

Abstract
Objectives:
The aim was to study the presentation, disease characteristics, operative outcome, and prognosis in patients with familial Pheochromocytoma associated with von Hippel-Lindau (VHL) disease.
Materials and Methods:
There were six patients belonging to two generations of a single family who developed features of VHL over a period of 13 years and were treated at our institute. Patients’ characteristics, that is, age, gender, presenting complaints and clinical signs, laboratory and biochemical evaluation, and the presence of associated conditions was gathered from medical records. The preoperative and postoperative radiological imaging and histopathological results were also collected.
Results:
Out of six cases, five were male, and one was female. The mean age at first presentation was 25 years (16-40). All patients presented with uncontrolled hypertension and were found to have Pheochromocytoma on workup. Three patients had unilateral adrenal tumor, and three had bilateral disease. None of the patients had extra-adrenal Pheochromocytoma. All patients were managed with adrenalectomy and had benign pathology. Two patients subsequently had craniotomy for excision of cerebellar hemangioma, and one patient had bilateral partial nephrectomy at the time of adrenalectomy. There was no peri- post-operative mortality and all patients are being followed by the surgeon(s) and endocrinologist.
Conclusion:
Pheochromocytoma can be a part of familial conditions including VHL. Other associated features should be suspected, investigated, and treated in these patients that can influence patients’ clinical course and prognosis. Family members should also be screened to achieve early diagnosis.
Keywords: Adrenal, familial, Pheochromocytoma, von Hippel-Lindau disease
INTRODUCTION
Pheochromocytoma is one of the rare tumors and is present in about 0.2% of patients presenting with hypertension.[1] Majority of them are sporadic, however, 15-20% are inherited as a part of the familial disorder. In these patients, the catecholamine secreting tumors are more likely bilateral adrenal Pheochromocytoma or paragangliomas and present at an early age than their sporadic counterparts.[2] Familial Pheochromocytoma are associated with von Hippel-Lindau (VHL) disease, multiple endocrine neoplasia II (MEN II) and neuro-ectodermal syndromes such as neurofibromatosis type 1 which is part of von Recklinghausen disease.[3] All of these conditions are inherited as autosomal dominant trait. Frequencies of Pheochromocytoma in these familial disorders are 10-20% in VHL, 50% in MEN II, and 0.1-5.7% with neurofibromatosis type 1.[4,5] We report six cases of familial pheochromocytoma with associated clinical manifestations of VHL.
MATERIALS AND METHODS
There were six patients belonging to two generations of a single family from northern areas of Pakistan, who developed features of VHL over a period of 13 years and were treated at our institute. All six patients presented with high blood pressure and were optimized prior to surgery and were managed by multidisciplinary team in perioperative period and follow-up. Patients’ characteristics, that is, age, gender, presenting complaints and clinical signs, laboratory and biochemical evaluation, and presence of associated conditions were gathered from medical records. The preoperative and postoperative radiological imaging and histo-pathological results were also collected. All patients were followed on regular interval by surgeon(s) and endocrinologists with the aim to identify and treat subsequent manifestations of disease as soon as possible.
RESULTS
A total of six patients were identified in the same family. Family history also revealed death from the brain tumor in three members of the affected family [Figure 1].
Figure 1.
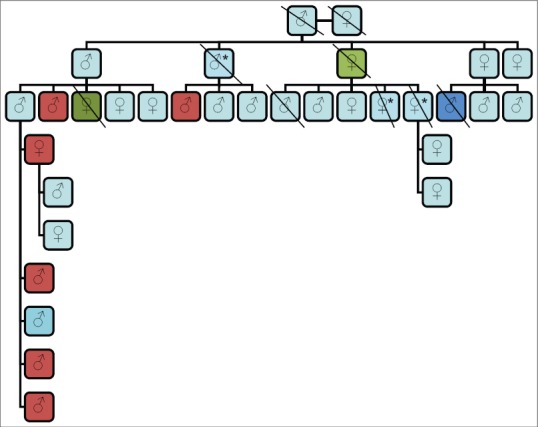
Pedigree of family with pheochromocytoma associated with von Hippel-Lindau disease: Red boxes - Patients with pheochromocytoma, *Died of brain tumor (not managed in our institute), Green boxes - Cause of death not known, Dark blue box - Died of renal failure (had nephrectomy in past)
Mean age at presentation was 25 years. There were three brothers and one sister. All patients presented with uncontrolled blood pressure, abdominal pain, and one with ataxia.
Initial radiological investigation was abdominal ultrasonography followed by contrast enhanced computerized tomography of the abdomen in five patients [Table 1]. Magnetic resonance imaging (MRI) was done in two patients, for evaluation of renal masses with pheochromocytoma in one patient and to look for suspected pancreatic mass in another one. MRI head was also done subsequently in patients.
Table 1.
Patients’ summary
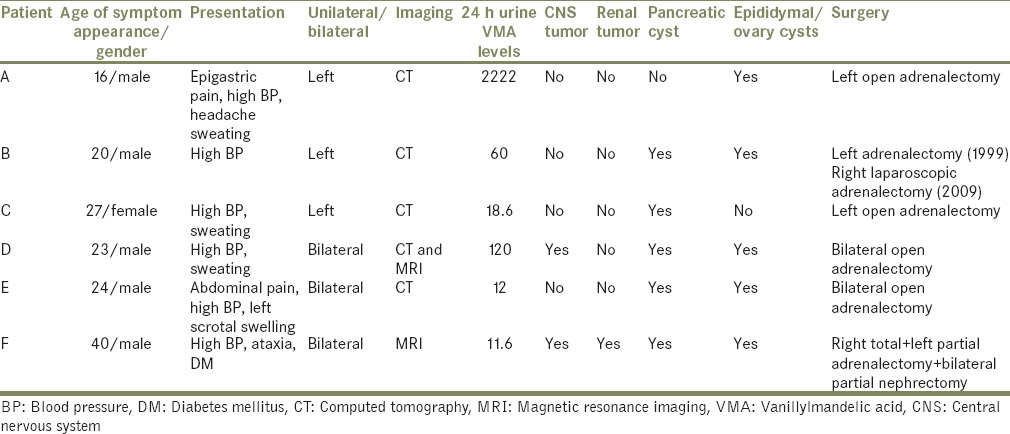
We performed 24 h urinary vanillylmandelic acid levels in all patients, and median value was 39.3 mg/24 h (Range: 11.6-2222 mg/24 h). All male patients had epididymal cyst and five patients had pancreatic cysts as well. None of our patients had retinal involvement, while renal involvement was seen in only one patient who was diagnosed with bilateral renal tumors on imaging.
Three patients (ABC) had unilateral adrenal tumor and underwent open adrenalectomy via retroperitoneal approach while rest of three (DEF) had bilateral tumors and were treated by trans-abdominal bilateral adrenalectomy. One patient (E) had partial adrenalectomy on the left side [Table 1].
Mean size of the tumor was 5.6 cm (range: 2-13.8 cm). All tumors were benign on final histopathology.
Two patients had central nervous system (CNS) manifestations. Patient D was found to have cerebellar and cervical lesions on subsequent follow-up and was operated at our institute. Histopathology reported these lesions to be hemangioblastoma. Patient F presented with high blood pressures, uncontrolled blood sugars, and ataxic gait. MRI revealed bilateral adrenal and bilateral renal tumors, cerebellar hemangioblastoma, and syringomyelia of spine [Figure 2]. He underwent bilateral partial nephrectomy, right adrenalectomy, and left partial adrenalectomy through midline laparotomy incision. After complete recovery, he underwent posterior fossa craniotomy for hemangioblastoma. Histopathology showed bilateral clear cell carcinoma Fuhrman's nuclear grade II, bilateral pheochromocytoma, and hemangioblastoma. Currently, he is off any mineralocorticoid replacement.
Figure 2.
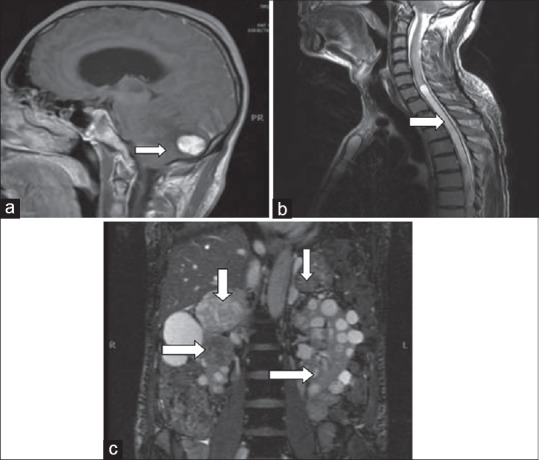
Images from patient E: (a) T1-weighted images of magnetic resonance imaging (MRI) brain showing hyperintence lesion (arrow) in cerebellum (hemangioblastoma). (b) T2-weighted images of MRI spine showing syringomyelia at thoracic spine (arrow). (c) T2-weighted images of MRI upper abdomen showing cystic bilateral renal cell carcinoma (horizontal arrows) with bilateral adrenal pheochromocytoma (vertical arrows)
During the follow-up, patient B developed another tumor after 5 years of adrenalectomy at left para-aortic lymph node that was encasing left renal hilum. Left nephrectomy with mass excision was done and histopathology confirmed pheochromocytoma. He developed another tumor in right adrenal gland and underwent right laparoscopic adrenalectomy in 2009. He is on mineralocorticoid replacement.
All 6 patients are under regular follow-up with the surgeon (s) and endocrinologists.
DISCUSSION
Von Hippel-Lindau disease is autosomal dominant disorder with high penetrance and variable expression. It has a population prevalence ranging from one in 39,000-53,000.[6]
Von Hippel-Lindau disease results by mutation in VHL tumor suppressor gene located on chromosome 3p25-26.[7] It is a multi-system disorder characterized by development of multiple benign and malignant tumors in the brain, spinal cord, kidneys, pancreas, adrenals, and epididymis. The syndrome demonstrates marked phenotypic variability and is suggested to account for approximately a third of patients with CNS hemangioblastoma and 50% of patients with apparently isolated familial pheochromocytoma.[8] The intra-familial variation may be marked.
Familial pheochromocytoma appears clinically in younger patients than their sporadic counterparts. Mean age of patients in our study is 25 years. In our study, based on manifestation and development of symptoms and involvement of both sexes, the pattern of inheritance of this disease is clearly autosomal dominant.
Bissada et al. reported 29 patients with familial pheochromocytoma with a mean age of 30.8 years. Mean tumor size in their series was 4.3 cm (range: 1.0-9.5 cm). Of 29 patients, 26 had an adrenal tumor, two had extra-adrenal, and one had combined adrenal and extra-adrenal tumors. Eight patients had VHL syndrome.[9] In our series, three patients had bilateral adrenal tumor, two patients had CNS involvement, and mean tumor size was 5.6 cm (range: 2-13.8 cm). All of our patients are alive and are on surveillance. Violaris et al. reported series of seven patients with VHL from three generations with CNS manifestations. Only two patients had renal, pancreatic, and epididymal cysts and three had retinal involvement. None of their patients had pheochromocytoma.[10] In our study, five out of six patients had pancreatic and epididymal cysts.
Von Hippel-Lindau patients are at risk of developing bilateral adrenal tumors, and these subset of patients will require lifelong mineralocorticoid supplementation if bilateral adrenalectomy is carried out. Diner et al. from national cancer institute in his series of 33 patients with familial pheochromocytoma managed with partial adrenalectomy reported that only five patients required mineralocorticoid supplement.[11] In our series, one patient with bilateral adrenal tumor was managed with left open partial adrenalectomy, he did not require mineralocorticoid supplement.
Hwang et al. assessed the surgical feasibility of single surgical approach for multi-organ visceral tumors in 29 patients with VHL disease and found it to be a viable option. 41% of their cases had pancreatic operations, and 55% had surgery involving kidney/adrenal.[12]
Two of our patients had bilateral open adrenalectomy and one had bilateral partial nephrectomy + adrenal surgery.
Genetic testing for VHL gene screening and radiological screening tests may be considered to establish early diagnosis in other members of familial pheochromocytoma.[13,14] The screening recommendations can be accessed through the VHL Family Alliance Website (www.vhl.org). However, prior to investigation, the details of the disease, testing methods, further consequences after diagnoses for both the patient and family as well as potential treatment options should be thoroughly discussed. The likelihood of false positive and false negative results and need for additional testing should also be discussed.[8] We have not done VHL gene screening so far, but the family is currently under surveillance, and they are informed and counseled about the potential of developing the manifestation of the syndrome and follow-up for the rest of their lives.
CONCLUSION
Pheochromocytoma can be a part of the familial syndrome including VHL. Other associated conditions should be suspected, investigated, and treated in these patients which can influence patients’ clinical course and prognosis. Family members should also be screened to achieve early diagnosis.
Footnotes
Source of Support: Nil
Conflict of Interest: None.
REFERENCES
- 1.Stein PP, Black HR. A simplified diagnostic approach to pheochromocytoma. A review of the literature and report of one institution's experience. Medicine (Baltimore) 1991;70:46–66. doi: 10.1097/00005792-199101000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, et al. Germ-line mutations in non syndromic pheochromocytoma. N Engl J Med. 2002;346:1459–66. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 3.Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc. 1981;56:354–60. [PubMed] [Google Scholar]
- 4.Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen's disease and pheochromocytomas. J Urol. 1999;162:1582–6. [PubMed] [Google Scholar]
- 5.Dluhy RG. Pheochromocytoma - death of an axiom. N Engl J Med. 2002;346:1486–8. doi: 10.1056/NEJM200205093461911. [DOI] [PubMed] [Google Scholar]
- 6.O’ Brien FJ, Danapal M, Jairam S, Lalani AK, Cunningham J, Morrin M, et al. Manifestations of Von Hippel- Lindau syndrome: A retrospective national review. QJM. 2014;107:291–6. doi: 10.1093/qjmed/hct249. [DOI] [PubMed] [Google Scholar]
- 7.Maher ER, Bentley E, Yates JR, Barton D, Jennings A, Fellows IW, et al. Mapping of von Hippel-Lindau disease to chromosome 3p confirmed by genetic linkage analysis. J Neurol Sci. 1990;100:27–30. doi: 10.1016/0022-510x(90)90008-b. [DOI] [PubMed] [Google Scholar]
- 8.Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: A clinical and scientific review. Eur J Hum Genet. 2011;19:617–23. doi: 10.1038/ejhg.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bissada MA, Safwat AS, Seyam RM, Al Sobhi S, Hanash KA, Bissada NK. Familial pheochromocytoma. Urol Oncol. 2008;26:361–3. doi: 10.1016/j.urolonc.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 10.Violaris K, Siozos T, Skoulios N, Sakellariou P. A case report of a family with 7 patients of the Von Hippel-Lindau disease. Surg Neurol. :2007–4. doi: 10.1016/j.surneu.2006.11.040. [DOI] [PubMed] [Google Scholar]
- 11.Diner EK, Franks ME, Behari A, Linehan WM, Walther MM. Partial adrenalectomy: The National Cancer Institute experience. Urology. 2005;66:19–23. doi: 10.1016/j.urology.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Hwang JJ, Uchio EM, Pavlovich CP, Pautler SE, Libutti SK, Linehan WM, et al. Surgical management of multi-organ visceral tumors in patients with von Hippel-Lindau disease: A single stage approach. J Urol. 2003;169:895–8. doi: 10.1097/01.ju.0000049518.15260.1f. [DOI] [PubMed] [Google Scholar]
- 13.Tsirlin A, Oo Y, Sharma R, Kansara A, Gliwa A, Banerji MA. Pheochromocytoma: A review. Maturitas. 2014;77:229–38. doi: 10.1016/j.maturitas.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Tong AL, Zeng ZP, Zhou YR, Yuan T, Cao CX, Zhang J, et al. Bilateral pheochromocytoma as first presentation of von Hippel-Lindau disease in a Chinese family. Chin Med Sci J. 2009;24:197–201. doi: 10.1016/s1001-9294(10)60001-6. [DOI] [PubMed] [Google Scholar]