factors in the etiology of squamous cell carcinoma of the head and neck. Arch Otolaryngol Head Neck Surg. Feb 1995;121(2):157–160. doi: 10.1001/archotol.1995.01890020019005. [DOI] [PubMed] [Google Scholar]
- 20.Foulkes W.D., Brunet J.S., Sieh W., Black M.J., Shenouda G., Narod S.A. Familial risks of squamous cell carcinoma of the head and neck: retrospective case-control study. BMJ. Sep 21 1996;313(7059):716–721. doi: 10.1136/bmj.313.7059.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sturgis E.M., Wei Q., Spitz M.R. Descriptive epidemiology and risk factors for head and neck cancer. Semin Oncol. Dec 2004;31(6):726–733. doi: 10.1053/j.seminoncol.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 22.Sturgis E.M., Wei Q. Genetic susceptibility–molecular epidemiology of head and neck cancer. Curr Opin Oncol. May 2002;14(3):310–317. doi: 10.1097/00001622-200205000-00010. [DOI] [PubMed] [Google Scholar]
- 23.Wang L.E., Sturgis E.M., Eicher S.A., Spitz M.R., Hong W.K., Wei Q. Mutagen sensitivity to benzo(a)pyrene diol epoxide and the risk of squamous cell carcinoma of the head and neck. Clin Cancer Res. Jul 1998;4(7):1773–1778. [PubMed] [Google Scholar]
- 24.Cheng L., Eicher S.A., Guo Z., Hong W.K., Spitz M.R., Wei Q. Reduced DNA repair capacity in head and neck cancer patients. Cancer Epidemiol Biomarkers Prev. Jun 1998;7(6):465–468. [PubMed] [Google Scholar]
- 25.Hashibe M., McKay J.D., Curado M.P. Multiple ADH genes are associated with upper aerodigestive cancers. Nat Genet. Jun 2008;40(6):707–709. doi: 10.1038/ng.151. [DOI] [PubMed] [Google Scholar]
- 26.da Silva S.D., Ferlito A., Takes R.P. Advances and applications of oral cancer basic research. Oral Oncol. Sep 2011;47(9):783–791. doi: 10.1016/j.oraloncology.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 27.Cadoni G., Boccia S., Petrelli L. A review of genetic epidemiology of head and neck cancer related to polymorphisms in metabolic genes, cell cycle control and alcohol metabolism. Acta Otorhinolaryngol Ital. Feb 2012;32(1):1–11. [PMC free article] [PubMed] [Google Scholar]
- 28.Brunotto M., Zarate A.M., Bono A., Barra J.L., Berra S. Risk genes in head and neck cancer: a systematic review and meta-analysis of last 5 years. Oral Oncol. Mar 2014;50(3):178–188. doi: 10.1016/j.oraloncology.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 29.McKay J.D., Truong T., Gaborieau V. A genome-wide association study of upper aerodigestive tract cancers conducted within the INHANCE consortium. PLoS Genet. Mar 2011;7(3):e1001333. doi: 10.1371/journal.pgen.1001333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung C.H., Parker J.S., Karaca G. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell. May 2004;5(5):489–500. doi: 10.1016/s1535-6108(04)00112-6. [DOI] [PubMed] [Google Scholar]
- 31.Walter V., Yin X., Wilkerson M.D. Molecular subtypes in head and neck cancer exhibit distinct patterns of chromosomal gain and loss of canonical cancer genes. PLoS One. 2013;8(2):e56823. doi: 10.1371/journal.pone.0056823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perou C.M., Sorlie T., Eisen M.B. Molecular portraits of human breast tumours. Nature. Aug 17 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 33.Verhaak R.G., Hoadley K.A., Purdom E. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. Jan 19 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roepman P., Wessels L.F., Kettelarij N. An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet. Feb 2005;37(2):182–186. doi: 10.1038/ng1502. [DOI] [PubMed] [Google Scholar]
- 35.Winter S.C., Buffa F.M., Silva P. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res. Apr 1 2007;67(7):3441–3449. doi: 10.1158/0008-5472.CAN-06-3322. [DOI] [PubMed] [Google Scholar]
- 36.Toustrup K., Sorensen B.S., Nordsmark M. Development of a hypoxia gene expression classifier with predictive impact for hypoxic modification of radiotherapy in head and neck cancer. Cancer Res. Sep 1 2011;71(17):5923–5931. doi: 10.1158/0008-5472.CAN-11-1182. [DOI] [PubMed] [Google Scholar]
- 37.Buffa F.M., Harris A.L., West C.M., Miller C.J. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br J Cancer. Jan 19 2010;102(2):428–435. doi: 10.1038/sj.bjc.6605450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eschrich S.A., Pramana J., Zhang H. A gene expression model of intrinsic tumor radiosensitivity: prediction of response and prognosis after chemoradiation. Int J Radiat Oncol Biol Phys. Oct 1 2009;75(2):489–496. doi: 10.1016/j.ijrobp.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Onken M.D., Winkler A.E., Kanchi K.L. A surprising cross-species conservation in the genomic landscape of mouse and human oral cancer identifies a transcriptional signature predicting metastatic disease. Clin Cancer Res. Jun 1 2014;20(11):2873–2884. doi: 10.1158/1078-0432.CCR-14-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Overgaard J., Hansen H.S., Overgaard M. A randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother Oncol. Feb 1998;46(2):135–146. doi: 10.1016/s0167-8140(97)00220-x. [DOI] [PubMed] [Google Scholar]
- 41.Toustrup K., Sorensen B.S., Lassen P. Gene expression classifier predicts for hypoxic modification of radiotherapy with nimorazole in squamous cell carcinomas of the head and neck. Radiother Oncol. Jan 2012;102(1):122–129. doi: 10.1016/j.radonc.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 42.Agrawal N., Frederick M.J., Pickering C.R. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. Aug 26 2011;333(6046):1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stransky N., Egloff A.M., Tward A.D. The mutational landscape of head and neck squamous cell carcinoma. Science. Aug 26 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.India Project Team of the International Cancer Genome C Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat Commun. 2013;4:2873. doi: 10.1038/ncomms3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pickering C.R., Zhang J., Yoo S.Y. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. Jul 2013;3(7):770–781. doi: 10.1158/2159-8290.CD-12-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alexandrov L.B., Nik-Zainal S., Wedge D.C. Signatures of mutational processes in human cancer. Nature. Aug 22 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burns M.B., Lackey L., Carpenter M.A. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. Feb 21 2013;494(7437):366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mroz E.A., Rocco J.W. MATH, a novel measure of intratumor genetic heterogeneity, is high in poor-outcome classes of head and neck squamous cell carcinoma. Oral Oncol. Mar 2013;49(3):211–215. doi: 10.1016/j.oraloncology.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Killela P.J., Reitman Z.J., Jiao Y. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. Apr 9 2013;110(15):6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poeta M.L., Manola J., Goldwasser M.A. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. Dec 20 2007;357(25):2552–2561. doi: 10.1056/NEJMoa073770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boyle J.O., Hakim J., Koch W. The incidence of p53 mutations increases with progression of head and neck cancer. Cancer Res. Oct 1 1993;53(19):4477–4480. [PubMed] [Google Scholar]
- 52.Balz V., Scheckenbach K., Gotte K., Bockmuhl U., Petersen I., Bier H. Is the p53 inactivation frequency in squamous cell carcinomas of the head and neck underestimated? Analysis of p53 exons 2-11 and human papillomavirus 16/18 E6 transcripts in 123 unselected tumor specimens. Cancer Res. Mar 15 2003;63(6):1188–1191. [PubMed] [Google Scholar]
- 53.Somers K.D., Merrick M.A., Lopez M.E., Incognito L.S., Schechter G.L., Casey G. Frequent p53 mutations in head and neck cancer. Cancer Res. Nov 1 1992;52(21):5997–6000. [PubMed] [Google Scholar]
- 54.Brosh R., Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. Oct 2009;9(10):701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 55.Olivier M., Hollstein M., Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. Jan 2010;2(1):a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Braakhuis B.J., Leemans C.R., Brakenhoff R.H. A genetic progression model of oral cancer: current evidence and clinical implications. J Oral Pathol Med. Jul 2004;33(6):317–322. doi: 10.1111/j.1600-0714.2004.00225.x. [DOI] [PubMed] [Google Scholar]
- 57.Petitjean A., Mathe E., Kato S. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. Jun 2007;28(6):622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 58.Dittmer D., Pati S., Zambetti G. Gain of function mutations in p53. Nat Genet. May 1993;4(1):42–46. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 59.Rothenberg S.M., Ellisen L.W. The molecular pathogenesis of head and neck squamous cell carcinoma. J Clin Invest. Jun 1 2012;122(6):1951–1957. doi: 10.1172/JCI59889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kyzas P.A., Loizou K.T., Ioannidis J.P. Selective reporting biases in cancer prognostic factor studies. J Natl Cancer Inst. Jul 20 2005;97(14):1043–1055. doi: 10.1093/jnci/dji184. [DOI] [PubMed] [Google Scholar]
- 61.Taylor D., Koch W.M., Zahurak M., Shah K., Sidransky D., Westra W.H. Immunohistochemical detection of p53 protein accumulation in head and neck cancer: correlation with p53 gene alterations. Hum Pathol. Oct 1999;30(10):1221–1225. doi: 10.1016/s0046-8177(99)90041-2. [DOI] [PubMed] [Google Scholar]
- 62.Loyo M., Li R.J., Bettegowda C. Lessons learned from next-generation sequencing in head and neck cancer. Head Neck. Mar 2013;35(3):454–463. doi: 10.1002/hed.23100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lindenbergh-van der Plas M., Brakenhoff R.H., Kuik D.J. Prognostic significance of truncating TP53 mutations in head and neck squamous cell carcinoma. Clin Cancer Res. Jun 1 2011;17(11):3733–3741. doi: 10.1158/1078-0432.CCR-11-0183. [DOI] [PubMed] [Google Scholar]
- 64.Koch W.M., Brennan J.A., Zahurak M. p53 mutation and locoregional treatment failure in head and neck squamous cell carcinoma. J Natl Cancer Inst. Nov 6 1996;88(21):1580–1586. doi: 10.1093/jnci/88.21.1580. [DOI] [PubMed] [Google Scholar]
- 65.Skinner H.D., Sandulache V.C., Ow T.J. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. Jan 1 2012;18(1):290–300. doi: 10.1158/1078-0432.CCR-11-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gudkov A.V., Komarova E.A. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. Feb 2003;3(2):117–129. doi: 10.1038/nrc992. [DOI] [PubMed] [Google Scholar]
- 67.van der Riet P., Nawroz H., Hruban R.H. Frequent loss of chromosome 9p21-22 early in head and neck cancer progression. Cancer Res. Mar 1 1994;54(5):1156–1158. [PubMed] [Google Scholar]
- 68.Cerami E., Gao J., Dogrusoz U. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. May 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Center BITGDA . 2014. Analysis Overview for Head and Neck Squamous Cell Carcinoma (Primary Solid Tumor Cohort) Accessed 18.04.14. [Google Scholar]
- 70.Perez-Sayans M., Suarez-Penaranda J.M., Gayoso-Diz P., Barros-Angueira F., Gandara-Rey J.M., Garcia-Garcia A. p16(INK4a)/CDKN2 expression and its relationship with oral squamous cell carcinoma is our current knowledge enough? Cancer Lett. Jul 28 2011;306(2):134–141. doi: 10.1016/j.canlet.2011.02.039. [DOI] [PubMed] [Google Scholar]
- 71.Finn R., Crown J., Lang I. 2014. Final Results of a Randomized Phase II Study of PD 0332991, a Cyclin-dependent Kinase (CDK)-4/6 Inhibitor, in Combination with Letrozole vs Letrozole Alone for First-line Treatment of ER+/HER2- advanced Breast Cancer (PALOMA-1; TRIO-18). Paper Presented at: AACR; 2014. San Diego, CA. [Google Scholar]
- 72.Musgrove E.A., Caldon C.E., Barraclough J., Stone A., Sutherland R.L. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. Aug 2011;11(8):558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 73.Rogers S.J., Harrington K.J., Rhys-Evans P., OC P., Eccles S.A. Biological significance of c-erbB family oncogenes in head and neck cancer. Cancer Metastasis Rev. Jan 2005;24(1):47–69. doi: 10.1007/s10555-005-5047-1. [DOI] [PubMed] [Google Scholar]
- 74.Kalyankrishna S., Grandis J.R. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. Jun 10 2006;24(17):2666–2672. doi: 10.1200/JCO.2005.04.8306. [DOI] [PubMed] [Google Scholar]
- 75.Hynes N.E., Lane H.A. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. May 2005;5(5):341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 76.Ciardiello F., Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. Mar 13 2008;358(11):1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 77.Schmitz S., Ang K.K., Vermorken J. Targeted therapies for squamous cell carcinoma of the head and neck: current knowledge and future directions. Cancer Treat Rev. Apr 2014;40(3):390–404. doi: 10.1016/j.ctrv.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 78.Bonner J.A., Harari P.M., Giralt J. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. Feb 9 2006;354(6):567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 79.Vermorken J.B., Mesia R., Rivera F. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. Sep 11 2008;359(11):1116–1127. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 80.Riaz N., Sherman E.J., Fury M., Lee N. Should cetuximab replace cisplatin for definitive chemoradiotherapy in locally advanced head and neck cancer? J Clin Oncol. Jan 10 2013;31(2):287–288. doi: 10.1200/JCO.2012.46.9049. [DOI] [PubMed] [Google Scholar]
- 81.Riaz N., Sherman E., Koutcher L. Concurrent chemoradiotherapy with cisplatin versus cetuximab for squamous cell carcinoma of the head and neck. Am J Clin Oncol. Jan 7 2014 doi: 10.1097/COC.0000000000000006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vermorken J.B., Stohlmacher-Williams J., Davidenko I. Cisplatin and fluorouracil with or without panitumumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck (SPECTRUM): an open-label phase 3 randomised trial. Lancet Oncol. Jul 2013;14(8):697–710. doi: 10.1016/S1470-2045(13)70181-5. [DOI] [PubMed] [Google Scholar]
- 83.Machiels J.P., Subramanian S., Ruzsa A. Zalutumumab plus best supportive care versus best supportive care alone in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck after failure of platinum-based chemotherapy: an open-label, randomised phase 3 trial. Lancet Oncol. Apr 2011;12(4):333–343. doi: 10.1016/S1470-2045(11)70034-1. [DOI] [PubMed] [Google Scholar]
- 84.Giralt J., Trigo J., S N Phase 2, randomized trial (CONCERT-2) of Panitumumab (PMAB) plus radiotherapy (PRT) compared with chemoradiotherapy (CRT) in patients (PTS) with unresected, locally advanced squamous cell carcinoma of the head and neck (LASCCHN) Ann Oncol. 2012;23(suppl 9):x334–347. [Google Scholar]
- 85.Argiris A., Ghebremichael M., Gilbert J. Phase III randomized, placebo-controlled trial of docetaxel with or without gefitinib in recurrent or metastatic head and neck cancer: an eastern cooperative oncology group trial. J Clin Oncol. Apr 10 2013;31(11):1405–1414. doi: 10.1200/JCO.2012.45.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martins R.G., Parvathaneni U., Bauman J.E. Cisplatin and radiotherapy with or without erlotinib in locally advanced squamous cell carcinoma of the head and neck: a randomized phase II trial. J Clin Oncol. Apr 10 2013;31(11):1415–1421. doi: 10.1200/JCO.2012.46.3299. [DOI] [PubMed] [Google Scholar]
- 87.Gregoire V., Hamoir M., Chen C. Gefitinib plus cisplatin and radiotherapy in previously untreated head and neck squamous cell carcinoma: a phase II, randomized, double-blind, placebo-controlled study. Radiother Oncol. Jul 2011;100(1):62–69. doi: 10.1016/j.radonc.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 88.Molinolo A.A., Hewitt S.M., Amornphimoltham P. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res. Sep 1 2007;13(17):4964–4973. doi: 10.1158/1078-0432.CCR-07-1041. [DOI] [PubMed] [Google Scholar]
- 89.Grandis J.R., Tweardy D.J. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. Aug 1 1993;53(15):3579–3584. [PubMed] [Google Scholar]
- 90.Temam S., Kawaguchi H., El-Naggar A.K. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J Clin Oncol. Jun 1 2007;25(16):2164–2170. doi: 10.1200/JCO.2006.06.6605. [DOI] [PubMed] [Google Scholar]
- 91.Ang K.K., Berkey B.A., Tu X. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. Dec 15 2002;62(24):7350–7356. [PubMed] [Google Scholar]
- 92.Hansen A.R., Siu L.L. Epidermal growth factor receptor targeting in head and neck cancer: have we been just skimming the surface? J Clin Oncol. Apr 10 2013;31(11):1381–1383. doi: 10.1200/JCO.2012.47.9220. [DOI] [PubMed] [Google Scholar]
- 93.Sharma S.V., Bell D.W., Settleman J., Haber D.A. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. Mar 2007;7(3):169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 94.Licitra L., Mesia R., Rivera F. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first-line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: EXTREME study. Ann Oncol. May 2011;22(5):1078–1087. doi: 10.1093/annonc/mdq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bonner J.A., Harari P.M., Giralt J. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. Jan 2010;11(1):21–28. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 96.Yang X., Zhang X., Mortenson E.D., Radkevich-Brown O., Wang Y., Fu Y.X. Cetuximab-mediated tumor regression depends on innate and adaptive immune responses. Mol Ther. Jan 2013;21(1):91–100. doi: 10.1038/mt.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Srivastava R.M., Lee S.C., Andrade Filho P.A. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. Apr 1 2013;19(7):1858–1872. doi: 10.1158/1078-0432.CCR-12-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yonesaka K., Zejnullahu K., Okamoto I. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. Sep 7 2011;3(99) doi: 10.1126/scitranslmed.3002442. 99ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kandoth C., McLellan M.D., Vandin F. Mutational landscape and significance across 12 major cancer types. Nature. Oct 17 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Engelman J.A. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. Aug 2009;9(8):550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 101.Rogers S.J., Box C., Harrington K.J., Nutting C., Rhys-Evans P., Eccles S.A. The phosphoinositide 3-kinase signalling pathway as a therapeutic target in squamous cell carcinoma of the head and neck. Expert Opin Ther Targets. Aug 2005;9(4):769–790. doi: 10.1517/14728222.9.4.769. [DOI] [PubMed] [Google Scholar]
- 102.Zhao L., Vogt P.K. Class I PI3K in oncogenic cellular transformation. Oncogene. Sep 18 2008;27(41):5486–5496. doi: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fruman D.A., Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. Feb 2014;13(2):140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dotto G.P. Notch tumor suppressor function. Oncogene. Sep 1 2008;27(38):5115–5123. doi: 10.1038/onc.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ranganathan P., Weaver K.L., Capobianco A.J. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. May 2011;11(5):338–351. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 106.Nicolas M., Wolfer A., Raj K. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. Mar 2003;33(3):416–421. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 107.Wang N.J., Sanborn Z., Arnett K.L. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci USA. Oct 25 2011;108(43):17761–17766. doi: 10.1073/pnas.1114669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Weng A.P., Ferrando A.A., Lee W. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. Oct 8 2004;306(5694):269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 109.Welcker M., Clurman B.E. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. Feb 2008;8(2):83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 110.Stacy D.R., Ely K., Massion P.P. Increased expression of nuclear factor E2 p45-related factor 2 (NRF2) in head and neck squamous cell carcinomas. Head Neck. Sep 2006;28(9):813–818. doi: 10.1002/hed.20430. [DOI] [PubMed] [Google Scholar]
- 111.Jaramillo M.C., Zhang D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. Oct 15 2013;27(20):2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shibata T., Ohta T., Tong K.I. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci USA. Sep 9 2008;105(36):13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Martinez V.D., Vucic E.A., Thu K.L., Pikor L.A., Lam S., Lam W.L. Disruption of KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex components by multiple genetic mechanisms is associated with poor prognosis in head and neck cancer. Head Neck. Mar 5 2014 doi: 10.1002/hed.23663. [DOI] [PubMed] [Google Scholar]
- 114.Solis L.M., Behrens C., Dong W. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. Jul 15 2010;16(14):3743–3753. doi: 10.1158/1078-0432.CCR-09-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jiang T., Chen N., Zhao F. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. Jul 1 2010;70(13):5486–5496. doi: 10.1158/0008-5472.CAN-10-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ohta T., Iijima K., Miyamoto M. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. Mar 1 2008;68(5):1303–1309. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 117.Abazeed M.E., Adams D.J., Hurov K.E. Integrative radiogenomic profiling of squamous cell lung cancer. Cancer Res. Oct 15 2013;73(20):6289–6298. doi: 10.1158/0008-5472.CAN-13-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dawson M.A., Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. Jul 6 2012;150(1):12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 119.Krivtsov A.V., Armstrong S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. Nov 2007;7(11):823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 120.Chung Y.R., Schatoff E., Abdel-Wahab O. Epigenetic alterations in hematopoietic malignancies. Int J Hematol. Oct 2012;96(4):413–427. doi: 10.1007/s12185-012-1181-z. [DOI] [PubMed] [Google Scholar]
- 121.Wang F., Marshall C.B., Ikura M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol Life Sci. Nov 2013;70(21):3989–4008. doi: 10.1007/s00018-012-1254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Morris L.G., Ramaswami D., Chan T.A. The FAT epidemic: a gene family frequently mutated across multiple human cancer types. Cell Cycle. Apr 1 2013;12(7):1011–1012. doi: 10.4161/cc.24305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Morris L.G., Kaufman A.M., Gong Y. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat Genet. Mar 2013;45(3):253–261. doi: 10.1038/ng.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. Jun 2007;35(4):495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zack T.I., Schumacher S.E., Carter S.L. Pan-cancer patterns of somatic copy number alteration. Nat Genet. Sep 26 2013;45(10):1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Negrini S., Gorgoulis V.G., Halazonetis T.D. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol. Mar 2010;11(3):220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 127.Patmore H.S., Cawkwell L., Stafford N.D., Greenman J. Unraveling the chromosomal aberrations of head and neck squamous cell carcinoma: a review. Ann Surg Oncol. Oct 2005;12(10):831–842. doi: 10.1245/ASO.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 128.Bockmuhl U., Wolf G., Schmidt S. Genomic alterations associated with malignancy in head and neck cancer. Head Neck. Mar 1998;20(2):145–151. doi: 10.1002/(sici)1097-0347(199803)20:2<145::aid-hed8>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 129.Califano J., van der Riet P., Westra W. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. Jun 1 1996;56(11):2488–2492. [PubMed] [Google Scholar]
- 130.Lee D.J., Schonleben F., Banuchi V.E. Multiple tumor-suppressor genes on chromosome 3p contribute to head and neck squamous cell carcinoma tumorigenesis. Cancer Biol Ther. Oct 1 2010;10(7):689–693. doi: 10.4161/cbt.10.7.12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Song Y., Li L., Ou Y. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. Mar 16 2014;509:91–95. doi: 10.1038/nature13176. [DOI] [PubMed] [Google Scholar]
- 132.Siebers T.J., Bergshoeff V.E., Otte-Holler I. Chromosome instability predicts the progression of premalignant oral lesions. Oral Oncol. Dec 2013;49(12):1121–1128. doi: 10.1016/j.oraloncology.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 133.Giaretti W., Monteghirfo S., Pentenero M., Gandolfo S., Malacarne D., Castagnola P. Chromosomal instability, DNA index, dysplasia, and subsite in oral premalignancy as intermediate endpoints of risk of cancer. Cancer Epidemiol Biomarkers Prev. Jun 2013;22(6):1133–1141. doi: 10.1158/1055-9965.EPI-13-0147. [DOI] [PubMed] [Google Scholar]
- 134.Giaretti W., Pentenero M., Gandolfo S., Castagnola P. Chromosomal instability, aneuploidy and routine high-resolution DNA content analysis in oral cancer risk evaluation. Future Oncol. Oct 2012;8(10):1257–1271. doi: 10.2217/fon.12.116. [DOI] [PubMed] [Google Scholar]
- 135.Carter S.L., Eklund A.C., Kohane I.S., Harris L.N., Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. Sep 2006;38(9):1043–1048. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- 136.Smeets S.J., Brakenhoff R.H., Ylstra B. Genetic classification of oral and oropharyngeal carcinomas identifies subgroups with a different prognosis. Cell Oncol. 2009;31(4):291–300. doi: 10.3233/CLO-2009-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bhattacharya A., Roy R., Snijders A.M. Two distinct routes to oral cancer differing in genome instability and risk for cervical node metastasis. Clin Cancer Res. Nov 15 2011;17(22):7024–7034. doi: 10.1158/1078-0432.CCR-11-1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mooren J.J., Kremer B., Claessen S.M. Chromosome stability in tonsillar squamous cell carcinoma is associated with HPV16 integration and indicates a favorable prognosis. Int J Cancer. Apr 15 2013;132(8):1781–1789. doi: 10.1002/ijc.27846. [DOI] [PubMed] [Google Scholar]
- 139.van den Broek G.B., Wreesmann V.B., van den Brekel M.W., Rasch C.R., Balm A.J., Rao P.H. Genetic abnormalities associated with chemoradiation resistance of head and neck squamous cell carcinoma. Clin Cancer Res. Aug 1 2007;13(15 Pt 1):4386–4391. doi: 10.1158/1078-0432.CCR-06-2817. [DOI] [PubMed] [Google Scholar]
- 140.Munger K., Baldwin A., Edwards K.M. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. Nov 2004;78(21):11451–11460. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rautava J., Syrjanen S. Biology of human papillomavirus infections in head and neck carcinogenesis. Head Neck Pathol. Jul 2012;6(suppl 1):S3–S15. doi: 10.1007/s12105-012-0367-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.D'Souza G., Kreimer A.R., Viscidi R. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. May 10 2007;356(19):1944–1956. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 143.Tang K.W., Alaei-Mahabadi B., Samuelsson T., Lindh M., Larsson E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun. 2013;4:2513. doi: 10.1038/ncomms3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Klussmann J.P., Weissenborn S.J., Wieland U. Prevalence, distribution, and viral load of human papillomavirus 16 DNA in tonsillar carcinomas. Cancer. Dec 1 2001;92(11):2875–2884. doi: 10.1002/1097-0142(20011201)92:11<2875::aid-cncr10130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 145.Thorland E.C., Myers S.L., Gostout B.S., Smith D.I. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene. Feb 27 2003;22(8):1225–1237. doi: 10.1038/sj.onc.1206170. [DOI] [PubMed] [Google Scholar]
- 146.Scheffner M., Werness B.A., Huibregtse J.M., Levine A.J., Howley P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. Dec 21 1990;63(6):1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 147.Mirghani H., Amen F., Moreau F., Guigay J., Hartl D.M., Lacau St Guily J. Oropharyngeal cancers: relationship between epidermal growth factor receptor alterations and human papillomavirus status. Eur J Cancer. Apr 2014;50(6):1100–1111. doi: 10.1016/j.ejca.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 148.Reimers N., Kasper H.U., Weissenborn S.J. Combined analysis of HPV-DNA, p16 and EGFR expression to predict prognosis in oropharyngeal cancer. Int J Cancer. Apr 15 2007;120(8):1731–1738. doi: 10.1002/ijc.22355. [DOI] [PubMed] [Google Scholar]
- 149.Hong A., Dobbins T., Lee C.S. Relationships between epidermal growth factor receptor expression and human papillomavirus status as markers of prognosis in oropharyngeal cancer. Eur J Cancer. Jul 2010;46(11):2088–2096. doi: 10.1016/j.ejca.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 150.Rosenthal D., Harari P., Giralt J. 2014. Impact of P16 Status on the Results of the Phase III Cetuximab (Cet)/radiotherapy (RT) p. 2014. Paper presented at: ASCO. Chicago, Il. [Google Scholar]