

Abstract
Cannabis has potential therapeutic use but tetrahydrocannabinol (THC), its main psychoactive component, appears as a risk factor for ischemic stroke in young adults. We therefore evaluate the effects of THC on brain mitochondrial function and oxidative stress, key factors involved in stroke. Maximal oxidative capacities V max (complexes I, III, and IV activities), V succ (complexes II, III, and IV activities), V tmpd (complex IV activity), together with mitochondrial coupling (V max/V 0), were determined in control conditions and after exposure to THC in isolated mitochondria extracted from rat brain, using differential centrifugations. Oxidative stress was also assessed through hydrogen peroxide (H2O2) production, measured with Amplex Red. THC significantly decreased V max (−71%; P < 0.0001), V succ (−65%; P < 0.0001), and V tmpd (−3.5%; P < 0.001). Mitochondrial coupling (V max/V 0) was also significantly decreased after THC exposure (1.8±0.2 versus 6.3±0.7; P < 0.001). Furthermore, THC significantly enhanced H2O2 production by cerebral mitochondria (+171%; P < 0.05) and mitochondrial free radical leak was increased from 0.01±0.01 to 0.10±0.01% (P < 0.001). Thus, THC increases oxidative stress and induces cerebral mitochondrial dysfunction. This mechanism may be involved in young cannabis users who develop ischemic stroke since THC might increase patient's vulnerability to stroke.
1. Introduction
Cannabis is now largely considered for its therapeutic potential and is the most widely used recreational drug in the world [1–4]. The principal psychoactive cannabinoid in cannabis is tetrahydrocannabinol (THC), and although cannabis is considered by many people as having few negative side-effects, severe cardiovascular complications were described such as myocardial infarction, sudden death, peripheral arteritis, and stroke [5–7]. Concerning stroke, we described in a prospective series that there was a link between ischemic stroke (IS) and cannabis use in young patients [8].
However, little is known regarding the potential mechanisms implied in cannabis-related stroke. Besides vascular alterations, cannabis might directly affect brain mitochondrial function and oxidative stress, both factors previously shown to be involved in stroke [9–11]. Indeed, mitochondria are a main source of adenosine triphosphate (ATP) production and are particularly involved in the balance between cell survival and cell death. Most cellular energy is obtained through oxidative phosphorylation, a process requiring the action of a set of respiratory enzyme complexes located in the inner mitochondrial membrane. It appears pertinent to study the activities of the different mitochondrial respiratory chain complexes, since mitochondrial respiratory chain complexes activities impairment is observed in many acute and chronic diseases [12–17]. This study is also justified because THC has been previously shown to decrease brain energetic metabolism [18].
Further and interestingly, increased reactive oxygen species (ROS) production leading to oxidative stress is involved in acute IS [9–11, 19, 20]. Oxidative stress not only is a major cause of endothelium dysfunction in the cerebral circulation [21] but can also directly affect mitochondrial function. Thus, as a vital organ rich in mitochondria and with high oxygen use, the brain is prone to oxidative stress and might be particularly sensitive to mitochondrial damage [22].
To challenge the hypothesis that THC might participate to cannabis-related stroke, we first determined the effects of THC on maximal brain mitochondrial respiration rate, and then analyzed precisely complexes I, II, III, and/or IV activities of the mitochondrial respiratory chain together with mitochondrial coupling. We also determined whether THC might increase ROS production in brain mitochondria and measured the mitochondrial free radical leak.
2. Material and Methods
2.1. Material, Reagents and Animals
Synthetic THC (25 mg/mL in ethanol), mitochondrial complexes substrates and/or inhibitors such as glutamate, malate, amytal, ADP, succinate, ascorbate, antimycine, and N,N,N′,N′-tetramethyl-p-phenylenediaminedihydrochloride (tmpd) were acquired from Sigma-Aldrich, France. THC was successively diluted in ethanol as needed and Amplex Red and horseradish peroxidase (HRP) were acquired by Invitrogen. All other chemicals used were of the highest grade commercially available.
Ten male Wistar rats (weight 438 to 500 grams; age 13 weeks) were housed in a neutral temperature environment (22° ± 2°C), on a 12 : 12 hours photoperiod, and were provided food and water ad libitum. This investigation was carried out in accordance with the Helsinki accords for human treatment of animals during experimentation. Rats were submitted to general anesthesia with 3% isoflurane and oxygen (2 L/min) in an induction chamber (Minerve, Esternay, France) and were then decapitated. Brains were excised and cleaned and then immediately used for the study of respiratory parameters.
2.2. Extraction of Brain Mitochondria
Extraction of brain mitochondria was performed as previously reported [23, 24]. All experimental steps were carried on ice. A piece of brain was placed into buffer A containing 50 mM tris, 1 mM ethylene glycol tetraacetic acid (EGTA), 70 mM sucrose, 210 mM mannitol, pH 7.40 at +4°C. Tissues were finely minced with scissors, placed in buffer A and homogenized with a gentleMACS Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany). Then, the homogenate was centrifuged at 1300 ×g for 3 minutes, 4°C. The supernatant was centrifuged at 10000 ×g for 10 minutes, 4°C, to sediment mitochondria. Finally, the mitochondrial pellet was washed twice and then suspended in 50 mM Tris, 70 mM sucrose, and 210 mM mannitol, pH 7.4 at +4°C. Protein content was routinely assayed with a Bradford assay using bovine serum albumin (BSA) as a standard [24].
2.3. Dose-Effect Curve of THC on Brain Mitochondria Maximal Oxidative Capacity
Before measurement, 3 mL of solution M containing 100 mM KCl, 50 mM Mops, 1 mM EGTA, 5 mMKpi, and 1 mg/mL defatted BSA was added to the oxygraph chambers for 10 minutes. Then, isolated mitochondria of brain (0.30 mg) were placed in the oxygraph chambers (using Clark oxygen electrodes) with glutamate (5 mM) and malate (5 mM) as substrates. The temperature was maintained at +25°C.
Adenosine diphosphate (ADP, 2 mM) was then added to measure maximal oxidative capacity (V max). After 1.5 minutes, increasing concentrations of THC were added to the respiration solution. The ranges of THC concentrations were 10−5, 2∗10−5, 3∗10−5, 4∗10−5, 5∗10−5, and 6∗10−5 M. The objective was to determine the dilution of THC in ethanol needed to inhibit at least 50% of the maximal mitochondrial respiration (V max) and the half maximal inhibitory concentration (IC50).
2.4. Mitochondrial Coupling Determination
After the determination of the basal oxygen consumption (V 0), the maximal brain mitochondrial respiration rates were measured in the presence of ADP as a phosphate acceptor (V max). The degree of coupling between oxidation and phosphorylation was inferred from the acceptor control ratio V max/V 0 (ACR).
2.5. Mitochondrial Respiratory Chain Complexes Activities
When V max was recorded, electron flow went through complexes I, III, and IV [25]. Complex I was blocked with amytal (0.02 mM) and complex II was stimulated with succinate (25 mM). Mitochondrial respiration in these conditions allowed determining complexes II, III, and IV activities (V succ).
After that, complex III was blocked by antimycine (1.8 μg/mL) and tmpd (0.5 mM) and ascorbate (0.5 mM) were added as artificial electron donors to cytochrome c. In these conditions, the activity of cytochrome c oxydase (complex IV) was determined as an isolated step of the respiratory chain (V tmpd).
In order to evaluate the effect of THC on the different mitochondrial respiratory chain complexes activities, THC was added 90 sec respectively after V max, V succ, and V tmpd. We used the dilution of THC determined by the dose-effect curve which inhibited at least 50% of V max. Forty-five independent analyses of mitochondrial respiration were performed. Each mean value was obtained from 15 independent measurements in triplo.
2.6. Measurements of the Production of H2O2 by Brain Mitochondria
Production of H2O2 with and without THC was measured with Amplex Red which reacted with H2O2 in a 1 : 1 stoichiometry catalyzed by HRP to yield the fluorescent compound resorufin and a molar equivalent of O2 [26, 27]. Resorufin has excitation and emission wavelengths of 563 nm and 587 nm, respectively, and is extremely stable once formed. Fluorescence was measured continuously with a Fluoromax 3 (Jobbin Yvon) spectrofluorometer with temperature control and magnetic stirring. After a baseline (reactants only) was established, the reaction was initiated by adding brain isolated mitochondria (0.30 mg) to 600 μL of a buffer (KCl 125 mM, KH2PO4 4 mM, NaCl 14 mM, HEPES-NaOH 20 mM, MGCl2 1 mM, EGTA 0.020 mM, and 0.2% fatty acids free BSA, pH 7.2).
H2O2 production was first determined with glutamate (10 mM) and malate (2 mM), with ADP (2 mM) used as substrates. We added THC and compared H2O2 production without and with THC. The results were reported in pmol/min/mg protein. Values are the means ± SEM of 12 independent experiments.
2.7. Measurement of the Mitochondrial Free Radical Leak (FRL)
H2O2 production and O2 consumption were measured in parallel in the same sample under similar experimental conditions. This allowed the calculation of the fraction of electrons out of sequence which reduce O2 to ROS in the respiratory chain (the percent of free radical leak) instead of reaching cytochrome oxidase to reduce O2 to water [12, 26, 27]. Because 2 electrons are needed to reduce 1 mole of O2 to H2O2, whereas 4 electrons are transferred in the reduction of 1 mole of O2 to water, the percent free radical leak was calculated as the rate of H2O2 production divided by two times the rate of O2 consumption, and the result was multiplied by 100. The FRL was calculated before and after THC exposure.
2.8. Statistical Analysis
Results are expressed as mean ± SEM. Statistical analyses were performed using Student's t-test, one-way repeated measures, or two-way ANOVA followed by Tukey's posttest (GraphPad Prism 5, Graph Pad Software, Inc., San Diego, CA, USA). Statistical significance was displayed as * P < 0.05 or ** P < 0.01 or *** P < 0.001.
3. Results
3.1. Dose-Dependent Inhibition of Maximal Brain Mitochondrial Respiration by THC
Dose-dependent inhibition of respiration by THC was demonstrated in the maximal oxidative capacities (V max) study and data were fit to a 4-parameter sigmoidal dose-response model to determine half-maximal inhibitory concentration (IC50) values. THC exhibited an apparent IC50 value of about 2∗10−5 M (Figure 1).
Figure 1.
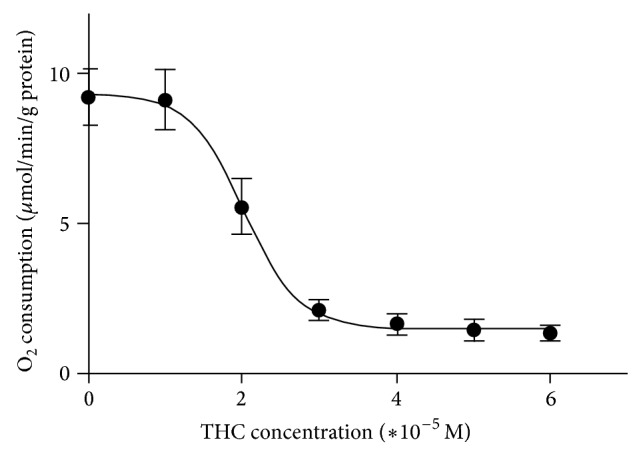
THC decreased brain mitochondrial maximal oxidative capacity: dose-response curve. Effects of ranges concentrations of THC (10−5 to 6∗10−5 M) on brain mitochondrial maximal oxygen consumption, measured using glutamate and malate as substrates. Values are expressed in μmol/min/g protein.
3.2. THC Impaired All Complexes of the Brain Mitochondrial Respiratory Chain and Decreases Mitochondrial Coupling
Figure 2 represents brain mitochondrial respiratory chain complexes activities before and after 3∗10−5 M of THC exposure. Baseline activities are represented by V max, V succ, and V tmpd and mitochondrial coupling. The maximal oxidative capacities, V max (n = 15), reflecting complexes I, III, and IV activities was 13.9 ± 1.3 μmol O2/min/g protein. V succ (n = 15), reflecting complexes II, III, and IV activities, was 8.7 ± 1.4 μmol O2/min/g protein. V tmpd (n = 15), reflecting complex IV activity, was 29.9 ± 0.9 μmol O2/min/g protein.
Figure 2.
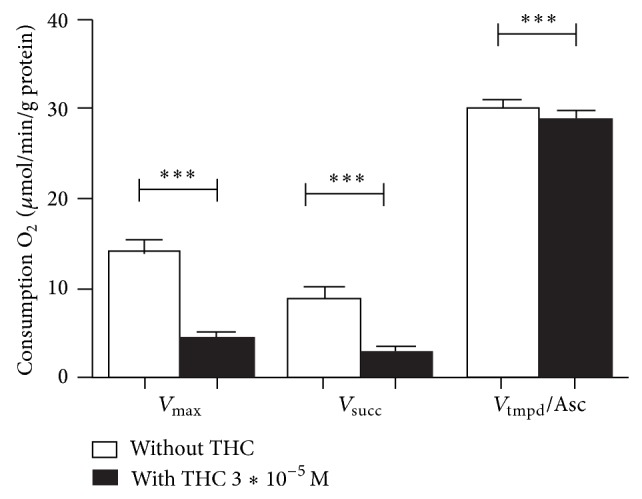
THC impaired complexes I, II, III, and IV activities of the brain mitochondrial respiratory chain. Effects of 3∗10−5 M of THC (black graphs) on brain mitochondrial respiratory chain complexes activities as compared to control values (white graphs). V max reflects complexes I, III, and IV activities and is measured using ADP. V succ reflects complexes II, III, and IV activities and is measured using succinate. V tmpd reflects complex IV activity and is measured using tmpd and ascorbate as mitochondrial substrates. THC: tetrahydrocannabinol. Data are means ± SEM and *** P < 0.001.
V max (n = 15), V succ (n = 15), and V tmpd (n = 15) were significantly decreased after exposure to THC 3∗10−5 M in brain isolated mitochondria (resp., 4.4 ± 0.7 μmol O2/min/g protein versus 13.9 ± 1.3; P < 0.001; 2.8 ± 0.5 versus 8.7 ± 1.4 μmol O2/min/g protein; P < 0.001; 28.8 ± 1 versus 29.9 ± 0.9 μmol O2/min/g protein; P < 0.001 with and without exposure, resp.).
The effect of THC on the respiratory chain might be linked to an effect on complexes I, II, and III rather than on complex IV, because THC reduced V max by 71%, which reflects I, III, and IV activities, reduced V succ by 68%, which reflects complexes II, III, and IV activities and reduced V tmpd by 3.5% which reflects complex IV activity.
Finally, mitochondrial coupling (V max/V o) was also significantly decreased after adjunction of THC (1.8 ± 0.2 versus 6.3 ± 0.7; P < 0.001).
3.3. THC Increased Brain H2O2 Production
To assess if the decreased brain mitochondria respiration induced by THC was related to the generation of ROS, we determined H2O2 production by brain mitochondria without and with THC in a concentration of 3∗10−5 M or 10−4 M on 12 independent experiments. The addition of 3∗10−5 M of THC and 10−4 M of THC significantly increased H2O2 by, respectively, 171% and 371% in comparison with baseline production (6.7 ± 0.8 versus 3.9 ± 0.4 pmol/min/mg; P < 0.05 and 14.1 ± 1.4 versus 3.9 ± 0.4 pmol/min/mg; P < 0.001) (Figure 3).
Figure 3.
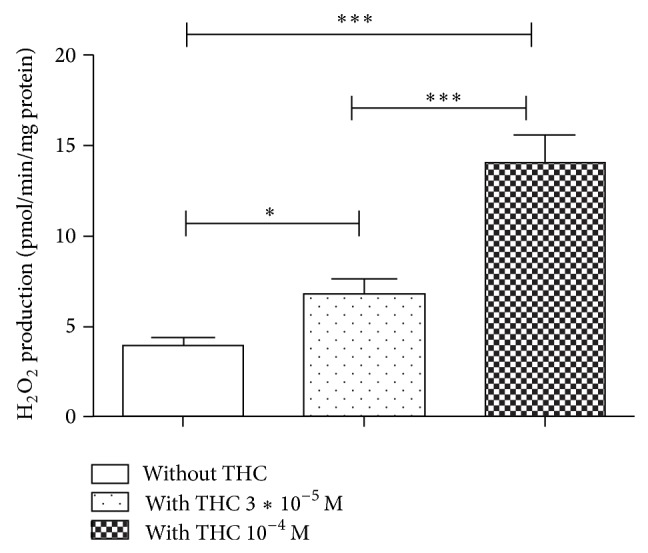
THC increased brain mitochondrial H2O2 production. Effects of 3∗10−5 M and 10−4 M of THC on brain mitochondria H2O2 production, as compared to control values (white graph). THC: tetrahydrocannabinol. Values are expressed in pmol/min/mg protein.
3.4. THC Increased the Mitochondrial Free Radical Leak in the Brain
The mitochondrial free radical leak (FRL) was increased after THC addition from 0.01 ± 0.01 to 0.10 ± 0.01%, P < 0.001 (Figure 4).
Figure 4.
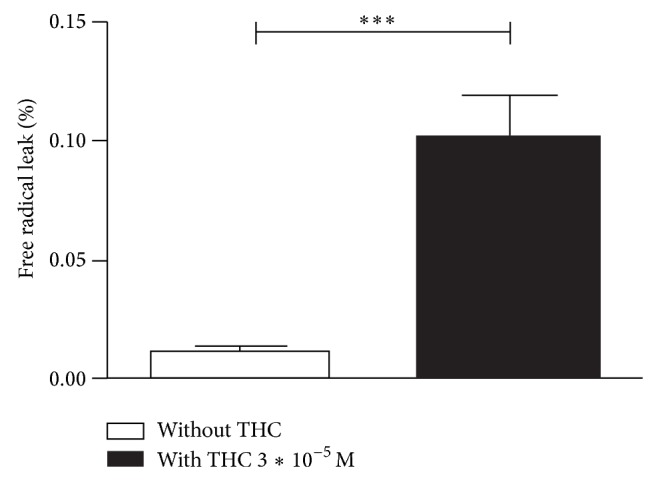
THC increased the free radical leak in brain mitochondria. Effect of 3∗10−5 M of THC on the free radical leak (FRL). FRL corresponds to the fraction of electrons out of sequence which reduces O2 to ROS in the respiratory chain (the percent of free radical leak) instead of reaching cytochrome oxidase. THC: tetrahydrocannabinol. Control FRL without THC (white graph) and FRL after THC exposure (black graph).
4. Discussion
The main results of this study are to show that THC has a direct dose-dependent toxic effect on brain mitochondria and to demonstrate for the first time that THC mainly inhibits complexes I, II, and III of the mitochondrial respiratory chain and decreases mitochondrial coupling. Furthermore, THC increases ROS production by the brain, which likely participates in its toxicity.
Cannabis is the most frequent illicit substance used in the world and has been associated with cardiovascular complications, especially stroke in young adults [6, 7]. Vascular effects might be involved and cerebral arterial stenoses have been observed but this could not be the sole mechanism and the precise actions of cannabis on brain in patients who develop a stroke are not determined. Particularly, THC, which is the main component of cannabis, has been shown to decrease mitochondrial oxygen consumption in oral cancer cells [28] and in human sperm [29]. In only few experimental reports, THC has been described to induce mitochondrial dysfunction in vitro, reducing oxygen consumption on several organs including the heart [30, 31], the liver [32], the skeletal muscle [33], or the brain [31, 33].
Recently, synthetic cannabinoids (known as K2) has been shown to result in ischemic stroke [34], supporting the need to further evaluate the potential toxicity of synthetic THC on brain mitochondria. In the present study, isolated mitochondria were incubated with different concentrations of THC and we demonstrated that THC globally impaired mitochondrial respiratory chain complexes activities. This is in accordance with the literature, similar results being observed in vitro on the brain of mice and rat [31, 33].
To go further, we analyzed the specific cellular effect of THC on different complexes of the mitochondrial respiratory chain. We showed for the first time that THC significantly reduced V max (71% inhibition) reflecting complexes I, III, and IV activities, V succ (68% inhibition) reflecting II, III, and IV activities, and more slightly V tmpd reflecting complex IV activity. Thus, THC has a main deleterious effect on complexes I, II, and III of the mitochondrial respiratory chain. This is globally consistent with the data of Athanasiou et al. showing on heart mitochondria in rat that THC may affect complexes I, II, and III depending on the concentration of THC used [30].
Additionally, we observed that mitochondrial coupling was decreased after THC adjunction, further supporting its toxic effects on the brain.
To get further knowledge on the mechanism potentially involved in the deleterious effect of THC on brain mitochondria, we determined ROS production as inferred by hydrogen peroxide change. Interestingly, THC induced a significant production of ROS (+171%). Since mitochondria are both causes and targets of ROS, mitochondrial production of H2O2 might be increased as a result of cannabis-related mitochondrial dysfunction.
Accordingly, the free radical leak increased after THC exposure, supporting that the fraction of electrons which reduce O2 to ROS in the respiratory chain [35] were greater in presence of cannabis. Thus, mitochondria likely participated in the ROS overproduction seen in the presence of THC.
This appears important since oxidative stress is a pathophysiological mechanism involved in stroke and since the brain is particularly vulnerable to oxidative stress with few protective antioxidant mechanisms [22]. From another point of view, Bartova and Birmingham reported that THC reduces NADPH activity, suggesting a decreased ROS production [31]. However the author did not measure directly ROS synthesis in the mitochondria. In our study we found both an altered function of the mitochondrial respiratory chain and an increased synthesis of H2O2 when using THC. Taken all data in consideration, one could speculate that THC might have different effects on various sources of ROS production and/or on ROS antioxidant defense and that the balance leads to an increased oxidative stress.
Our data are likely to be pertinent in the clinical setting since THC and its main metabolite 11-hydroxy-delta9-THC are highly lipophilic and cross the blood-brain barrier [36]. Thus, THC concentration has been observed in animals [36] and human brains [37, 38]. Accordingly, although the effects of THC on brain respiration is controversial after a single intraperitoneal dose [18, 39, 40], Costa and Colleoni reported a decrease in oxygen consumption when repeated doses of THC were administrated [18]. Such decrease in oxygen consumption in chronic treatment by THC indicated low ATP production, little disposable energy and consequent neuronal damage. In favor of this hypothesis, we observed in our previous prospective study that all patients who developed a stroke related to cannabis use were chronic abusers [8]. Further, inhaled marijuana smoke has been shown to disrupt mitochondrial energetics in pulmonary epithelial cells in vivo [41].
Taken together, these data suggest that the deleterious effect of THC on brain mitochondria could be linked to its ability to generate oxidative stress and that this mechanism may be involved in young cannabis users who develop a stroke.
This study presents some limitations. Although the in vitro approach gives a better control of the amount of THC that actually reaches the mitochondria itself, to approach real life conditions of cannabis consumption needs THC administration in vivo. Accordingly, intraperitoneal administration of THC allowed assessing attention functions or addiction in rats [42, 43]. Such route may nevertheless lead to controversial results when mitochondrial function is examined [18, 39, 40]. Thus, further studies aiming to closely resemble the effects of cannabis in the brain in vivo are needed, investigating not only intraperitoneal but also intravenous and/or inhalation routes often used by humans.
5. Conclusion
THC exposure alters brain maximal oxidative capacity. It impairs mainly the complexes I, II, and III of the mitochondrial respiratory chain and mitochondrial coupling. THC also increases brain ROS production and mitochondrial free radical leak.
Both mitochondrial dysfunction and oxidative stress are key events during stroke, suggesting that THC might increase patient's vulnerability to stroke and that further investigations would be helpful to determine whether mitochondrial protection and antioxidants might decrease THC-related neuronal damage in cannabis-induced stroke.
Acknowledgments
The authors thank Fabienne Goupilleau, Isabelle Bentz, and Anne-Marie Kasprowicz for their expert biological, technical, and secretarial assistances. They also thank Rodrigue Galani Ph.D. holder for writing assistance.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Volkow N. D., Baler R. D., Compton W. M., Weiss S. R. B. Adverse health effects of marijuana use. The New England Journal of Medicine. 2014;370(23):2219–2227. doi: 10.1056/NEJMra1402309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ben Amar M. Cannabinoids in medicine: a review of their therapeutic potential. Journal of Ethnopharmacology. 2006;105(1-2):1–25. doi: 10.1016/j.jep.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Fowler C. J. Plant-derived, synthetic and endogenous cannabinoids as neuroprotective agents: non-psychoactive cannabinoids, “entourage” compounds and inhibitors of N-acyl ethanolamine breakdown as therapeutic strategies to avoid pyschotropic effects. Brain Research Reviews. 2003;41(1):26–43. doi: 10.1016/S0165-0173(02)00218-7. [DOI] [PubMed] [Google Scholar]
- 4.Caldicott D. G. E., Holmes J., Roberts-Thomson K. C., Mahar L. Keep off the grass: marijuana use and acute cardiovascular events. European Journal of Emergency Medicine. 2005;12(5):236–244. doi: 10.1097/00063110-200510000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Thanvi B. R., Treadwell S. D. Cannabis and stroke: Is there a link? Postgraduate Medical Journal. 2009;85(1000):80–83. doi: 10.1136/pgmj.2008.070425. [DOI] [PubMed] [Google Scholar]
- 6.Wolff V., Armspach J.-P., Lauer V., et al. Cannabis-related stroke: myth or reality? Stroke. 2013;44(2):558–563. doi: 10.1161/STROKEAHA.112.671347. [DOI] [PubMed] [Google Scholar]
- 7.Thomas G., Kloner R. A., Rezkalla S. Adverse cardiovascular, cerebrovascular, and peripheral vascular effects of marijuana inhalation: what cardiologists need to know. The American Journal of Cardiology. 2014;113(1):187–190. doi: 10.1016/j.amjcard.2013.09.042. [DOI] [PubMed] [Google Scholar]
- 8.Wolff V., Lauer V., Rouyer O., et al. Cannabis use, ischemic stroke, and multifocal intracranial vasoconstriction: a prospective study in 48 consecutive young patients. Stroke. 2011;42(6):1778–1780. doi: 10.1161/STROKEAHA.110.610915. [DOI] [PubMed] [Google Scholar]
- 9.Chen H., Yoshioka H., Kim G. S., et al. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxidants & Redox Signaling. 2011;14(8):1505–1517. doi: 10.1089/ars.2010.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olmez I., Ozyurt H. Reactive oxygen species and ischemic cerebrovascular disease. Neurochemistry International. 2012;60(2):208–212. doi: 10.1016/j.neuint.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Sims N. R., Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochimica et Biophysica Acta: Molecular Basis of Disease. 2010;1802(1):80–91. doi: 10.1016/j.bbadis.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Kindo M., Gerelli S., Bouitbir J., et al. Pressure overload-induced mild cardiac hypertrophy reduces left ventricular transmural differences in mitochondrial respiratory chain activity and increases oxidative stress. Frontiers in Physiology. 2012;3(article 332) doi: 10.3389/fphys.2012.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mansour Z., Charles A. L., Kindo M., et al. Remote effects of lower limb ischemia-reperfusion: impaired lung, unchanged liver and stimulated kidney oxidative capacities. BioMed Research International. 2014;2014:7. doi: 10.1155/2014/392390.392390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charles A.-L., Guilbert A.-S., Bouitbir J., et al. Effect of postconditioning on mitochondrial dysfunction in experimental aortic cross-clamping. British Journal of Surgery. 2011;98(4):511–516. doi: 10.1002/bjs.7384. [DOI] [PubMed] [Google Scholar]
- 15.Meyer A., Zoll J., Charles A. L., et al. Skeletal muscle mitochondrial dysfunction during chronic obstructive pulmonary disease: Ccentral actor and therapeutic target. Experimental Physiology. 2013;98(6):1063–1078. doi: 10.1113/expphysiol.2012.069468. [DOI] [PubMed] [Google Scholar]
- 16.Duteil D., Chambon C., Ali F., et al. The transcriptional coregulators TIF2 and SRC-1 regulate energy homeostasis by modulating mitochondrial respiration in skeletal muscles. Cell Metabolism. 2010;12(5):496–508. doi: 10.1016/j.cmet.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garnier A., Zoll J., Fortin D., et al. Control by circulating factors of mitochondrial function and transcription cascade in heart failure: a role for endothelin-1 and angiotensin II. Circulation: Heart Failure. 2009;2(4):342–350. doi: 10.1161/CIRCHEARTFAILURE.108.812099. [DOI] [PubMed] [Google Scholar]
- 18.Costa B., Colleoni M. Changes in rat brain energetic metabolism after exposure to anandamide or delta9-tetrahydrocannabinol. European Journal of Pharmacology. 2000;395(1):1–7. doi: 10.1016/S0014-2999(00)00170-9. [DOI] [PubMed] [Google Scholar]
- 19.Cojocaru I. M., Cojocaru M., Sapira V., Ionescu A. Evaluation of oxidative stress in patients with acute ischemic stroke. Romanian Journal of Internal Medicine. 2013;51(2):97–106. [PubMed] [Google Scholar]
- 20.Manzanero S., Santro T., Arumugam T. V. Neuronal oxidative stress in acute ischemic stroke: sources and contribution to cell injury. Neurochemistry International. 2013;62(5):712–718. doi: 10.1016/j.neuint.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 21.Chrissobolis S., Miller A. A., Drummond G. R., Kemp-Harper B. K., Sobey C. G. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Frontiers in Bioscience. 2011;16(5):1733–1745. doi: 10.2741/3816. [DOI] [PubMed] [Google Scholar]
- 22.Popa-Wagner A., Mitran S., Sivanesan S., Chang E., Buga A.-M. ROS and brain diseases: the good, the bad, and the ugly. Oxidative Medicine and Cellular Longevity. 2013;2013:14. doi: 10.1155/2013/963520.963520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baratli Y., Charles A.-L., Wolff V., et al. Impact of iron oxide nanoparticles on brain, heart, lung, liver and kidneys mitochondrial respiratory chain complexes activities and coupling. Toxicology in Vitro. 2013;27(8):2142–2148. doi: 10.1016/j.tiv.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 24.Baratli Y., Charles A.-L., Wolff V., et al. Age Modulates Fe3O4 nanoparticles liver toxicity: dose-dependent decrease in mitochondrial respiratory chain complexes activities and coupling in middle-aged as compared to young rats. BioMed Research International. 2014;2014:10. doi: 10.1155/2014/474081.474081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thaveau F., Zoll J., Rouyer O., et al. Ischemic preconditioning specifically restores complexes I and II activities of the mitochondrial respiratory chain in ischemic skeletal muscle. Journal of Vascular Surgery. 2007;46(3):541–547. doi: 10.1016/j.jvs.2007.04.075. [DOI] [PubMed] [Google Scholar]
- 26.Anderson E. J., Neufer P. D. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H2O2 generation. The American Journal of Physiology—Cell Physiology. 2006;290(3):C844–C851. doi: 10.1152/ajpcell.00402.2005. [DOI] [PubMed] [Google Scholar]
- 27.Picard M., Ritchie D., Thomas M. M., Wright K. J., Hepple R. T. Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell. 2011;10(6):1047–1055. doi: 10.1111/j.1474-9726.2011.00745.x. [DOI] [PubMed] [Google Scholar]
- 28.Whyte D. A., Al-Hammadi S., Balhaj G., Brown O. M., Penefsky H. S., Souid A.-K. Cannabinoids inhibit cellular respiration of human oral cancer cells. Pharmacology. 2010;85(6):328–335. doi: 10.1159/000312686. [DOI] [PubMed] [Google Scholar]
- 29.Badawy Z. S., Chohan K. R., Whyte D. A., Penefsky H. S., Brown O. M., Souid A.-K. Cannabinoids inhibit the respiration of human sperm. Fertility and Sterility. 2009;91(6):2471–2476. doi: 10.1016/j.fertnstert.2008.03.075. [DOI] [PubMed] [Google Scholar]
- 30.Athanasiou A., Clarke A. B., Turner A. E., et al. Cannabinoid receptor agonists are mitochondrial inhibitors: a unified hypothesis of how cannabinoids modulate mitochondrial function and induce cell death. Biochemical and Biophysical Research Communications. 2007;364(1):131–137. doi: 10.1016/j.bbrc.2007.09.107. [DOI] [PubMed] [Google Scholar]
- 31.Bartova A., Birmingham M. K. Effect of Δ9 tetrahydrocannabinol on mitochondrial NADH oxidase activity. Journal of Biological Chemistry. 1976;251(16):5002–5006. [PubMed] [Google Scholar]
- 32.Mahoney J. M., Harris R. A. Effect of delta9-tetrahydrocannabinol on mitochondrial processes. Biochemical Pharmacology. 1972;21(9):1217–1226. doi: 10.1016/0006-2952(72)90283-3. [DOI] [PubMed] [Google Scholar]
- 33.Chiu P., Karler R., Craven C. The influence of Δ9 tetrahydrocannabinol, cannabinol and cannabidiol on tissue oxygen consumption. Research Communications in Chemical Pathology and Pharmacology. 1975;12(2):267–286. [PubMed] [Google Scholar]
- 34.Freeman M. J., Rose D. Z., Myers M. A., Gooch C. L., Bozeman A. C., Scott Burgin W. Ischemic stroke after use of the synthetic marijuana ‘spice’. Neurology. 2013;81(24):2090–2093. doi: 10.1212/01.wnl.0000437297.05570.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson E. J., Lustig M. E., Boyle K. E., et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. Journal of Clinical Investigation. 2009;119(3):573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ameri A. The effects of cannabinoids on the brain. Progress in Neurobiology. 1999;58(4):315–348. doi: 10.1016/S0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- 37.Brunet B., Hauet T., Hébrard W., Papet Y., Mauco G., Mura P. Postmortem redistribution of THC in the pig. International Journal of Legal Medicine. 2010;124(6):543–549. doi: 10.1007/s00414-009-0403-2. [DOI] [PubMed] [Google Scholar]
- 38.Giroud C., Michaud K., Sporkert F., et al. A fatal overdose of cocaine associated with coingestion of marijuana, buprenorphine, and fluoxetine. Body fluid and tissue distribution of cocaine and its metabolites determined by hydrophilic interaction chromatography-mass spectrometry (HILIC-MS) Journal of Analytical Toxicology. 2004;28(6):464–474. doi: 10.1093/jat/28.6.464. [DOI] [PubMed] [Google Scholar]
- 39.Dembert M. L., Harclerode J. Effects of 1-Δ9-tetrahydrocannabinol, dl-amphetamine and pentobarbital on oxygen consumption by mouse brain and heart homogenates. Biochemical Pharmacology. 1974;23(5):947–956. doi: 10.1016/0006-2952(74)90025-2. [DOI] [PubMed] [Google Scholar]
- 40.Nazar B. L., Harclerode J., Roth R. I., Butler R. C. Acquisition of tolerance to Δ 9 THC as measured by the response of a cellular function. Life Sciences. 1974;14(12):2513–2520. doi: 10.1016/0024-3205(74)90148-9. [DOI] [PubMed] [Google Scholar]
- 41.Sarafian T. A., Habib N., Oldham M., et al. Inhaled marijuana smoke disrupts mitochondrial energetics in pulmonary epithelial cells in vivo. The American Journal of Physiology—Lung Cellular and Molecular Physiology. 2006;290(6):L1202–L1209. doi: 10.1152/ajplung.00371.2005. [DOI] [PubMed] [Google Scholar]
- 42.Verrico C. D., Jentsch J. D., Roth R. H., Taylor J. R. Repeated, intermittent Δ9-tetrahydrocannabinol administration to rats impairs acquisition and performance of a test of visuospatial divided attention. Neuropsychopharmacology. 2004;29(3):522–529. doi: 10.1038/sj.npp.1300316. [DOI] [PubMed] [Google Scholar]
- 43.Panlilio L. V., Zanettini C., Barnes C., Solinas M., Goldberg S. R. Prior exposure to THC increases the addictive effects of nicotine in rats. Neuropsychopharmacology. 2013;38(7):1198–1208. doi: 10.1038/npp.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]