

Abstract
The Ras-ERK pathway is deregulated in approximately a third of human cancers, particularly those of epithelial origin. In aggressive, triple-negative, basal-like breast cancers, most tumors display increased MEK and ERK phosphorylation and exhibit a gene expression profile characteristic of Kras or EGFR mutant tumors; however, Ras family genetic mutations are uncommon in triple-negative breast cancer and EGFR mutations account for only a subset of these tumors. Therefore, the upstream events that activate MAPK signaling and promote tumor aggression in triple-negative breast cancers remain poorly defined. We have previously shown that a secreted TGF-β family signaling ligand, Nodal, is expressed in breast cancer in correlation with disease progression. Here we highlight key findings demonstrating that Nodal is required in aggressive human breast cancer cells to activate ERK signaling and downstream tumorigenic phenotypes both in vitro and in vivo. Experimental knockdown of Nodal signaling downregulates ERK activity, resulting in loss of c-myc, upregulation of p27, G1 cell cycle arrest, increased apoptosis and decreased tumorigenicity. The data suggest that ERK activation by Nodal signaling regulates c-myc and p27 proteins post-translationally and that this cascade is essential for aggressive breast tumor behavior in vivo. As the MAPK pathway is an important target for treating triple-negative breast cancers, upstream Nodal signaling may represent a promising target for breast cancer diagnosis and combined therapies aimed at blocking ERK pathway activation.
Keywords: Nodal, breast cancer, p27, c-myc, ERK
1.0 Nodal is an oncofetal protein
Normally expressed during early development, Nodal is essential for supporting an undifferentiated state in embryonic stem (ES) cells and for endodermal and mesodermal differentiation, including epithelial-to-mesenchymal transition (EMT) [1], processes which are reactivated in aggressive epithelial-derived cancer cells [2,3,4]. Recently, Nodal expression has been shown to reemerge in a number of cancerous tumors, including melanoma, breast, pancreas and prostate, where its expression level appears to correlate with tumor grade [5–8], although the mechanisms by which Nodal signaling promotes cancerous growth are not well defined.
Canonically, Nodal signals by partnering with its co-receptor, Cripto-1, to activate Smad2 and Smad3, thereby stimulating downstream gene transcription and promoting specific developmental outcomes. During development, Nodal signaling is restricted by an inhibitor, termed Lefty, which is co-expressed in ES cells and attenuates Nodal signaling as differentiation begins[1]. Importantly, unlike developing embryonic cells, breast carcinoma cells do not express Lefty, thus emergent Nodal signaling and the Nodal-driven plasticity program proceed unchecked during tumor growth. Furthermore, culturing cancer cells in the presence of Lefty is sufficient to inhibit both Nodal expression and tumorigenicity [9].
While we have previously shown that Nodal expression correlates with tumor stage, the mechanistic requirement for Nodal in breast cancer development has not been defined. Interestingly, Nodal and other TGFβ family members have been shown to cooperate with MAPK cascades to regulate developmental and pathological processes [6]. Further, the mechanisms underlying MAPK activation in breast cancer are incompletely understood, yet critically important for treating the disease. In this review, we report that Nodal signaling is fundamentally required for the aggressive phenotype of human breast cancer cells, both in vitro and in a mouse xenograft model. Most noteworthy, our results indicate that Nodal signaling maintains breast carcinoma tumoriogenicity through activation of ERK and downstream regulation of c-myc and p27 protein expression.
2.0 Nodal knockdown impairs growth in breast cancer
Previous studies from our laboratory have demonstrated that human breast tumors and aggressive breast cancer cell lines express Nodal in correlation with tumor grade [9]. More recently, we found very high expression of Nodal by immunohistochemistry in a series of 20 triple negative breast cancer biopsy samples compared to weak or almost undetectable Nodal staining in biopsy samples of benign breast disease used as control (Figure 1A). To investigate the functional requirement for Nodal signaling in triple negative breast cancer cell behavior, we used shRNAs to knock down Nodal expression in MDA-MB-231 and MDA-MB-468 aggressive breast cancer cell lines and observed decreased Nodal protein expression accompanied by decreased Smad3 phosphorylation at Serine 423/425 in Nodal knockdown cells, indicating both effective knockdown and that Nodal signaling is essential to maintain Smad3 phosphorylation in these breast cancer cell lines (Figure 1B). Further, Nodal knockdown led to growth suppression in both cell lines when compared with controls (Figure 1C), increased apoptosis and decreased overall viability (Figure 1D). Additionally, Nodal knockdown cells incorporated less BrdU than controls, demonstrating that reduced proliferation (Figure 1E) accompanied by increased apoptosis reduces overall cell growth in the absence of Nodal signaling.
Figure 1.
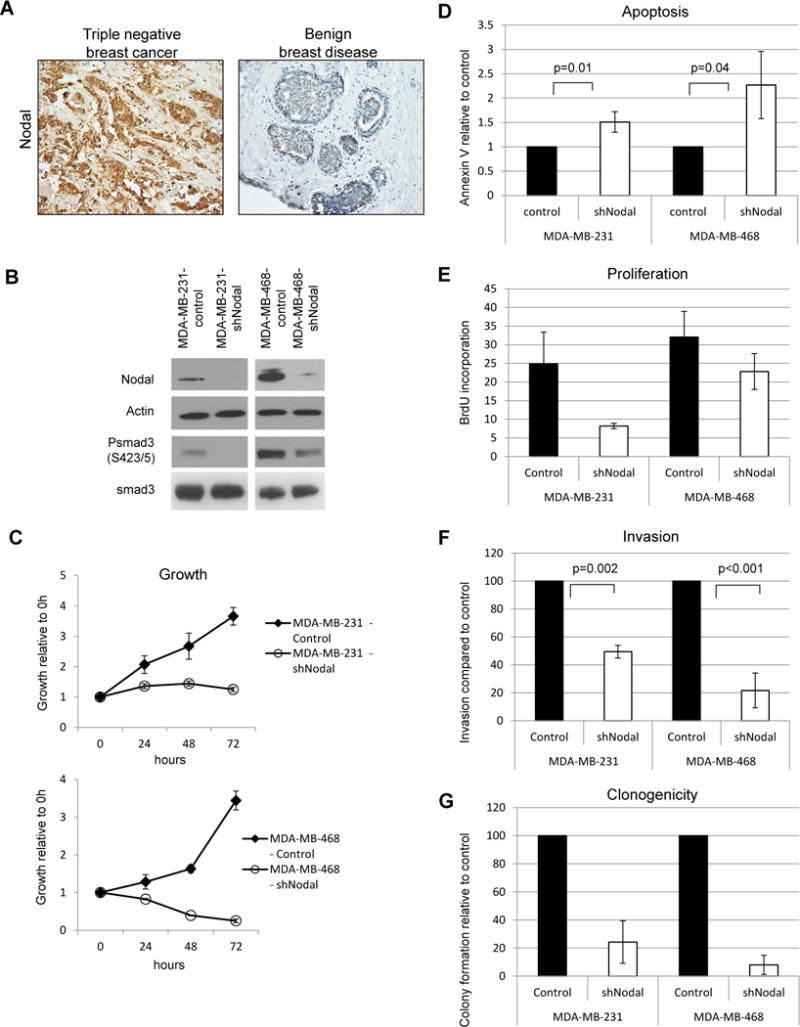
Nodal knockdown impairs growth and aggressive behavior in human breast cancer cell lines. A) Representative immunohistochemistry results show increased expression (brown staining) of Nodal in a triple negative breast cancer biopsy section compared to a biopsy section of benign breast disease (200X original magnification). A) Western blot demonstrating Nodal knockdown and smad 3 phosphorylation status in human breast cancer cell lines. Data are representative of 3 experiments. B) Cell growth curves for knockdown and control cell lines. Error bars represent standard deviation. C) Apoptosis in Nodal knockdown and control cells was measured by Annexin V staining and flow cytometry. Error bars represent standard deviation. D) Proliferation in Nodal knockdown and control cells. Cells were cultured in the presence of BrdU for 30 minutes prior to fixation and analyzed by flow cytometry. N=3 for each cell type; error bars represent standard deviation. E) Invasive potential of Nodal knockdown and control cells. Percent invasion was calculated based on the number of cells seeded and normalized to controls. F) Colony formation in soft agar after 3 weeks. Colonies were counted and normalized to controls.
To determine whether Nodal signaling is required for other aggressive characteristics of breast cancer cells, we performed invasion assays in a laminin IV/collagen matrix and found that Nodal knockdown cells were significantly less invasive than controls for both MDA-MB-231 and MDA-MB-468 cells (Figure 1F). Finally, Nodal knockdown impaired clonogenicity in soft-agar assays, indicating a loss of breast cancer cell self-renewal in the absence of Nodal signaling (Figure 1G). Together, these data suggest that Nodal signaling is required for viability, proliferation, invasion and self-renewal of triple negative breast cancer cells lines.
To assess whether Nodal is essential for breast cancer progression in vivo, we used a mouse mammary fat pad xenograft model to measure tumorigenicity of MDA-MB-231 or MDA-MB-468 cells transduced either with shNodal or control in immunocompromised Nude mice. For both cell lines, knockdown of Nodal expression dramatically slowed tumor growth compared with controls (Figure 2A, B). For the MDA-MB-231 cell line, Nodal knockdown also suppressed tumor engraftment, with only 5/20 injections giving rise to tumors as compared with 19/20 for control cells (Figure 2C). Additionally, tumors that did form were histologically less organized in the absence of Nodal expression (Figure 2D). For MDA-MB-468 cells, Nodal knockdown dramatically suppressed tumor growth, although, interestingly, overall tumor engraftment was not affected during the period of observation, potentially highlighting differences in genetic mutations between the two cell lines.
Figure 2.
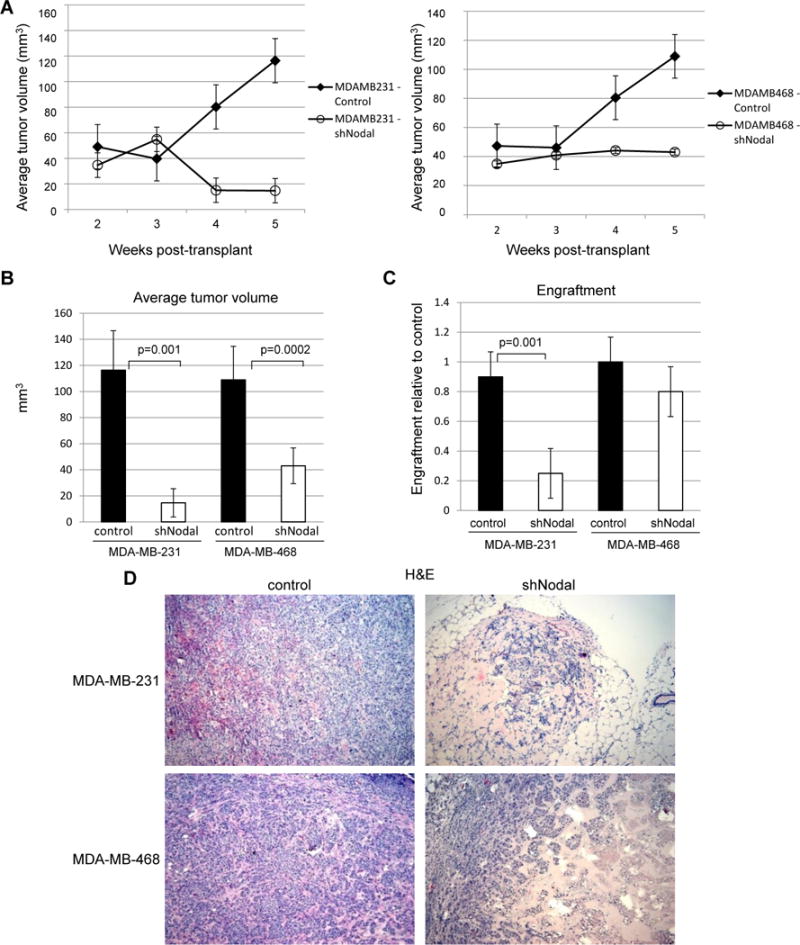
Nodal knockdown impairs breast tumor growth in vivo. A) Nodal knockdown or control cell xenograft growth. Error bars represent standard error (line graphs). N=20 (MDA-MB-231) and n=10 (MDA-MB-468). B) Proportion of mice forming tumors. Error bars represent standard deviation. N=20 (MDA-MB-231) and n=10 (MDA-MB-468). C) Reduced incidence of tumors generated from Nodal knockdown cells. D) H&E staining of Nodal knockdown and control tumors after 5 weeks. (10× original magnification)
3.0 Downregulation of Nodal alters cell cycling in breast cancer cells
Since a decrease in proliferation was observed in the absence of Nodal, cell cycle analysis was performed and revealed a striking upward shift in DNA content in Nodal knockdown cells after several passages (Figure 3A) accompanied by substantially larger nuclei (Figure 3B). To better characterize a possible cell cycle defect, we synchronized knockdown or control cells immediately after viral transduction by double thymidine block and nocodozole arrest, followed by release and DNA content analysis every 2 hours by flow cytometry. Control cells displayed a prominent 4N peak immediately after release from nocodozole arrest, and then proceeded to shift over time to 2N DNA content consistent with progression to G1 after mitotic arrest (Figure 3C). In contrast, MDA-MB-231 Nodal knockdown cells produced a 2N peak after nocodozole arrest, which never shifted toward 4N content over 14 hours of observation, suggesting G1 arrest. These data appear at first to conflict with increased DNA content observed after several passages; however, polyploidization has been shown to accompany G1 arrest in breast cancer cells in some circumstances, including increased expression of the cell cycle inhibitor p27 [10]. Interestingly, MDA-MB-468 Nodal knockdown cells displayed a peak at 4N upon release from nocodozole arrest, indicating synchronization in G2/M and ability to cycle; however, these cells were much slower to progress into G1 and through G1 than controls, possibly indicating mild cell cycle defects. These data are consistent with the observed incorporation of BrdU in MDA-MB-468 Nodal knockdown cells and, together with growth and apoptosis data (Figure 1), may indicate Nodal loss produces a strong apoptotic response and mild cell cycle defect in MDA-MB-468 cells, while MDA-MB-231 cells respond to Nodal loss with cell cycle arrest more than apoptosis.
Figure 3.
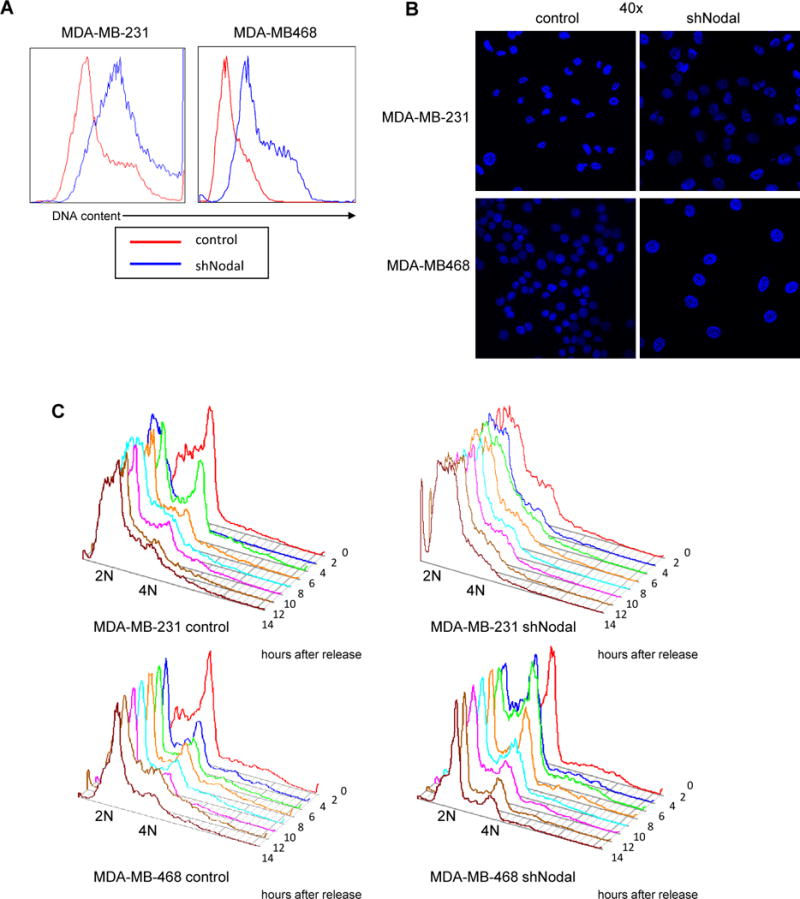
Nodal knockdown alters DNA content and cell cycling in breast cancer cells. A) Representative DNA content analysis of Nodal knockdown or control cells after 2 weeks in culture. B) DAPI staining of control or knockdown nuclei. C) Flow cytometric analysis of cell cycle progression after G2 synchronization by double-thymidine block and nocodozole arrest. Data are representative of 2 independent experiments.
3.1 Changes occur in cell cycle regulators in the absence of Nodal signaling
The observed changes in cell cycle regulators in the absence of Nodal signaling required further validation. Therefore, to confirm the observed G1 arrest, we performed Western blot analyses and observed diminished Cyclin B1 and Mad2 proteins in both cell lines, with concurrent upregulation of p27. We further observed decreased phosphorylation of Rb on residues 780 and 807/811 in the MDA-MB-231 knockdown cell line (Figure 4A). All of these data are consistent with G1 cell cycle arrest. Interestingly, p15, another cell cycle regulator that can be targeted by the TGF-β family, was unaffected. As increased p27 expression is associated with G1 arrest and increased nuclear size, our results correlate with G1 cell cycle arrest in Nodal knockdown cells mediated in part by increased p27 expression. Protein levels of the primary p27 ubiquitin ligase, Skp2, which targets p27 for ubiquitin-mediated degradation, were not changed, suggesting that p27 protein levels are increased by other mechanisms in Nodal knockdown cells.
Figure 4.
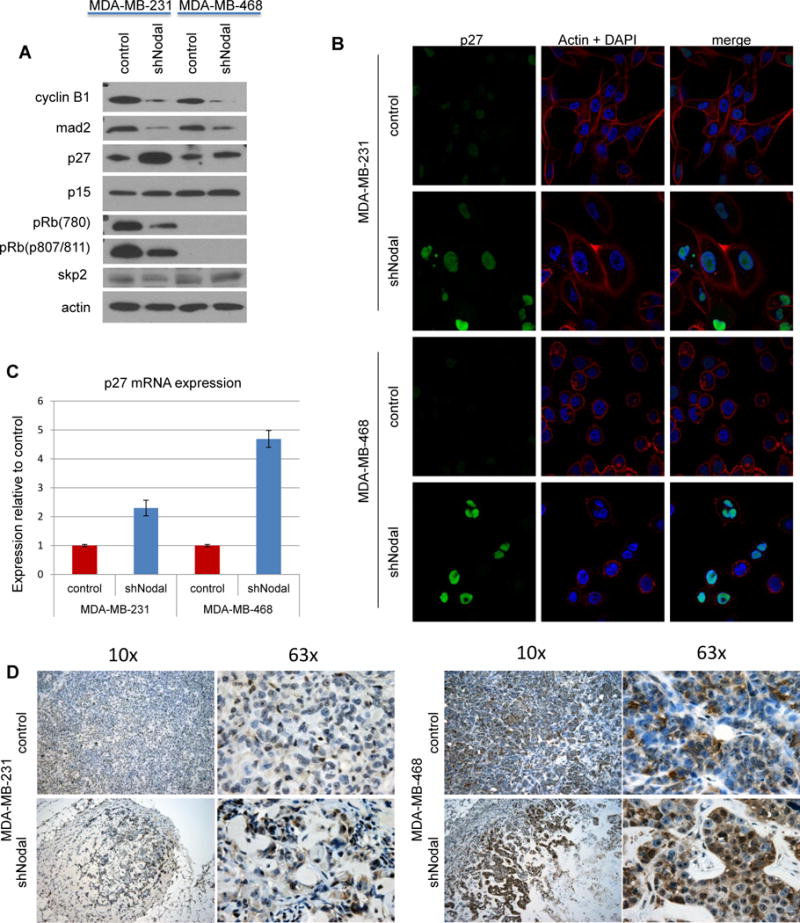
Nodal knockdown cells are arrested in G1 with increased p27 expression. A) Western blot analysis of cell cycle regulatory protein levels. Data are representative of at least 3 experiments. B) Immunofluorescent analysis of p27 localization in breast cancer cells; p27 (green), actin (red) and DAPI (Blue) (40× original magnification). C) QPCR for p27 mRNA expression, normalized to controls. D) Nodal-knockdown and control xenograft tumors were stained for p27 (brown) and counterstained with hematoxylin. (original magnification 10× and 63×)
Additional investigation focused on subcellular localization of p27, which can regulate its stability and function [11]. Immunofluorescence labeling suggests that p27 protein is indeed increased in Nodal knockdown cells and localizes to the nucleus, similar to the localization observed in control cells, suggesting that mislocalization of the protein is not responsible for its increased expression (Figure 4B). Quantitative real-time PCR analysis of p27 mRNA expression demonstrates that increased p27 mRNA in Nodal knockdown cells may contribute to the increased p27 expression observed in the absence of Nodal signaling (Figure 4C). Interestingly, our findings indicate that breast cancer cells may require Nodal signaling to suppress p27 protein levels in vitro, thereby obscuring cell cycle control and permitting proliferation. To determine whether Nodal signaling may regulate p27 during tumorigenesis in vivo, we performed immunohistochemistry for p27 on breast tumors generated in immunocompromised mice with control or Nodal knockdown human breast cancer cells. Consistent with in vitro data, tumors from mice xenografted with Nodal knockdown cells display increased protein expression of p27 when compared with controls, suggesting that Nodal may be required to suppress p27 expression during in vivo tumorigenesis (Figure 4D).
3.2 An inverse regulation of c-myc and p27 occurs in Nodal signaling
P27 functions as a cell cycle inhibitor by sequestering cyclin A/cdk2 and cyclin E/cdk2 complexes, which are then unable to promote progression through G1 and into S phas [11]. To assess whether increased p27 interacts with cyclin E in Nodal knockdown cells, we immunoprecipitated p27 from control and Nodal knockdown cells and performed Western blotting for co-precipitating cyclin E and cdk2 proteins (Figure 5A). Although protein levels of cyclin E and cdk2 are similar between control and knockdown cells, the amount of cyclin E and cdk2 associated with p27 is significantly greater in Nodal knockdown cells than in controls, suggesting that increased sequestration of Cyclin E/cdk2 by p27 may prevent entry into S phase in the absence of Nodal siganling.
Figure 5.
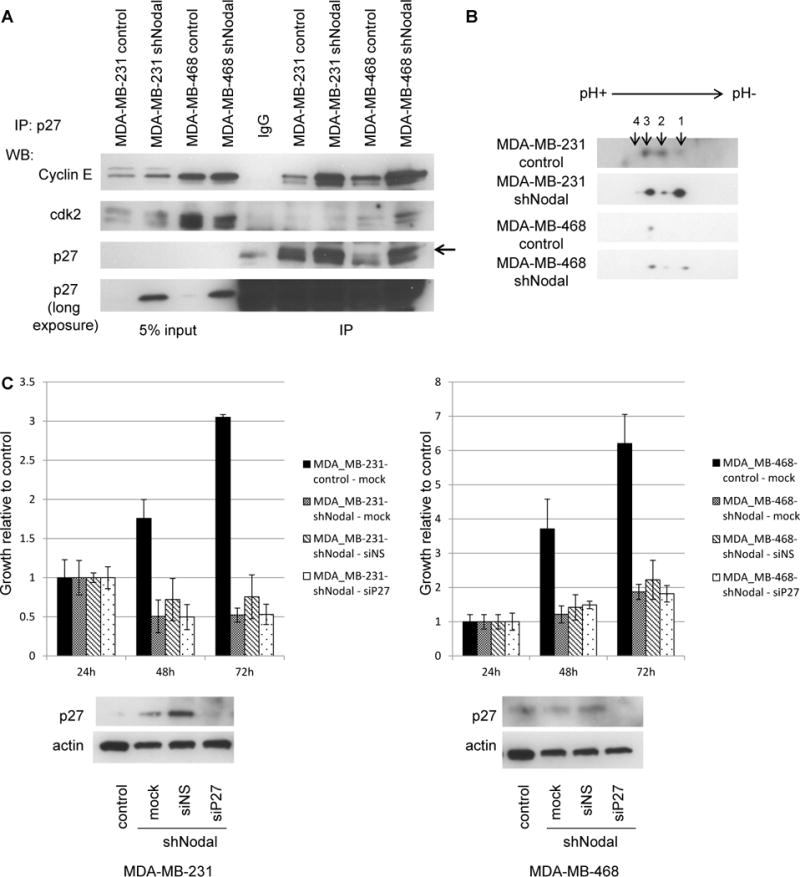
P27 sequesters Cyclin E in Nodal knockdown cells. A) Western blot demonstrating co-immunoprecipitation of p27 with Cdk2 and Cyclin E. B) Two-dimensional western blotting for p27. C) Growth of control or Nodal-knockdown cells in response to siRNA-mediated loss of p27. Lower panels: Western blot analysis of p27 protein levels. All data are representative of 3 experiments.
As a potent cell cycle inhibitor, p27 protein function is regulated by many mechanisms, including transcription, degradation, protein interactions and multiple, complex combinations of post-translational modifications (PTMs). To assess whether Nodal signaling may modulate p27 function post-translationally, in additional to its observed transcriptional effect, we performed two-dimensional Western blot analyses for p27 in control and Nodal knockdown cells (Figure 5B). These experiments demonstrate that p27 PTMs are indeed altered in Nodal knockdown cells, most likely through changes in phosphorylation events [12]. Additionally, a lower ratio of p27 phospho-isoforms 1 and 3 has been shown to correlate with aggressive breast cancer cell behavior [12]. As Nodal knockdown produces an inverted relationship between isoforms 1 and 3 when compared with controls, these results are consistent with our functional data and suggest that Nodal signaling functionally regulates p27 through multiple mechanisms to permit a cancerous phenotype.
To determine whether p27 downregulation is the primary pro-tumor function of Nodal signaling, we attempted to rescue cell growth in Nodal knockdown cells by downregulating p27 expression with siRNAs. Although p27 protein was reduced by siRNA transfection, growth was not restored in Nodal knockdown cells (Figure 5C). The inability of p27 knockdown to rescue cell growth in Nodal knockdown cells may indicate that knockdown cells have entered states of apoptosis or senescence and are thereby unable to respond to p27 knockdown during the period of observation. Alternatively, Nodal signaling may promote a pro-growth program further upstream of p27 and through additional downstream effectors.
Especially noteworthy in this cascade is c-myc, the most commonly activated oncogene in human cancers that regulates thousands of genes involved in tumorigenesis, including p27, by acting as an amplifier of transcriptional change [13]. Because c-myc is also a known transcriptional target of TGFβ family signaling [14], we assessed whether c-myc protein levels are altered in Nodal knockdown breast cancer cells and observed marked downregulated in Nodal knockdown cells (Figure 6A). Interestingly, c-myc mRNA levels are not significantly altered between control and Nodal knockdown cells (Figure 6B), suggesting c-myc may be regulated by post-transcriptional mechanisms downstream of Nodal signaling in breast cancer cells. C-myc is indeed an extremely labile protein and is dynamically regulated by PTMs, including multiple phosphorylation events that control targeting for degradation. By 2-dimensional Western blotting we observed a difference in the c-myc protein profile in Nodal knockdown cells when compared with controls (Figure 6C), evident as leftward shift, and suggesting that Nodal signaling may affect c-myc protein phosphorylation and stability.
Figure 6.
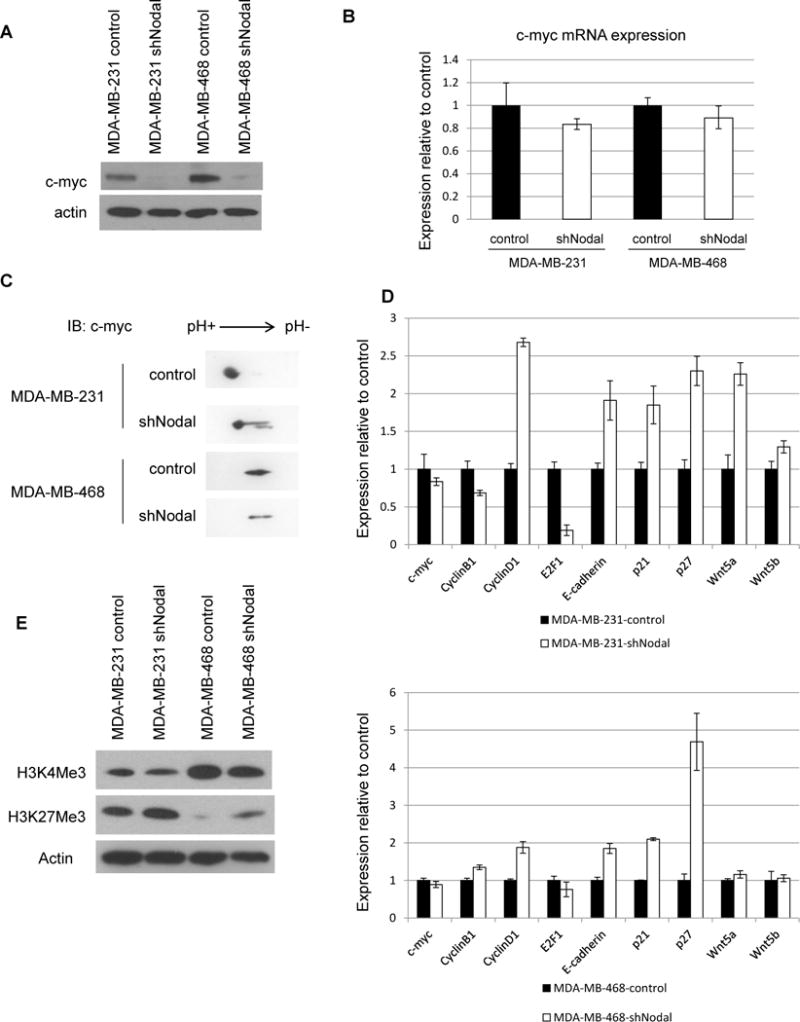
Nodal signaling regulates c-myc expression. A) Western blot analysis of c-myc protein, B) QPCR analysis of c-myc mRNA expression, C) Two-dimensional protein analysis of c-myc, D) Expression of c-myc target genes. Upper panels: MDA-MB-231; lower panel: MDA-MB-468, E) Western blot analysis of c-myc targeted histone modifications. Data are representative of 3 experiments.
To correlate increased c-myc protein downstream of Nodal signaling with transcriptional changes, we performed QPCR analysis of a subset of reported c-myc target genes in breast cancer and observed deregulation of multiple c-myc targets, including CYCLIN D1, p21, p27 and WNT5A in Nodal knockdown cells (Figure 6D). The dramatic impact of c-myc on gene regulation stems in part from its ability to modify chromatin structure. Myc proteins have been shown to recruit many histone modifying enzymes to modulate global transcription in cancer and development [15–17]. To determine whether c-myc-associated histone marks are altered in the absence of Nodal, we performed Western blotting for histone H3 tri-methylated at lysine 4 (H3K4Me3) or lysine 27 (H3K27Me3), marks associated with gain and loss of c-myc transcriptional activity, respectively [18]. We found that H3K4Me3 decreased in the absence of Nodal signaling, while H3K27Me3 was upregulated. These results demonstrate global changes in chromatin structure downstream of Nodal signaling in breast cancer cells, consistent with increased c-myc expression and maintenance of an open, stem-like chromatin landscape (Figure 6E).
To determine the relationship behind c-myc stabilization and p27 destabilization in the absence of Nodal signaling, we investigated well-known relationships between these events and activation of the ERK pathway [12, 19] in breast cancer cells. Active ERK can phosphorylate c-myc protein to stabilize it against degradation and likewise phosphorylate p27 protein to promote its destruction. Additionally, TGFβ family members have been shown to activate ERK signaling. In fact, ERK activity can be detected in breast cancer biopsy samples by immunohistochemistry in areas that also express Nodal (Figure 7A). To assess whether Nodal signaling might affect c-myc and p27 protein stability through the stimulation of the ERK pathway, we stimulated control or Nodal-knockdown cells with Nodal in the presence or absence of U0126, an ERK inhibitor, and examined the effects on c-myc and p27 protein levels by Western blotting (Figure 7B). In both MDA-MB-231 and MDA-MB-468 control cells, Nodal stimulation stabilized c-myc protein and repressed p27, but not in the presence of U0126. In MDA-MB-231 knockdown cells, Nodal stimulation also stabilized c-myc and repressed p27 in an ERK-dependent manner. In contrast to the effect on MDA-MB-468 control cells, Nodal stimulation did not activate ERK or stabilize c-myc in MDA-MB-468 Nodal-knockdown cells, again indicating that with the absence of Nodal, these cells may become progressively less able to respond to Nodal signaling. Interestingly, MDA-MB-468 cells did activate ERK and stabilize c-myc in response to EGF stimulation. Collectively, these data indicate that Nodal signaling stabilizes c-myc and destabilizes p27 by activating the ERK pathway in breast cancer cells.
Figure 7.
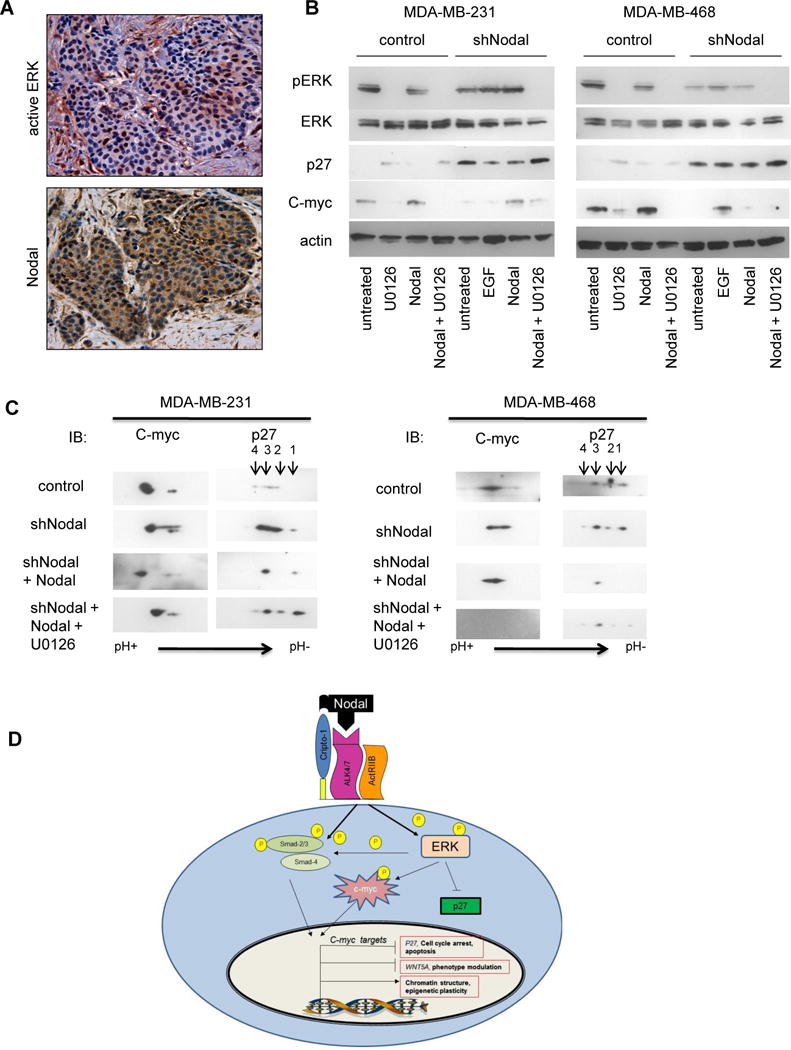
Nodal regulates p27 and c-myc protein levels and post-translational modifications through ERK activation in breast cancer cells. A) A representative immunohistochemistry result shows increased expression (red brown staining) of active ERK in a Nodal positive (brown staining) breast cancer biopsy section (200× original magnification). B) Indicated cells were stimulated with rhNodal (100 ng/ml), U0126 (2μM) or EGF (50 ng/ml) for 6 hours as indicated and protein expression determined by Western blotting. C) Cells were stimulated as indicated and lysates subjected to isoelectric focusing and two-dimensional western blotting. Data are representative of two experiments. D) Targeting Nodal in breast cancer cells disables multiple pro-tumor processes, including ERK activation, downstream stabilization of c-myc, destabilization of p27 and modification of c-myc pro-tumor target gene expression.
In order to correlate Nodal activation of ERK signaling with post-translational modifications to p27 and c-myc proteins, we stimulated Nodal knockdown cells with Nodal in the presence or absence of U0126 and performed two-dimensional Western blotting (Figure 7C). Consistent with our hypothesis, Nodal stimulation produced a shift in isoelectric point, suggesting increased phosphorylation of p27 and c-myc, which was dependent on ERK activation. Interestingly, c-myc protein is completely lost in MDA-MB-468 Nodal knockdown cells treated with U0126, perhaps indicating that c-myc expression is solely dependent on a combination of Nodal and ERK signaling in these cells.
Taken together, our studies demonstrate that Nodal signaling is required for the full malignant potential of triple-negative, basal-like human breast cancer cells, both in vitro and in vivo. Further, our studies suggest that this embryonic morphogen may have widespread impact in coordinating tumor cell plasticity and cell cycle programs through ERK activation and downstream regulation of c-myc and p27 proteins (Figure 7D).
4.0 Molecular cross-talk between Nodal signaling and the Ras-ERK pathway
The Ras-ERK pathway is activated in many human cancers, either by genetic mutations or by overexpression of growth factor receptors and ligands. Once activated, Ras-ERK signaling participates in virtually all of the acquired phenotypes of self-renewal, plasticity and resistance to therapy that characterize aggressive breast cancer. In regulating transcriptional programming, active ERK phosphorylates c-myc at serine 62 to stabilize an otherwise short half-life of the protein. Because c-myc activates transcription of many of the growth factors that stimulate ERK, an autocrine/paracrine loop can be established during cancer progression to autonomously support proliferation and survival. Inhibition of ERK in breast cancer cells leads to rapid loss of c-myc by proteasome-mediated degradation and attenuation of the c-myc driven transcriptional program [31, 33, 34], similar to that observed upon loss of Nodal. While c-myc can be regulated transcriptionally by TGFβ signaling [30], our studies did not reveal significant changes in c-myc mRNA levels. Rather, we found that c-myc is regulated post-translationally by Nodal signaling through the ERK pathway. Interestingly, fewer than 25% of human cancers with high c-myc protein expression harbor mutations in the CMYC gene itself or upregulation of the transcript, implicating post-translational c-myc stabilization mechanisms in cancer [31, 32].
Cell growth and survival are also regulated by Ras-ERK through downstream targets. ERK can activate p15, p16 and p21 to arrest the cell cycle, or cooperate with the PI3K-mTOR pathway to suppress p27 and allow activity of cyclinE/cdk2 complexes and Rb phosphorylation to promote cell cycle entry. P27 is a target of both c-myc and ERK activity and P27 expression correlates negatively with breast tumor grade and prognosis [11]. Like c-myc, p27 seldom harbors mutations in cancer, suggesting cancer cells may require p27 at some point in their development, but may misappropriate control of its activity to permit growth [40]. Notably, another TGF-β family member, TGF-β itself, has been shown to positively regulate p27 protein levels in the early stages of breast cancer development, consistent with the inhibitory role of TGF-β signaling at this stage[44]. In later stages of tumor progression, TGF-β assumes an oncogenic function and cells no longer respond to TGF-β signaling with p27-mediated growth arrest and apoptosis. Although the mechanism underlying this switch in activity remains unknown, changes in ERK activation is a possibility.
In addition to regulating growth, ERK can repress apoptosis in cancer cells by activating BCL2 and BCL-xl and repressing Bim and Bad, stimulate invasion through Rho/Rac activation and mediate cancer cell interactions with the stroma that stimulate angiogenesis. While both MDA-MB-231 and MDA-MB468 cells require Nodal-stimulated ERK activation for growth, their disparate responses to Nodal loss may highlight different oncogenic pathways activated in the two cell types. MDA-MB-468 cells respond to Nodal knockdown with proportionally more apoptosis than do MDA-MB-231 cells, but less inhibited proliferation as seen by BrdU incorporation. As MDA-MB-468 cells harbor a homozygous deletion of Rb and oncogenic activation of PTEN, they may be less sensitive to cell cycle arrest in G1, but dependent on Nodal signaling to integrate ERK and AKT activated pathways and suppress apoptosis.
Conversely, Nodal knockdown in MDA-MB-231 cells slows proliferation and reduces in vivo engraftment capability more than in MDA-MB-468 cells. An explanation may be that p16 is inactivated in MDA-MB-231 cells along with KRAS activation, which may indicate a dependence on circumventing cell cycle inhibitors in G1 and vulnerability to the cell cycle inhibitory function of p27. Furthermore, c-myc signaling is known to regulate WNT5A/B in MDA-MB-231 cells [26], with downstream effects on plasticity and self-renewal, which may be reflected by the diminished ability of Nodal knockdown MDA-MB-231 cells to engraft in vivo.
5.0 Conclusion
ERK activation is in fact essential for the tumorigenicity of triple-negative breast cancers, yet mutations in the pathway are uncommon and the root cause of ERK activation in many of these tumors remains obscure. Our data uniquely demonstrate that Nodal signaling activates the ERK pathway in triple negative breast cancer cells and is required for downstream ERK-dependent phenotypes, including c-myc stabilization, histone modifications and suppression of p27. In the absence of Nodal signaling, breast cancer cells undergo cell cycle arrest, apoptosis and decreased invasiveness, similar to that observed during ERK inhibition [35, 36]. Our working model (Figure 7D) demonstrates the ability of Nodal signaling to activate the Ras-ERK oncogenic signaling pathway in breast cancer and suggests that Nodal may be a widely-relevant therapeutic target to limit the plasticity and renewal of breast cancer cells and additional cancers where Nodal is reactivated.
Acknowledgments
The authors would like to gratefully acknowledge Z. Khalkhali-Ellis, K. Hardy, T. Bodenstine, R.E.B. Seftor and J. Koblinski for helpful discussions. This work was supported by a Susan G. Komen for the Cure post-doctoral fellowship (GK), NIH Oncogenesis and Developmental Biology Training Grant (GK), Eisenberg Research Scholar Fund (LS), the PSOC U54CA143869 and CA121205 (MH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
MH and ES hold a patent for the use of Nodal in cancer diagnostics and Nodal antibodies in cancer therapeutics.
References
- 1.Shen MM. Nodal signaling: developmental roles and regulation. Development. 2007;134(6):1023–34. doi: 10.1242/dev.000166. [DOI] [PubMed] [Google Scholar]
- 2.Al-Hajj M, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charafe-Jauffret E, et al. Cancer stem cells in breast: current opinion and future challenges. Pathobiology. 2008;75(2):75–84. doi: 10.1159/000123845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shipitsin M, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11(3):259–73. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Strizzi L, et al. Potential for the embryonic morphogen Nodal as a prognostic and predictive biomarker in breast cancer. Breast Cancer Res. 14(3):R75. doi: 10.1186/bcr3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strizzi L, et al. Nodal as a biomarker for melanoma progression and a new therapeutic target for clinical intervention. Expert Rev Dermatol. 2009;4(1):67–78. doi: 10.1586/17469872.4.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lonardo E, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 9(5):433–46. doi: 10.1016/j.stem.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Lawrence MG, et al. Reactivation of embryonic nodal signaling is associated with tumor progression and promotes the growth of prostate cancer cells. Prostate. 71(11):1198–209. doi: 10.1002/pros.21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Postovit LM, et al. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc Natl Acad Sci U S A. 2008;105(11):4329–34. doi: 10.1073/pnas.0800467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kossatz U, et al. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev. 2004;18(21):2602–7. doi: 10.1101/gad.321004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nature Reviews Cancer. 2008;8(4):253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 12.Donovan JC, Milic A, Slingerland JM. Constitutive MEK/MAPK activation leads to p27(Kip1) deregulation and antiestrogen resistance in human breast cancer cells. J Biol Chem. 2001;276(44):40888–95. doi: 10.1074/jbc.M106448200. [DOI] [PubMed] [Google Scholar]
- 13.Adhikary S, Eilers M. Transcriptional regulation and transformation by MYC proteins. Nature Reviews Molecular Cell Biology. 2005;6(8):635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 14.Chen CR, et al. E2F4/5 and p107 as Smad cofactors linking the TGF beta receptor to c-myc repression. Cell. 2002;110(1):19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 15.Knoepfler PS, et al. Myc influences global chromatin structure. Embo Journal. 2006;25(12):2723–2734. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin CH, et al. Gene Regulation and Epigenetic Remodeling in Murine Embryonic Stem Cells by c-Myc. PLoS One. 2009;4(11) doi: 10.1371/journal.pone.0007839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang XY, et al. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol Cell. 2008;29(1):102–11. doi: 10.1016/j.molcel.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin CH, et al. Gene regulation and epigenetic remodeling in murine embryonic stem cells by c-Myc. PLoS One. 2009;4(11):e7839. doi: 10.1371/journal.pone.0007839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duncan JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149(2):307–21. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong DJ, Segal E, Chang HY. Stemness, cancer and cancer stem cells. Cell Cycle. 2008;7(23):3622–4. doi: 10.4161/cc.7.23.7104. [DOI] [PubMed] [Google Scholar]
- 21.Ben-Porath I, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40(5):499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143(2):313–24. doi: 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole MD, Henriksson M. 25 years of the c-Myc oncogene. Semin Cancer Biol. 2006;16(4):241. doi: 10.1016/j.semcancer.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Olopade OI. MYC in breast tumor progression. Expert Rev Anticancer Ther. 2008;8(10):1689–98. doi: 10.1586/14737140.8.10.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8(4):253–67. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 26.Roarty K, et al. Loss of TGF-beta or Wnt5a results in an increase in Wnt/beta-catenin activity and redirects mammary tumour phenotype. Breast Cancer Res. 2009;11(2):R19. doi: 10.1186/bcr2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cappellen D, et al. Novel c-MYC target genes mediate differential effects on cell proliferation and migration. EMBO Rep. 2007;8(1):70–6. doi: 10.1038/sj.embor.7400849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinato F, et al. Analysis of Myc-induced histone modifications on target chromatin. PLoS One. 2008;3(11):e3650. doi: 10.1371/journal.pone.0003650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amente S, Lania L, Majello B. Epigenetic reprogramming of Myc target genes. Am J Cancer Res. 1(3):413–418. [PMC free article] [PubMed] [Google Scholar]
- 30.Chen CR, et al. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell. 2002;110(1):19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 31.Vervoorts J, Luscher-Firzlaff J, Luscher B. The ins and outs of MYC regulation by posttranslational mechanisms. J Biol Chem. 2006;281(46):34725–9. doi: 10.1074/jbc.R600017200. [DOI] [PubMed] [Google Scholar]
- 32.Eisenman RN. Deconstructing myc. Genes Dev. 2001;15(16):2023–30. doi: 10.1101/gad928101. [DOI] [PubMed] [Google Scholar]
- 33.Kerkhoff E, et al. Regulation of c-myc expression by Ras/Raf signalling. Oncogene. 1998;16(2):211–6. doi: 10.1038/sj.onc.1201520. [DOI] [PubMed] [Google Scholar]
- 34.Eckert LB, et al. Involvement of Ras activation in human breast cancer cell signaling, invasion, and anoikis. Cancer Res. 2004;64(13):4585–92. doi: 10.1158/0008-5472.CAN-04-0396. [DOI] [PubMed] [Google Scholar]
- 35.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 13(10):616–30. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quail DF, et al. Nodal promotes invasive phenotypes via a mitogen-activated protein kinase-dependent pathway. Oncogene. 2013 doi: 10.1038/onc.2012.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Viglietto G, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8(10):1136–44. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- 38.Komatsu Y, Kaartinen V, Mishina Y. Cell cycle arrest in node cells governs ciliogenesis at the node to break left-right symmetry. Development. 138(18):3915–20. doi: 10.1242/dev.068833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egozi D, et al. Regulation of the cell cycle inhibitor p27 and its ubiquitin ligase Skp2 in differentiation of human embryonic stem cells. FASEB J. 2007;21(11):2807–17. doi: 10.1096/fj.06-7758com. [DOI] [PubMed] [Google Scholar]
- 40.Nickeleit I, et al. p27kip1: a target for tumor therapies? Cell Div. 2007;2:13. doi: 10.1186/1747-1028-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muraoka RS, et al. ErbB2/Neu-induced, cyclin D1-dependent transformation is accelerated in p27-haploinsufficient mammary epithelial cells but impaired in p27-null cells. Mol Cell Biol. 2002;22(7):2204–19. doi: 10.1128/MCB.22.7.2204-2219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy M, et al. Hypoxia regulation of the cell cycle in malignant melanoma: putative role for the cyclin-dependent kinase inhibitor p27. J Cutan Pathol. 2004;31(7):477–82. doi: 10.1111/j.0303-6987.2004.00205.x. [DOI] [PubMed] [Google Scholar]
- 43.Menchon C, Edel MJ, Izpisua Belmonte JC. The cell cycle inhibitor p27Kip(1) controls self-renewal and pluripotency of human embryonic stem cells by regulating the cell cycle, Brachyury and Twist. Cell Cycle. 10(9):1435–47. doi: 10.4161/cc.10.9.15421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Padua D, Massague J. Roles of TGFbeta in metastasis. Cell Res. 2009;19(1):89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 45.Liao DJ, Dickson RB. c-Myc in breast cancer. Endocr Relat Cancer. 2000;7(3):143–64. doi: 10.1677/erc.0.0070143. [DOI] [PubMed] [Google Scholar]