

Abstract
Neurofibromas are tumors of neural origin. They can be solitary or may be associated with neurofibromatosis type-1 (NF-1). These are mostly seen in the head and neck region, upper extremities or along the nerves. Visceral neurofibromas are extremely rare. In this paper, we present an unusual case of solitary neurofibroma of the adrenal gland not associated with NF-1.
Keywords: Adrenal, neurofibroma, solitary
INTRODUCTION
Neurofibromas are benign nerve sheath tumors originating from the perineural and Schwann cells of the normal peripheral nerve. These may present either as solitary lesions or may coexist with neurofibromatosis type-1 (NF-1), an autosomal dominant disease. Sites of predilection are the head and neck region, upper extremities or along the nerves. Retroperitoneal, mesenteric and paraspinal location of neurofibromas have also been mentioned, but the splanchnic location is very unusual. Solitary neurofibroma arising in the adrenal gland is extremely rare and only one isolated case report has been published till date in the English literature.[1] Herein, we describe an interesting case of adrenal neurofibroma in a 51-year-old woman.
CASE REPORT
A 51-year-old woman presented in the outpatient department of a tertiary care hospital with a 2-week history of left upper abdominal pain. The pain was constant, dull aching in nature. It was not accompanied by fever, vomiting or bowel disturbance. She had a similar history of episodic abdominal pain in the past, which has subsided after taking routine analgesics. On physical examination, her abdomen was soft and tender without any palpable mass. Rest of the systemic examination was within normal limits. Computed tomography (CT) scan of the abdomen revealed a 10.0 cm heterogenously enhancing left adrenal mass with stippled calcification within it [Figure 1]. Plasma and urine catecholamine levels and serum cortisol levels were within normal limits. Routine hematological investigations were normal. Based on clinical examination and radiological assessment, a diagnosis of left adrenal mass was suggested. Subsequently, laparoscopic left adrenalectomy was performed. Intraoperatively, an oblong 10 cm × 8 cm tumor was seen to extend from the left suprarenal region up to the esophageal hiatus of the diaphragm. It was lying posterior to the stomach and medial to the spleen. The tumor could be separated from the tail of the pancreas, splenic vein and superomedial aspect of the spleen by gentle dissection. It was well encapsulated and compressed adrenal tissue could be seen toward the inferolaterolateral aspect of the tumor. At no stage of the operative procedure was the capsule violated.
Figure 1.
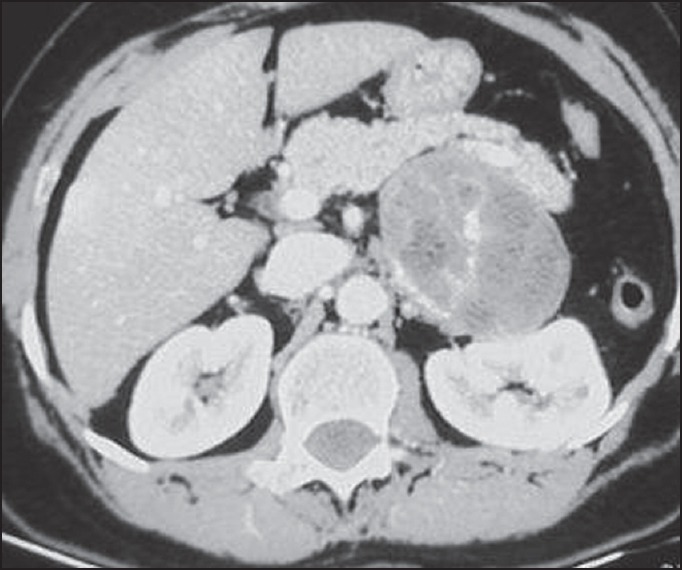
Computed tomography scan of the abdomen showing a heterogenously enhancing left adrenal mass with stippled calcification within it
Gross examination of the surgical specimen revealed replacement of adrenal gland by a well-circumscribed tumor measuring 10.0 cm in greatest dimension. Cut-surface of the tumor was grey-white and lobulated with focal areas of cystic degeneration and hemorrhage. Compressed adrenal tissue was identified at the periphery of the tumor [Figure 2]. Histologically, the tumor comprised of interlacing fascicles of elongated or plump spindle cells with cytologically bland wavy nuclei in a collagenous matrix [Figure 3a]. Areas of myxoid degeneration and calcification were noted. No necrosis, mitotic activity or infiltrative growth pattern was observed. On immunohistochemistry, the tumor cells were diffusely and strongly positive for S-100 [Figure 3b].
Figure 2.
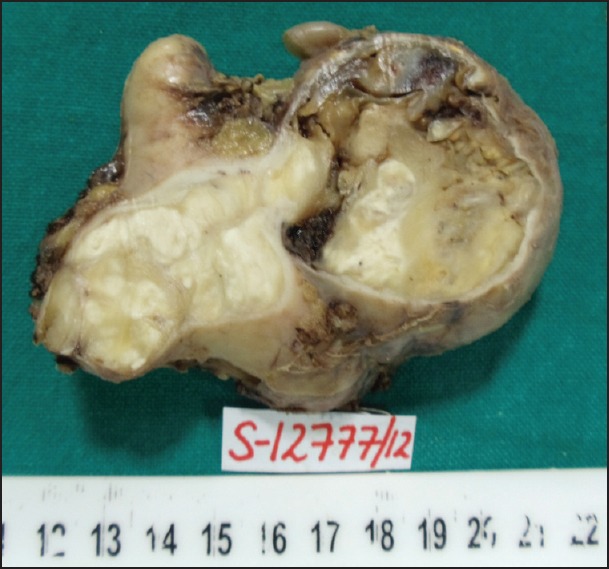
Gross specimen showing replacement of the adrenal gland by a well-circumscribed tumor. Cut-surface of the tumor was grey-white and lobulated with focal areas of cystic degeneration and hemorrhage
Figure 3a.
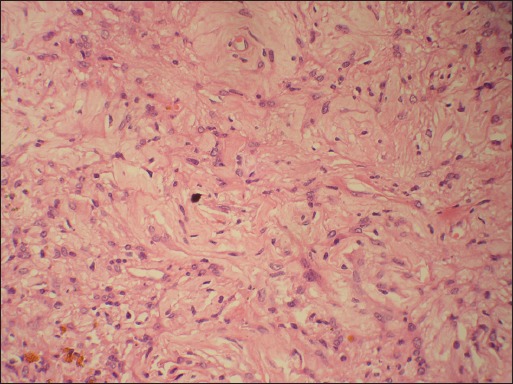
Microphotograph showing tumor comprised of interlacing fascicles of elongated or plump spindle cells with cytologically bland wavy nuclei in a collagenous matrix. (Hematoxylin and eosin, ×200 original magnification)
Figure 3b.
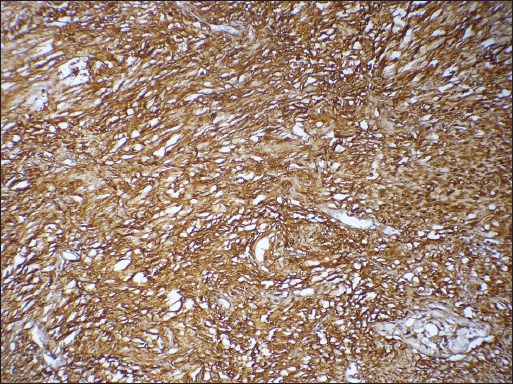
Microphotograph showing the tumor cells to be diffusely and strongly positive for S-100. (Immunohistochemistry, ×200 original magnification)
The postoperative course of the patient was uneventful and no recurrence was noted after 6 months of follow-up. Because neurofibromas are frequently associated with NF-1, the present case was extensively investigated, both clinically and radiologically, to rule out any such pathology. No clinical manifestations of neurofibromatosis were detected and she had no family history of NF-1 hence confirming that the mass was solitary.
DISCUSSION
The most common sites of neurogenic tumors of the abdomen are retroperitoneum, paraspinal regions and adrenal glands.[2] Few cases of solitary neurofibroma, not associated with neurofibromatosis, have also been reported in the wall of the abdomen,[3] kidney,[4] colon,[5] anal canal,[6] common bile duct[7] and pancreas.[8] We report a rare case of solitary neurofibroma of the adrenal gland.
Adrenal tumors are derived mostly from the constituent cortical, chromaffin and neuronal cells. The four most common adrenal masses are adrenal adenoma, adrenocortical carcinoma, phaeochromocytoma and neuroblastoma. Nerve sheath tumors, although extremely rare, must be considered in the differential diagnosis of the adrenal masses.
Clinically, depending upon the site, neurofibromas may cause various symptoms. Chang et al.[9] reported abdominal neurofibroma presenting as an inguinal hernia, while in the ileum and colon[5] these caused intussusception and lower gastrointestinal bleeding. In our case, the tumor was discovered incidentally while investigating the cause of abdominal pain. Falahatkar et al. reported a similar case of adrenal neurofibroma in a 24-year-old woman who presented with discomfort in her right flank.[1] Because of the widespread use of computed tomography (CT), magnetic resonance imaging (MRI) and ultrasonography for the evaluation of diseases with abdominal discomfort, the incidence of detection of adrenal masses is increasing. Adrenal masses are detected accidentally in 1-5% of all abdominal CT scans performed. On CT scan, neurofibromas have a homogenous, smooth appearance with distinct outlines.[2] However, the preoperative imaging does not help to diagnose these tumors with certainty, and only histology is definite.
Neurofibromas are well-circumscribed, nonencapsulated tumors that may grow in a solitary, diffuse or plexiform pattern. The main constituents are Schwann cells, perineurial cells, axons, fibroblasts, mast cells and collagen. Solitary neurofibroma of the adrenal gland reveals the same morphology as seen at other anatomic sites in the form of spindle-shaped cells with wavy nuclei dispersed in an extracellular matrix containing variable amounts of mucin and collagen. By immunohistochemistry, these are strongly and diffusely positive for S-100 protein. Many variants of neurofibroma have been reported to date, such as myxoid, hyalinized, epithelioid, pacinian, pigmented, granular, lipomatous, dendritic cell neurofibroma with pseudorosettes and neurofibroma with rhabdomyomatous differentiation.[10] The risk of malignant degeneration, although very rare, is known to occur.
Sometimes, neurofibromas also exhibit pauci-cellular and cell-rich areas (cellular neurofibromas) similar to that seen in schwannoma and therefore it becomes difficult to distinguish the two. Nevertheless, neurofibromas are nonencapsulated with uniformly distributed nerve fibers while schwannomas are well encapsulated with subcapsular/peripherally placed nerve fibers.
Other adrenal gland neoplasm exhibiting spindle cell morphology include ganglioneuroma, leiomyoma, solitary fibrous tumor and, rarely, pheochromocytomas. The presence of ganglion cells in ganglioneuroma differentiates it from neurofibroma. Immunohistochemically, leiomyomas are smooth muscle actin and desmin positive, unlike neurofibromas. Solitary fibrous tumors can be distinguished from the latter as they are strong positivity for CD34 and lack reactivity for S-100 protein. In contrast to functionally inactive neurofibromas, pheochromocytomas are hormonally active tumors with uniform positivity for synaptophysin and chromogranin and lack of S-100 protein reactivity.
Laparoscopic adrenalectomy is considered as the primary treatment modality for many adrenal tumors. The advantages of this procedure over open adrenalectomy are less postoperative discomfort, a shorter hospital stay and a lower rate of complications.[10] Because neurofibromas do not have the potential to bleed and are well circumscribed, these can be easily dissected out from the adjacent soft tissue planes.
To conclude, we report a case of neurofibroma of the adrenal gland not associated with NF-1. Although extremely rare, neurofibroma should be considered in the differential diagnosis of primary adrenal tumors.
Footnotes
Source of Support: Nil
Conflict of Interest: None.
REFERENCES
- 1.Falahatkar S, Mohammadzadeh A, Nikpour S, Khoshrang H, Askari K. First reported case of adrenal neurofibroma from Iran. Urol J. 2007;4:242–4. [PubMed] [Google Scholar]
- 2.Rha SE, Byun JY, Jung SE, Chun HJ, Lee HG, Lee JM. Neurogenic tumors in the abdomen: Tumor types and imaging characteristics. RadioGraphics. 2003;23:29–43. doi: 10.1148/rg.231025050. [DOI] [PubMed] [Google Scholar]
- 3.Barajas-Gamboa JS, Flórez-Salamanca L. Solitary neurofibroma in the abdominal wall of a patient without neurofibromatosis: Case report. Biomedica. 2009;29:501–5. [PubMed] [Google Scholar]
- 4.Kostakopoulos A, Chorti M, Protogerou V, Kokkinou S. Solitary neurofibroma of kidney: Clinical, histological and chromosomal appeareance. Int Urol Nephrol. 2003;35:11–3. doi: 10.1023/a:1025943827621. [DOI] [PubMed] [Google Scholar]
- 5.Abramson LP, Orkin BA, Schwartz AM. Isolated colonic neurofibroma manifested by massive lower gastrointestinal bleeding and intussusception. South Med J. 1997;90:952–4. doi: 10.1097/00007611-199709000-00020. [DOI] [PubMed] [Google Scholar]
- 6.Frick E, Jr, Lapos L, Vargas HD. Solitary neurofibroma of the anal canal: Report of two cases. Dis Colon Rectum. 2000;43:109–12. doi: 10.1007/BF02237253. [DOI] [PubMed] [Google Scholar]
- 7.Li FY, Cheng JQ, He S, Li N, Zhang MM, Zhang XL, et al. Primary neurofibroma of the common bile duct as an unusual cause of obstructive jaundice: A case report. Dig Dis Sci. 2005;50:1166–8. doi: 10.1007/s10620-005-2726-2. [DOI] [PubMed] [Google Scholar]
- 8.Tsai PJ, Liu KY, Wang SE, Shyr YM, Su CH, Chen TH. Solitary neurofibroma of the pancreas body not associated with type 1 neurofibromatosis. J Chin Med Assoc. 2012;75:132–5. doi: 10.1016/j.jcma.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 9.Chang CG, Provost DA, LeVoyer T, Ellison RW. Abdominal wall neurofibroma presenting as an inguinal hernia. Mil Med. 2004;169:192–3. doi: 10.7205/milmed.169.3.192. [DOI] [PubMed] [Google Scholar]
- 10.Kanthan R, Senger JL, Kanthan S. Three uncommon adrenal incidentalomas: A 13-year surgical pathology review. World J Surg Oncol. 2012;10:64. doi: 10.1186/1477-7819-10-64. [DOI] [PMC free article] [PubMed] [Google Scholar]