

Abstract
Parkinson's disease (PD) is the world's second most common neurodegenerative disease and most common movement disorder. Characterised by a loss of dopaminergic neurons and the development of intraneuronal inclusions known as Lewy bodies, it has classically been thought of as a cell-autonomous disease. However, in 2008, two groups reported the startling observation of Lewy bodies within embryonic neuronal grafts transplanted into PD patients little more than a decade previously, suggesting that PD pathology can be propagated to neighbouring cells and calling basic assumptions of our understanding of the disease into question. Subsequent research has largely served to confirm this interpretation, pointing towards a prion-like intercellular transfer of misfolded α-synuclein, the main component of Lewy bodies, as central to PD. This shift in thinking offers a revolutionary approach to PD treatment, potentially enabling a transition from purely symptomatic therapy to direct targeting of the pathology that drives disease progression. In this short review, we appraise current experimental support for PD as a prion-like disease, whilst highlighting areas of controversy or inconsistency which must be resolved. We also offer a brief discussion of the therapeutic implications of these discoveries.
1. Introduction
First described as “the shaking palsy” in 1817, Parkinson's disease (PD) is the second most common neurodegenerative disease, prevalent in 1% of the population over the age of 60 [1]. The unprecedented rate of world population ageing is predicted to dramatically increase the number of PD sufferers, reducing overall quality of life and increasing healthcare burden [2]. In the late 1980s and early 1990s, the search for better treatment strategies prompted trials of embryonic neuronal transplants [3]. When, some years later, these patients came to autopsy, a startling observation was made: PD pathology, in the form of Lewy bodies and neurites, was now present in the grafts, raising the possibility of spread from diseased tissue to the young, transplanted neurons [4–6]. This unexpected discovery engendered a novel field of research that is leading to a radical shift in understanding. This review focuses on the evidence examining whether, contrary to classical thinking, PD does indeed progress through non-cell-autonomous mechanisms. The earliest hypotheses concerning these novel mechanisms suggested a prion-like mode of disease propagation, with misfolded α-synuclein (αsyn, the main component of Lewy pathology), the leading candidate for the transmissible protein agent [7]. As we will discuss, subsequent work has offered strong support for this model, leading to a novel view of PD pathogenesis and progression with exciting therapeutic implications, since targeting αsyn spread could lead to new therapies that prevent, halt, or slow PD progression, thereby alleviating both motor and nonmotor symptoms of PD.
2. PD Pathology
The traditional understanding of PD as a movement disorder centres on dopaminergic neuronal loss in the substantia nigra pars compacta (SNPC) [8], as typically demonstrated by a macroscopic reduction in neuromelanin pigmentation and microscopically confirmed by decreased immunoreactivity for dopaminergic neuronal markers: tyrosine hydroxylase (TH, an essential enzyme in dopamine synthesis) and dopamine transporter (DAT, responsible for dopamine reuptake and recycling) [9]. The loss of these neurons causes much of the characteristic motor disturbance that gives rise to the cardinal clinical signs of PD: bradykinesia, postural instability, muscle rigidity, and tremor [8]. Accordingly, most current therapies aim to restore motor function by raising dopamine levels within the remaining functioning dopaminergic neurons, thus boosting their effectiveness.
Intraneuronal inclusions, known as Lewy bodies or Lewy neurites, are also present and are considered the hallmark of microscopic PD pathology. The main component of these abnormal aggregates was an enigma until it was discovered that monoclonal antibodies against αsyn, a presynaptic protein suggested to regulate neurotransmission, intensely labelled Lewy bodies [10]. Both mutations and duplication or triplication of the αsyn gene cause an autosomal dominant form of PD [11], strongly implicating αsyn in PD pathogenesis. The αsyn present in Lewy pathology exhibits a conformational change from the native soluble protein to an insoluble, fibrillar form. These fibrils are rich in β-pleated sheets, shown clearly by intense staining with thioflavin S. Over 90% of this αsyn, compared to 5% normally, is phosphorylated at serine-129 (pser-129), which promotes fibril formation [12]. Pathological human αsyn shows ubiquitin immunoreactivity [13], indicating that at least some of it is targeted for degradation by the proteasome; however, the degree of ubiquitination is variable even within individual cases [14], which may reflect the fact that other processes have also been implicated in the degradation of αsyn, most notably the autophagy-lysosomal pathway [15].
3. Prion Diseases
The science of infectious disease was revolutionised in 1982 when Prusiner postulated that proteinaceous infectious particles (prions) devoid of nucleic acids cause the disease scrapie in animals [16]. This once-heretical theory is now generally well accepted due to vast experimental support [17]. Composed of misfolded forms of native cellular proteins, prions are capable of perpetuating infection by catalysing the conversion of their normally folded counterparts into the misfolded conformation. In humans, all known prionopathies are neurodegenerative and are caused by misfolded prion protein (PrP) [18]. Protein misfolding is a common theme in most of the major neurodegenerative diseases, and mechanistic similarities to prionopathies have been suggested for a number of these, although PD has attracted the most interest from this perspective. In order for a disease to qualify as a prionopathy, specific criteria at both microscopic and macroscopic levels have been proposed [19], which are shown in Figure 1.
Figure 1.

Diagram showing the criteria that must be satisfied for a disease to qualify as a prionopathy [19]. The most unique attribute of prion diseases is their transmissibility between individuals via transfer of pathological protein alone.
Currently, there is no evidence of interorganismal infectivity in PD; therefore it can not a priori be a true prion disease according to this definition [20]. Despite this, a consideration of the other criteria in light of our mechanistic understanding of PD and other neurodegenerative diseases suggests that the line between infectious disease and what is traditionally thought of as cell-autonomous neurodegeneration is much less clear than originally thought.
4. The Transplant Studies
Dopamine replacement therapy, the mainstay of current treatment for PD, can demonstrate limited efficacy and may be further complicated by drug-induced dyskinesias. Furthermore, PD leads to many nonmotor symptoms, such as autonomic dysfunction and cognitive and mood disturbances, all of which respond very poorly to dopamine replacement. These problems emphasise the need to target the cause of the progressive neuronal loss [21]. Consequently, cell replacement therapy was considered to be a logical treatment strategy for PD.
In a series of open-label trials in the 1990s [3], fetal mesencephalic dopaminergic neuronal grafts into the putamen, or the putamen and caudate nucleus of PD patients, were reported to induce long-term functional benefits. However, the questionable sensitivity of subjective outcome measures to placebo effects engendered two new double-blind trials which, unfortunately, did not find such benefits, suggesting placebo effects, and/or observer bias affected earlier trials [22, 23]. Nevertheless, in 2008 results from the postmortem analyses of 9 patients who died between 11 and 16 years after graft insertion were published. Incredibly, pser-129-αsyn-positive Lewy-like inclusions were observed within the mesencephalic grafts of 4 patients [4–6], a completely uncharacteristic finding in such young neurons. Reduced DAT and TH levels were also seen, indicating graft dysfunction. It was suggested that the appearance of pathology was time-dependent due to a lack of pathology in grafts younger than 4 years old and an increased percentage of αsyn-positive dopaminergic neurons in a 16-year-old graft compared to a 12-year-old graft from the same person [6].
In light of these observations, a hypothesis that PD was a transmissible disease with prion-like features was proposed [6]. However, Lewy pathology was not detected in all grafts [24], casting doubt upon the theory of host-to-graft transmission. Further studies were required to understand what was occurring. Over the subsequent six years, a substantial body of work has amassed dedicated to examining the prion-like hypothesis for PD. Below, this evidence is summarised with regard to the specific cellular, intercellular, and tissue level criteria for prionopathies introduced above [19]. As the remaining criterion, for interorganismal infectivity, has not been reported in PD, the term “prion-like” must strictly be used. It might reasonably be argued, however, that demonstrated host-graft spread might go at least some way towards meeting this requirement.
5. Cellular Level
An important characteristic of prion disease is that a pool of endogenous, natively folded PrP (PrPc) is required as a substrate for conversion by, and to, the pathological, misfolded form (PrPsc) [25]. Via the use of αsyn knockout tissues and animals, endogenous αsyn has been confirmed as similarly necessary for the development of αsyn pathology both in vitro [26] and in vivo [27]. Furthermore, the pathological forms of both αsyn and PrP are structurally similar, being rich in β-pleated sheets [28], prone to fibril formation, and resistant to normally denaturing agents.
There are two mainstream theories regarding the mechanism of conversion of PrP (PrPc) to the misfolded “scrapie” form (PrPsc): the refolding hypothesis and the currently more favoured seeding hypothesis [25] (Figure 2). There is evidence of αsyn seeding in Lewy body formation, which has best been demonstrated in vitro [28].
Figure 2.
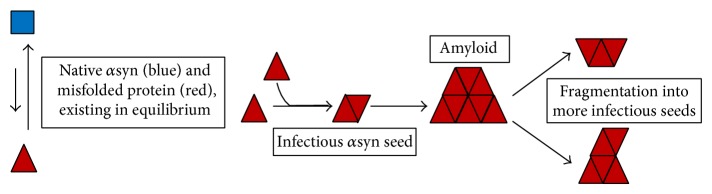
The seeding hypothesis of prion disease pathogenesis, adapted from [25].
One study added myc-tagged recombinant αsyn preformed fibrils (PFFs) to cells overexpressing αsyn, leading to the formation of Lewy-body-like inclusions. Anti-myc antibodies only labeled the centres of inclusions; endogenous αsyn had been recruited and converted to form the periphery [29]. Whilst this demonstrates seeding, nonphysiological lipofection agents were used to optimise intracellular delivery of PFFs. However, other studies have demonstrated seeding in immunofluorescence studies without lipofection agents. In one, cells expressing either green or red fluorescence-labeled αsyn were cocultured, generating inclusions with a central core of green-labeled αsyn surrounded by endogenous red-labeled αsyn [30]. A more recent example used a methodology for the synthesis of artificial prions to create short αsyn amyloid fibrils; a single exposure to these was sufficient to induce aggregation of endogenous αsyn in human neuroblastoma SH-SY5Y cells [31].
These similarities between prionopathies and PD on a molecular and cellular level would seem to create a stable foundation for the prion-like hypothesis.
6. Intercellular Level
The spread of Lewy pathology in graft patients is likely to be due to intercellular transfer of αsyn. There are numerous mechanisms by which this could occur, from cellular release and uptake of αsyn to direct transmission via nanotubes [7].
6.1. αsyn Secretion and Uptake
Despite a lack of secretory signal peptide sequence, αsyn is present in the cerebrospinal fluid of individuals with or without PD [32]. There is support for exocytosis-mediated αsyn release in vitro, shown to be independent of cell death or membrane leakage. This secretion is reduced by low temperature, which also slows vesicular exocytosis, but is unaffected by Brefeldin A, which inhibits conventional exocytosis. These data suggest that αsyn is secreted via an unconventional vesicular pathway [33], although current opinion on what form this could take is divided. Electron microscopy shows that αsyn can be found within large dense-core vesicles [33]. Alternatively, it has been suggested that exosomes, small membrane vesicles released into the extracellular space, are the carrier and that exosome-associated αsyn is more likely to be internalised than free αsyn [34]. Thus, it is possible that there may be more than one αsyn secretory pathway.
The need for lipofection agents in many experiments has highlighted the difficulty in demonstrating meaningful αsyn internalization [29], although unaided uptake both in vitro and in vivo has been demonstrated by Desplats and colleagues [35]. Fluorescently labeled mouse cortical neuronal stem cells (MCNSCs) were injected into the hippocampus of transgenic mice expressing human αsyn under the control of the brain-specific Thy-1 promoter. Four weeks later, 15% of grafted cells displayed human-αsyn immunoreactivity. Some cells developed αsyn-positive inclusions although, unexpectedly, fibrillar αsyn was not detected. This pathology was not observed in grafts inserted into nontransgenic mice: αsyn is therefore essential for host-to-graft transmission. However, the hippocampus is not a site relevant to grafts in clinical PD treatment, and MCNSCs are proliferative, unlike most neurons. Accordingly, Hansen and colleagues transplanted wild-type mouse embryonic mesencephalic neurons into the striatum of transgenic mice overexpressing human-αsyn under the control of the native mouse αsyn promoter. Six months later, 5% of transplanted neurons were human-αsyn-positive [30].
Both Desplats and Hansen give evidence for an endocytic mechanism of αsyn uptake via the use of inhibitors [30, 35]. Other studies find that this is true for aggregated forms of αsyn but that monomeric αsyn can be directly translocated across the plasma membrane, suggesting that at least two internalisation pathways may exist which are αsyn state-dependent [36]. However, one notable disparity between the Desplats and Hansen studies is the proportion of cells displaying αsyn immunoreactivity (15% in 4 weeks versus 5% in 6 months). This is likely due to differences in experimental design, which include different promoters driving αsyn expression, the types of transplanted cell utilized, and differing brain areas studied. Furthermore, Hansen and colleagues did not observe seeding after uptake in vivo, and a similar study in rats also reported little aggregation [37]. Despite these variations in experimental observations, we believe there is overall strong evidence for αsyn secretion and uptake.
6.2. Tunneling Nanotubes (TNTs)
TNTs are a recently discovered form of direct cell-to-cell communication. They are F-actin containing tubes with diameters of less than 200 nm that connect the cytoplasm of two cells. Intercellular transfer of PrPsc [38] has been confirmed and, recently, αsyn has been found within TNTs between glioblastoma cells [39]. This preliminary evidence will require further investigation to elucidate the role, if any, of TNTs in αsyn transmission in PD.
6.3. Intercellular Transfer: The Braak Hypothesis
The Braak hypothesis is perhaps the currently best-accepted model of PD progression and would be highly consistent with a prion-like mechanism of spread. Derived from the postmortem analyses of 100 αsyn-positive subjects, it suggests that, in PD, Lewy pathology affects regions in the brain in a stereotypic, topographical manner. Lewy pathology severity is also suggested to correlate with the clinical progression of PD symptoms [40]. Braak suggests that the stereotypical propagation of pathology depends partly on the vulnerability of specific neuron types. A potential explanation of the particular vulnerability of SNPC dopaminergic neurons might be furnished by the suggestion that oxidative stress and αsyn aggregation form a positive feedback loop [28], coupled with evidence that these neurons are particularly susceptible to oxidative stress, due to both metabolic demands particular to this population and dopamine oxidation with generation of reactive oxidative species [41]. Dopamine is also known to induce the formation of soluble toxic aggregates of αsyn, which have been shown to be capable of replication by self-propagation in vitro [42].
The Braak hypothesis proposes that αsyn pathology begins, due to environmental insult, within enteric epithelium. From here, it travels retrogradely via the axons of enteric nervous system (ENS) neurons to their somata within the intermediolateral column of the spinal cord and then to the dorsal motor nucleus of the vagus (DMNV) in the medulla. Thereafter, it is able to reach the pons and substantia nigra and, beyond that, vulnerable regions of the cortex [7] (Figure 3). There is some experimental support for this scheme: in rats, intragastric administration of rotenone, a mitochondrial complex I inhibitor that generates Lewy-like inclusions, generated Lewy pathology in the enteric nervous system (ENS) which spreads to the substantia nigra in agreement with the Braak hypothesis [43]. The pathology only appeared in synaptically connected areas, suggestive of transsynaptic transmission, and a later study using the same model showed that transection of autonomic axons was sufficient to halt the spread of enteric αsyn pathology [44]. However, the Braak hypothesis remains controversial, and challenges exist for this particular account of pathological αsyn spread, as well as for interneuronal spread more generally. These include several studies that find distributions of αsyn pathology in PD that would not be predicted by the stereotyped Braak pattern of disease spread and the observation that αsyn pathology is also the predominant histopathological characteristic of other diseases, most notably multiple system atrophy (MSA), which feature a completely different pattern of CNS involvement and spread than PD [7]. There is also the related observation that some forms of substantia nigra degeneration and clinical parkinsonism exhibit no Lewy pathology at all [45].
Figure 3.
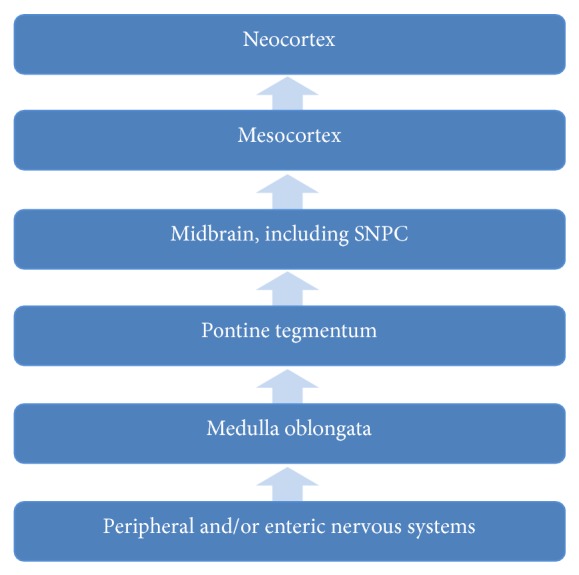
The 6 stages of PD according to the Braak hypothesis. Pathology begins in the enteric nervous system and progresses to the neocortex.
Most of the specific conflicts with the Braak hypothesis can be resolved by extending the latter to include the possibility of anterograde spread, which is still quite compatible with a prion-like model, which greatly broadens the possible patterns and sequences of involvement one could see [46]. Many of the remaining inconsistencies can be addressed by considering the role of deposited αsyn: is it a reliable marker of cell damage, therefore indicating the degree to which regions are affected by the disease, or is it part of a protective response which mitigates cellular damage? In the latter case, Lewy bodies might mark those neurons resisting degeneration, which might actually be most active elsewhere [7]. If the latter were true even some of the time, then the assumption of so many studies, that αsyn pathology correlates with degeneration, could be seriously flawed, with clear implications for study conclusions. In support of this idea, recent work examining the relationship between nigral cell loss, the duration of motor symptoms, and the distribution and density of Lewy pathology found no correlation [47]. It has also been shown that Lewy bodies are absent from the majority of cells showing apoptotic changes [48].
An imperfect correlation of pathological αsyn deposits with active disease may simply reflect the emerging theory that it is not visibly aggregated αsyn, but oligomeric αsyn, that is cytotoxic [49], a theory that has also found support in prion diseases [50]. In keeping with this, it has been shown that stabilisation of αsyn in oligomeric form increases cytotoxicity, whilst reduced oligomer formation decreases cytotoxicity [51].
7. Tissue Level
In prionopathies, the protein itself is toxic and leads to rapid neurodegeneration [18]. The prion-like hypothesis of PD and the Braak hypothesis both propose that αsyn can serve the same function. In one study, PFFs were injected intrastriatally into nontransgenic mice and αsyn spread to the dopaminergic cells of the SNPC was observed by 30 days of postinjection (dpi), with negligible cell loss. However, by 100 dpi, the total number of cells and striatal dopamine concentration had decreased, suggesting that αsyn Lewy pathology is cytotoxic [52]. This cell death correlated with declines in motor function. In vitro, αsyn has been shown to lead to caspase-3 activation [35]. Cell-cell spread in the opposite direction has also been elegantly demonstrated via the experimental induction of human-αsyn overexpression in the right substantia nigra of rats, along with bilateral striatal transplant of embryonic dopaminergic neurons. This led to αsyn immunoreactivity in the right striatal transplant but not the left [53]. Furthermore, different sites of injection of pathological αsyn-containing material elicited different and connectivity-dependent patterns of αsyn spread, although it must be noted that nonphysiologically high concentrations of αsyn were used to elicit widespread transfer in these experiments [52].
Overall, the evidence provides substantial support for prion-like transfer of αsyn, but controversies remain; in particular, one of the most prominent models for PD propagation, the Braak hypothesis, which would be entirely consistent with prion-like spread, has not been clearly proved. To properly address this question experimentally, a technique to longitudinally monitor αsyn spread will need to be developed, which will also allow prion-like transfer to be examined over time.
8. Therapeutic Strategies
The field of research into prion-like αsyn spread is very young; however, the therapeutic implications could be huge. Inhibition of αsyn transmission could offer a disease-modifying approach with the potential to prevent both motor and nonmotor decline in patients.
A summary of possible treatment strategies targeting αsyn transmission is represented in Figure 4. Whilst many of these remain theoretical, some have been trialled in vitro for PD or other protein misfolding diseases. One possibility is to increase the resistance of αsyn to seeding. In transthyretin amyloidosis, where transthyretin misfolds to cause disease, compounds have been developed to stabilise the protein as a functional tetramer [54]. As αsyn can exist as a folded tetramer that is resistant to aggregation [55], this strategy could be adopted for PD treatment. Another attractive strategy is to decrease the amount of αsyn available for misfolding, which may cause only few side effects since the protein shows a high degree of cellular redundancy in mammals. One study reported that interfering RNA against αsyn conferred resistance to MPP+ exposure, which usually causes PD-like degeneration [56]. Such strategies would be of particular interest due to their ability to halt the disease at the very start of the pathological cascade. Other strategies could directly or indirectly reduce the rate of intercellular transfer; while the mechanisms underlying this are currently not entirely clear, once they are better understood it is hoped that specific inhibitors of the relevant processes can be designed or may even exist already. Finally, neuronal transplants could still be useful if the benefits previously seen in clinical trials could be maximised, whilst simultaneously minimising graft dysfunction. An ongoing multicentre trial, TRANSEURO, aims to elucidate the factors affecting transplant efficacy to improve such cell replacement therapies [57].
Figure 4.

Possible therapeutic strategies to decrease αsyn transmission in PD. This is not an exhaustive representation; see [7, 57, 59].
9. Future Studies and Conclusions
While many studies have provided much good evidence for prion-like αsyn transfer, progress is hampered by many of the limitations of current experimental animal models, which are an imperfect replication of human PD. Of course, such problems are far from unique to this field. While many improvements would be desirable to ensure greater applicability to human disease, perhaps the most significant shortcomings are the chronicity of the studies, which take place in fairly short-lived organisms, and the nonphysiological levels of αsyn protein which have been used to elicit spread, both of which are clear confounds when considering a process which takes place over many years and with native levels of αsyn in PD. With regard to the latter problem, it is noteworthy that while the majority of studies use cells or animals overexpressing αsyn at high levels, this is not always necessary for PD pathology to develop if neurons are exposed to PFFs [26]. In vitro models also have an important role to play in allowing high throughput approaches and a level of access that in vivo models often cannot support. To this end, the use of human induced pluripotent stem cells is an exciting development which promises all the advantages of the in vitro approach coupled with the unprecedented ability to study dynamic disease processes in real time in living human neurons differentiated along the lineages of relevant populations such as SNPC dopaminergic neurons [58].
In summary, the role of prion-like spread in PD progression remains an exciting area of research. Whilst there are still unanswered questions, many studies corroborate the idea that αsyn spread via a prion-like infectious process is central to PD. The therapeutic implications of this conclusion are powerful indeed and have the potential to revolutionise treatment of both motor and nonmotor manifestations of this devastating disease.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.de Lau L. M., Breteler M. M. Epidemiology of Parkinson's disease. The Lancet Neurology. 2006;5(6):525–535. doi: 10.1016/s1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 2.Boland D. F., Stacy M. The economic and quality of life burden associated with Parkinson's disease: a focus on symptoms. The American Journal of Managed Care. 2012;18(7):S168–S175. [PubMed] [Google Scholar]
- 3.Dunnett S. B., Björklund A., Lindvall O. Cell therapy in Parkinson's disease—stop or go? Nature Reviews Neuroscience. 2001;2(5):365–369. doi: 10.1038/35072572. [DOI] [PubMed] [Google Scholar]
- 4.Kordower J. H., Chu Y., Hauser R. A., Freeman T. B., Olanow C. W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nature Medicine. 2008;14(5):504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 5.Kordower J. H., Chu Y., Hauser R. A., Olanow C. W., Freeman T. B. Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Movement Disorders. 2008;23(16):2303–2306. doi: 10.1002/mds.22369. [DOI] [PubMed] [Google Scholar]
- 6.Li J.-Y., Englund E., Holton J. L., et al. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nature Medicine. 2008;14(5):501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 7.Visanji N. P., Brooks P. L., Hazrati L. N., Lang A. E. The prion hypothesis in Parkinson's disease: braak to the future. Acta Neuropathologica Communications. 2013;1:p. 2. doi: 10.1186/2051-5960-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez-Oroz M. C., Jahanshahi M., Krack P., et al. Initial clinical manifestations of Parkinson's disease: features and pathophysiological mechanisms. The Lancet Neurology. 2009;8(12):1128–1139. doi: 10.1016/S1474-4422(09)70293-5. [DOI] [PubMed] [Google Scholar]
- 9.Kordower J. H., Olanow C. W., Dodiya H. B., et al. Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain. 2013;136(8):2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spillantini M. G., Schmidt M. L., Lee V. M. Y., Trojanowski J. Q., Jakes R., Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 11.Singleton A. B., Farrer M., Johnson J., et al. α-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302(5646):p. 841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 12.Fujiwara H., Hasegawa M., Dohmae N., et al. α-synuclein is phosphorylated in synucleinopathy lesions. Nature Cell Biology. 2002;4(2):160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 13.Hasegawa M., Fujiwara H., Nonaka T., et al. Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. Journal of Biological Chemistry. 2002;277(50):49071–49076. doi: 10.1074/jbc.m208046200. [DOI] [PubMed] [Google Scholar]
- 14.Sampathu D. M., Giasson B. I., Pawlyk A. C., Trojanowski J. Q., Lee V. M.-Y. Ubiquitination of α-synuclein is not required for formation of pathological inclusions in α-synucleinopathies. The American Journal of Pathology. 2003;163(1):91–100. doi: 10.1016/s0002-9440(10)63633-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebrahimi-Fakhari D., Cantuti-Castelvetri I., Fan Z., et al. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. Journal of Neuroscience. 2011;31(41):14508–14520. doi: 10.1523/jneurosci.1560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prusiner S. B. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 17.Prusiner S. B. Molecular biology of prion diseases. Science. 1991;252(5012):1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 18.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annual Review of Neuroscience. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 19.Guest W. C., Silverman J. M., Pokrishevsky E., O'Neill M. A., Grad L. I., Cashman N. R. Generalization of the prion hypothesis to other neurodegenerative diseases: an imperfect fit. Journal of Toxicology and Environmental Health Part A. 2011;74(22–24):1433–1459. doi: 10.1080/15287394.2011.618967. [DOI] [PubMed] [Google Scholar]
- 20.Irwin D. J., Abrams J. Y., Schonberger L. B., et al. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurology. 2013;70(4):462–468. doi: 10.1001/jamaneurol.2013.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rascol O., Payoux P., Ory F., Ferreira J. J., Brefel-Courbon C., Montastruc J. L. Limitations of current Parkinson's disease therapy. Annals of Neurology. 2003;53(supplement 3):S3–S12. doi: 10.1002/ana.10513. [DOI] [PubMed] [Google Scholar]
- 22.Freed C. R., Greene P. E., Breeze R. E., et al. Transplantation of embryonic dopamine neurons for severe Parkinson's disease. The New England Journal of Medicine. 2001;344(10):710–719. doi: 10.1056/nejm200103083441002. [DOI] [PubMed] [Google Scholar]
- 23.Olanow C. W., Goetz C. G., Kordower J. H., et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson's disease. Annals of Neurology. 2003;54(3):403–414. doi: 10.1002/ana.10720. [DOI] [PubMed] [Google Scholar]
- 24.Mendez I., Vĩuela A., Astradsson A., et al. Dopamine neurons implanted into people with Parkinson's disease survive without pathology for 14 years. Nature Medicine. 2008;14(5):507–509. doi: 10.1038/nm1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aguzzi A., Calella A. M. Prions: protein aggregation and infectious diseases. Physiological Reviews. 2009;89(4):1105–1152. doi: 10.1152/physrev.00006.2009. [DOI] [PubMed] [Google Scholar]
- 26.Volpicelli-Daley L. A., Luk K. C., Patel T. P., et al. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72(1):57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuda-Suzukake M., Nonaka T., Hosokawa M., et al. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathologica Communications. 2014;2(1):p. 88. doi: 10.1186/s40478-014-0088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dunning C. J. R., George S., Brundin P. What's to like about the prion-like hypothesis for the spreading of aggregated α-synuclein in Parkinson disease? Prion. 2013;7(1):92–97. doi: 10.4161/pri.23806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luk K. C., Song C., O'Brien P., et al. Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(47):20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen C., Angot E., Bergström A.-L., et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. Journal of Clinical Investigation. 2011;121(2):715–725. doi: 10.1172/jci43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aulic S., Le T. T. N., Moda F., et al. Defined alpha-synuclein prion-like molecular assemblies spreading in cell culture. BMC Neuroscience. 2014;15, article 69 doi: 10.1186/1471-2202-15-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borghi R., Marchese R., Negro A., et al. Full length α-synuclein is present in cerebrospinal fluid from Parkinson's disease and normal subjects. Neuroscience Letters. 2000;287(1):65–67. doi: 10.1016/s0304-3940(00)01153-8. [DOI] [PubMed] [Google Scholar]
- 33.Lee H.-J., Patel S., Lee S.-J. Intravesicular localization and exocytosis of α-synuclein and its aggregates. Journal of Neuroscience. 2005;25(25):6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danzer K. M., Kranich L. R., Ruf W. P., et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Molecular Neurodegeneration. 2012;7(1, article 42) doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Desplats P., Lee H.-J., Bae E.-J., et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(31):13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee H.-J., Suk J.-E., Bae E.-J., Lee J.-H., Paik S. R., Lee S.-J. Assembly-dependent endocytosis and clearance of extracellular α-synuclein. The International Journal of Biochemistry & Cell Biology. 2008;40(9):1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 37.Kordower J. H., Dodiya H. B., Kordower A. M., et al. Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiology of Disease. 2011;43(3):552–557. doi: 10.1016/j.nbd.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gousset K., Schiff E., Langevin C., et al. Prions hijack tunnelling nanotubes for intercellular spread. Nature Cell Biology. 2009;11(3):328–336. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- 39.Agnati L. F., Guidolin D., Balǔka F., et al. A new hypothesis of pathogenesis based on the divorce between mitochondria and their host cells: possible relevance for alzheimer's disease. Current Alzheimer Research. 2010;7(4):307–322. doi: 10.2174/156720510791162395. [DOI] [PubMed] [Google Scholar]
- 40.Braak H., del Tredici K., Rüb U., de Vos R. A. I., Jansen Steur E. N. H., Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiology of Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 41.Lotharius J., Brundin P. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nature Reviews. Neuroscience. 2002;3(12):932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- 42.Planchard M. S., Exley S. E., Morgan S. E., Rangachari V. Dopamine-induced alpha-synuclein oligomers show self- and cross-propagation properties. Protein Science. 2014;23:1369–1379. doi: 10.1002/pro.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan-Montojo F., Anichtchik O., Dening Y., et al. Progression of Parkinson's disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS ONE. 2010;5(1) doi: 10.1371/journal.pone.0008762.e8762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pan-Montojo F., Schwarz M., Winkler C., et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Scientific Reports. 2012;2, article 898 doi: 10.1038/srep00898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gasser T., Hardy J., Mizuno Y. Milestones in PD genetics. Movement Disorders. 2011;26(6):1042–1048. doi: 10.1002/mds.23637. [DOI] [PubMed] [Google Scholar]
- 46.Lerner A., Bagic A. Olfactory pathogenesis of idiopathic parkinson disease revisited. Movement Disorders. 2008;23(8):1076–1084. doi: 10.1002/mds.22066. [DOI] [PubMed] [Google Scholar]
- 47.Parkkinen L., O'Sullivan S. S., Collins C., et al. Disentangling the relationship between lewy bodies and nigral neuronal loss in Parkinson's disease. Journal of Parkinson's Disease. 2011;1(3):277–286. doi: 10.3233/jpd-2011-11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tompkins M. M., Hill W. D. Contribution of somal Lewy bodies to neuronal death. Brain Research. 1997;775(1-2):24–29. doi: 10.1016/S0006-8993(97)00874-3. [DOI] [PubMed] [Google Scholar]
- 49.Kalia L. V., Kalia S. K., McLean P. J., Lozano A. M., Lang A. E. α-synuclein oligomers and clinical implications for parkinson disease. Annals of Neurology. 2013;73(2):155–169. doi: 10.1002/ana.23746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simoneau S., Rezaei H., Salès N., et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathogens. 2007;3(8, article e125) doi: 10.1371/journal.ppat.0030125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Outeiro T. F., Putcha P., Tetzlaff J. E., et al. Formation of toxic oligomeric α-synuclein species in living cells. PLoS ONE. 2008;3(4) doi: 10.1371/journal.pone.0001867.e1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luk K. C., Kehm V., Carroll J., et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338(6109):949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Angot E., Steiner J. A., Tomé C. M., et al. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS ONE. 2012;7(6) doi: 10.1371/journal.pone.0039465.e39465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peterson S. A., Klabunde T., Lashuel H. A., Purkey H., Sacchettini J. C., Kelly J. W. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(22):12956–12960. doi: 10.1073/pnas.95.22.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bartels T., Choi J. G., Selkoe D. J. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477(7362):107–111. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fountaine T. M., Wade-Martins R. RNA interference-mediated knockdown of α-synuclein protects human dopaminergic neuroblastoma cells from MPP+ toxicity and reduces dopamine transport. Journal of Neuroscience Research. 2007;85(2):351–363. doi: 10.1002/jnr.21125. [DOI] [PubMed] [Google Scholar]
- 57.Petit G. H., Olsson T. T., Brundin P. Review: the future of cell therapies and brain repair: parkinson's disease leads the way. Neuropathology and Applied Neurobiology. 2014;40(1):60–70. doi: 10.1111/nan.12110. [DOI] [PubMed] [Google Scholar]
- 58.Hartfield E. M., Yamasaki-Mann M., Ribeiro Fernandes H. J., et al. Physiological characterisation of human iPS-derived dopaminergic neurons. PLoS ONE. 2014;9(2) doi: 10.1371/journal.pone.0087388.e87388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vekrellis K., Stefanis L. Targeting intracellular and extracellular alpha-synuclein as a therapeutic strategy in Parkinson's disease and other synucleinopathies. Expert Opinion on Therapeutic Targets. 2012;16(4):421–432. doi: 10.1517/14728222.2012.674111. [DOI] [PubMed] [Google Scholar]