

Abstract
An S. maltophilia strain named WJ66 was isolated from a patient; WJ66 showed resistance to more antibiotics than the other S. maltophilia strains. This bacteraemia is resistant to sulphonamides, or fluoroquinolones, while the representative strain of S. maltophilia, K279a, is sensitive to both. To explore drug resistance determinants of this strain, the draft genome sequence of WJ66 was determined and compared to other S. maltophilia sequences. Genome sequencing and genome-wide evolutionary analysis revealed that WJ66 was highly homologous with the strain K279a, but strain WJ66 contained additional antibiotic resistance genes. Further analysis confirmed that strain WJ66 contained an amino acid substitution (Q83L) in fluoroquinolone target GyrA and carried a class 1 integron, with an aadA2 gene in the resistance gene cassette. Homology analysis from the pathogen-host interaction database showed that strain WJ66 lacks raxST and raxA, which is consistent with K279a. Comparative genomic analyses revealed that subtle nucleotide differences contribute to various significant phenotypes in close genetic relationship strains.
1. Introduction
Stenotrophomonas maltophilia (S. maltophilia) is an aerobic Gram-negative bacillus ubiquitous in the natural and hospital environment [1]. In addition, S. maltophilia is emerging as a global multidrug resistant (MDR) human opportunistic pathogen. In a clinical environment, S. maltophilia has been isolated from several sources, including the suction system tubing of dental chair units, contaminated endoscopes, and tap water, all of which present possible patient exposure sources. S. maltophilia contaminated central venous catheters and tap water faucets have been implicated in cutaneous and soft tissue infections in patients with neutropenia [2].
S. maltophilia is most commonly associated with respiratory and other serious infections, especially in immunocompromised patients [3]. In polymicrobial infections, S. maltophilia was present in 45.8% of patients [4]. S. maltophilia is being isolated with increasing frequency from patients with intensive care unit (ICU) associated infections [5]. ICU-acquired infections related to S. maltophilia were independently associated with increased morbidity and mortality [6, 7]. Specifically, early reports showed that this important nosocomial pathogen is associated with crude mortality rates ranging from 14 to 69% in patients with bacteraemia and the ICU mortality rate attributed to bacteraemia was 16.7% according to an 11 years review [4, 8, 9].
Since the first genome of S. maltophilia K279a was reported in 2008 [10]; bioprojects of 48 S. maltophilia strains can be found in NCBI. However, the majority of these strains are not available.
Here, we isolated a S. maltophilia strain from a human blood sample, named WJ66. This strain exhibited resistance to more antibiotics than the other S. maltophilia strains. Drug resistance test showed that S. maltophilia WJ66 is resistant to nearly all drugs, including sulphonamides and fluoroquinolones. We present the draft genome sequence of S. maltophilia WJ66 and conduct a comparison genomic and phenotypic analysis. Genome sequencing revealed that strain WJ66 contained additional antibiotic resistance genes and an amino acid substitution (Q83L) in fluoroquinolone target GyrA and carried a class 1 integron, with an aadA2 gene in the resistance gene cassette. This paper may lead to a better understanding of the correlation between pathogenic traits and specific genes or gene clusters of S. maltophilia.
2. Results and Discussion
2.1. Bacterial Strains and Drug Resistance
Blood cultures and in vitro susceptibility testing confirmed the presence of a multidrug resistant S. maltophilia isolate, termed WJ66, in an infected patient. The patient was an elderly man in ICU with underlying diseases including type 2 diabetes, diabetic nephropathy, chronic renal insufficiency, uraemia, and a lung infection. Clinical tests showed that the isolates were resistant to nearly all drugs available for treatment, including sulphonamides and fluoroquinolones [8, 11]. Multilocus sequence typing (MLST) sequencing showed that S. maltophilia WJ66 belongs to ST31 (1, 4, 12, 6, 6, 22, and 7 for atpD, gapA, guaA, mutM, nuoD, ppsA, and recA).
In Vitro 96-well plate susceptibility results showed that S. maltophilia WJ66 was resistant to ampicillin-sulbactam (>64–32 μg/mL), piperacillin-tazobactam (>256–4 μg/mL), doxycycline (>32 μg/mL), trimethoprim-sulfamethoxazole (32–608 μg/mL), ciprofloxacin (>16 μg/mL), levofloxacin (>16 μg/mL), polymyxin (4 μg/mL), and amikacin (64 μg/mL) and moderately resistant to minocycline (8 μg/mL) and tigecycline (16 μg/mL). S. maltophilia K279a is sensitive to minocycline (0.25 μg/mL), trimethoprim-sulfamethoxazole (1–9 μg/mL), ciprofloxacin (2 μg/mL), and levofloxacin (2 μg/mL) and resistant to amikacin (64 μg/mL), gentamicin (16 μg/mL), piperacillin-tazobactam (256–4 μg/mL), and tetracycline (16 μg/mL) according to CLSI agar dilution MICs of antimicrobials [12].
We obtained S. maltophilia K279a from the ATCC and examined the drug resistant differences by Etest. Table 1 indicates that both K279a and WJ66 are resistant to imipenem, doxycycline, and tetracycline. K279a is sensitive to polymyxin, while WJ66 is moderately sensitive to polymyxin. Levofloxacin and trimethoprim-sulfamethoxazole are two of the few drugs that are generally efficacious against clinical S. maltophilia infections. K279a is sensitive to both these drugs, but WJ66 is resistant. The mechanisms of this resistance will be described in the following sections. Susceptibility results of control Escherichia coli strains (ATCC25922) and Pseudomonas aeruginosa (ATCC27853) are in line with CLSI standards.
Table 1.
Etest results.
| Isolate | MIC (μg/mL) of | |||||
|---|---|---|---|---|---|---|
| IPM | TC | LE | TS | DC | PO | |
| K279a | >32 | 32 | 1 | 0.125 | 16 | 1 |
| WJ66 | >32 | >256 | >32 | >32 | 48 | 4 |
| ATCC27853 | 1 | 32 | 0.5 | 32 | 64 | 2 |
| ATCC25922 | 0.19 | 1.5 | 0.016 | 0.047 | 1.5 | 0.5 |
IPM: imipenem; TC: tetracycline; LE: levofloxacin; TS: sulfamethoxazole-trimethoprim; DC: doxycycline; PO: polymyxin.
2.2. Total Genome Overview of S. maltophilia WJ66
The draft genomic sequence of S. maltophilia WJ66 was obtained by an Illumina Hiseq 2000. A total of 258 contigs, covering a total of 4,642,973 bp, were generated. The average length of a contig is 17,996 bp with a G + C content of 66.48%. The N50 contig size was 33,808 bp. The genome includes 4,392 predicted open reading frames (ORFs) and 3 ribosomal RNAs (rRNAs) and 61 transfer RNAs (tRNAs) (Table 2). The genome sequence of WJ66 makes it possible for a comparative and functional genomic analysis of S. maltophilia isolates.
Table 2.
S. maltophilia WJ66 genome assembly results.
| Type | WJ66 genome |
|---|---|
| Scaffold number | 75 |
| Scaffold length (bp) | 4,657,282 |
| Scaffold average length (bp) | 62,097 |
| Scaffold N50 (bp) | 143,182 |
| Contigs number | 258 |
| Contigs length (bp) | 4,642,973 |
| Contigs average length (bp) | 17,996 |
| Contigs N50 (bp) | 33,808 |
| Contigs G + C content (%) | 66.48 |
| ORFs number | 4,392 |
| ORFs length (bp) | 4,118,607 |
| ORFs average length (bp) | 937 |
| ORFs G + C content (%) | 66.88 |
| tRNA number | 61 |
| rRNA number | 3 |
2.3. Phylogenetic Relationships among Sequenced S. maltophilia Strains Using Whole Genome Comparisons
The S. maltophilia WJ66 genomic sequence was compared with 11 other S. maltophilia strains, whosegenomic sequence data are in the GenBank database. Figure 1 shows the phylogenetic relationships between different S. maltophilia isolate genomes. We took the Xanthomonas campestris pv. campestris (Xcc) stain 8004 as an outgroup construct for the phylogenetic tree. WJ66, K279A, EPM1, and Ab 55555 are on the same branch, indicating that they are highly homologous. Strain K279a is a blood sample isolate from the United Kingdom [10], strain WJ66 is also a blood sample isolate from China, strain Ab55555 is an American isolate, and strain EPM1 was found during the sequencing of two human-derived strains of Giardia duodenalis [13]. The environmental isolate MF89, an oyster-associated bacteria, shows potential for crude oil hydrocarbon degradation [14] and is the most closely related to the above four isolates. In addition, JV3, D457, and AU12-09 are close relatives with each other. D457 is a clinical isolate [15] and AU12-09 is an intravascular catheters isolate [16]. Homologous strains R551-3 and RA8 were isolated from the poplar Populus trichocarpa [17] and from sewage plant effluent [18], respectively. SKK35 is ulcer swab isolate [9]. Overall, the S. maltophilia strains can be divided into two groups: the clinical isolates and the environmental isolates. The mutual affinity within groups is closer than the relationship between the groups. As the Xcc strain 8004 is the only non-S. maltophilia strain, the genomic differences between Xcc and S. maltophilia are the largest.
Figure 1.

Evolutionary relationships among S. maltophilia strains. Neighbour-joining phylogram of S. maltophilia. Neighbour-joining phylogenetic tree was constructed based on a single copy gene family with the bootstrap set to 1000. Sequences from Xanthomonas campestris pv. campestris strain 8004 were used as an outgroup. The black spot indicates S. maltophilia WJ66. R551 and AU12 are short for R551-3 and AU12-09, respectively.
2.4. Comparing the Genomes of S. maltophilia WJ66 and K279a
Because S. maltophilia strains WJ66 and K279a exist in the same isolated background and are closely related, more comparison need to be performed. Global alignments were performed by the MUMER software [19], and the results showed a large homology between WJ66 and K279a (Figure 2).
Figure 2.

Genome alignment of S. maltophilia WJ66 and K279a. Line figures depict the results of whole genome alignment results using MUMmer. The query genomic sequence was the strain WJ66, and the subject genome was K279a. The red and blue lines represent direct reverse alignments, respectively.
2.5. The Comprehensive Antibiotic Resistance Database (CARD)
Within the CARD, 3411 genes are tagged for antibiotic molecule, biosynthesis, resistance, or inhibition of resistance, and 1859 genes were specifically annotated for antibiotic resistance. The Resistance Gene Identifier (RGI) version 2 provides an automated annotation of DNA sequences based upon the data available in the CARD, thus providing a prediction of antibiotic resistance genes [20].
The autoannotation results of the WJ66 and K279a contigs are shown in Figure 3. Most of the resistance genes are antibiotic efflux genes (32 and 31 genes in WJ66 and K279a, resp.), beta-lactam resistance genes (5 and 4 genes in WJ66 and K279a, resp.), aminoglycoside resistance genes (3 and 2 genes in WJ66 and K279a, resp.), lin/str/phe/lin/mac (linezolid/streptogramin/phenicol/lincosamide/macrolide) resistance gene (1 gene in WJ66 and K279a, resp.), a rifampin resistance gene (1 gene in WJ66 and K279a, resp.), a sulphonamide resistance gene (1 gene in WJ66), fluoroquinolone resistance genes (2 genes and 1 genes in WJ66 and K279a, resp.), and a peptide (1 gene in WJ66 and K279a). This analysis reveals the resistance strength of WJ66.
Figure 3.

Antibiotic resistance genes in the S. maltophilia WJ66 and K279a genomes. Contigs were uploaded to RGI, autoannotation results are as follows: most of the resistance genes are antibiotic efflux approximately (32/31), beta-lactam resistance (5/4), aminoglycoside resistance (3/3), lin/str/phe/lin/mac (1/1), rifampin resistance (1/1), sulphonamide resistance (1/0), fluoroquinolone resistance (2/1), and peptide (1/1). The reference website is http://arpcard.mcmaster.ca/.
Open reading frames (ORFs) obtained from the GeneMarks annotation were subjected to BLAST analysis against the CARD. This approach highlighted the presence of 255 genes related to antibiotic resistance. The largest proportion of homology to antibiotic resistance genes was found to be efflux pump genes (approximately 41 homologous genes), the beta-lactamase gene (33 homologous genes), and quinolone resistance related genes (7 homologous genes). By comparing with the CARD, we found that WJ66_04174 and WJ66_01359 codes for the qepA gene, whose gene product is a quinolone efflux pump, are homologous with an antibiotic resistance gene in E. coli 1520 genome. The protein QnrB encoded by WJ66_00023 is a S. maltophilia inherent quinolone resistance gene. WJ66_04112 (gyrA), WJ66_03278 (gyrB), WJ66_01551 (parC), WJ66_03996 (parC), and WJ66_03111 (parE) were annotated as fluoroquinolone target genes, and mutation in these genes causes resistance to quinolones. GMS_00385 encodes dihydropteroate synthetase type 1 (sul1); downstream sul1 is GMS_00386 (WJ66_01310) coding an aadA2 aminoglycoside acetyltransferase; these two genes are homologous with that found in Salmonella enterica subsp. enterica serovar Typhimurium DT104. Advanced analyses found that sul1 and aadA2 are members of a class 1 integron and were not predicted by GeneMark analysis. Moreover, the two genes are 100% identical with latter strain Typhimurium DT104, indicating that they were probably transferred from another bacterium by mobile elements.
2.6. The Trimethoprim-Sulfamethoxazole Associated Integron
In many Gram-negative bacteria, integrons carrying antibiotic resistance genes have been identified by mobile elements such as transposons and plasmids that facilitate the transfer of antibiotic resistance genes between different species. In the present study, the distribution of class 1 and 2 integrons was examined in a collection of clinical S. maltophilia isolates [21].
Integrons were initially described at the end of the 1980s [22]. This family of elements appears formally distinct from other known mobile DNA elements such as transposons. Class 1 integrons play an important role in the emergence and spread of antimicrobial resistance determinants. Typically, class 1 integrons have three distinct genetic regions of which two are highly conserved, the 5′-conserved segment (5′-CS) and the 3′-conserved segment (3′-CS) flank the central variable region where the gene cassettes are located. The 5′-CS includes the integrase intI1 gene, the recombination site attI1, and the promoters Pc and P2 when present. The 3′-CS consists of the qacEΔ1 gene, which encodes an incomplete version of a protein that mediates the resistance to certain detergents, the sul1 gene, which encodes resistance to sulphonamides, and an open reading frame of unknown function orf5 [23].
Although most of class 1 integrons have this classic structure, an increasing number of class 1 integrons, related to trimethoprim-sulphamethoxazole resistance, with different structures have been reported [24, 25]. Although trimethoprim-sulfamethoxazole is considered the drug of choice for treating S. maltophilia infections, 25% of the isolates were resistant [21].
The WJ66 strain carries a detectable class 1 integron, whereas class 2 and class 3 integrons were not identified. In addition to the sul1 gene encoding a sulphonamide-resistant dihydrofolate synthase, the aadA2 gene, which encodes an aminoglycoside acetyltransferase giving resistance to aminoglycosides including amikacin, was identified as the only other gene in the resistance gene cassette (Figure 4). More details about the integron can be found in supplementary data 1 (see Supplementary Materials available online at http://dx.doi.org/10.1155/2015/580240).
Figure 4.
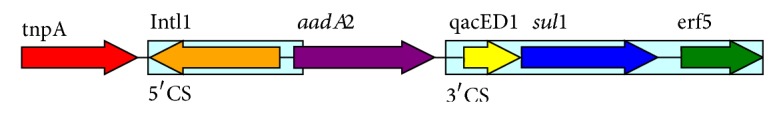
Class 1 integron in S. maltophilia WJ66. The S. maltophilia WJ66 genome carries a detectable class 1 integron; aadA2 (resistance to aminoglycosides) was found as the only gene in the resistance gene cassette.
There are 24 different S. maltophilia integrons reported in the NCBI. Therefore, we compared class 1 integron in WJ66 with other class 1 integrons of S. maltophilia (22/24). Gene orders in cassettes are as follows: aadB-aac(6)-II-blaCARB-8 (GQ866976.1), aadB-aadA1 (HQ914241.1), aacA4-blaIMP-25-blaOXA-30-blacatB3 (GU944726.1), blaVIM-2-aac(6′)-II (KF471098.1), qacL-aadB-cmlA-aadA2 (KF556708.1), qacK-aac6-aac6 (KF556707.1), aac6 (KF556705.1), qacK (KF556704.1), aadB-aadA2 (JN108896.1), dhfra1 (HQ584988.1), aac(6′)-ib-cr (HQ438047.1), arr-3-aac (GU137303.1), dfrA12-aadA2 (GQ981416.1), dfrA17-aadA5 (GQ924479.1), aacA 4-catB8-aadA1 (GQ906532.1), dfrA15 (JN108895.1), aac(6′)-Ib-cr (EF210035.1), arr3-dfrA27 (KC748137.1), aadB (JX560784.1), P2 promoter-aacA4-aadA2 (GQ896541.1), aacA4 (GQ502669.1), smr (AF406792.1), and aadA1 (HQ832470.1).
Because class 1 integron has had a major role in the spread of antibiotic resistance and has led to worldwide difficulties in controlling bacterial infection, understanding the origin of these elements is important for the practical control of antibiotic resistance and for exploring the means by which bacterial lateral gene transfer can seriously impact, and be impacted by, human activities.
2.7. The Fluoroquinolone Levofloxacin Resistance Associated Gene
Quinolones are synthetic antibacterial reagents with broad-spectrum activity. They inhibit the enzyme topoisomerase II, a DNA gyrase that is necessary for the replication of the microorganism. Topoisomerase II enzyme produces a negative DNA supercoil on DNA, which permits transcription or replication. By inhibiting this enzyme, DNA replication and transcription are blocked.
The earliest quinolone, nalidixic acid, was first used in a clinical setting in 1962. The targets of quinolone action are the essential bacterial enzymes DNA gyrase, as stated above, and DNA topoisomerase IV. Both are large, complex enzymes composed of 2 pairs of subunits. The subunits of DNA gyrase are GyrA, a 97-kDa protein encoded by the gyrA gene, and GyrB, a 90-kDa protein encoded by the gyrB gene. The subunits of topoisomerase IV are ParC (75 kDa) and ParE (70 kDa).
One of the mechanisms of quinolone resistance is the accumulation of mutations that alter the drug targets. In Gram-negative bacteria, DNA gyrase is more susceptible to inhibition by quinolones than is topoisomerase IV, whereas in Gram-positive bacteria, topoisomerase IV is usually the prime target and DNA gyrase is intrinsically less susceptible. Consequently, resistant mutations are found in gyrA in Gram-negative bacteria and in parC in Gram-positive bacteria [26].
The MIC of levofloxacin for K279a is 1 μg/mL, the MIC of norfloxacin for D457 is 6 μg/mL [27], and the MIC of levofloxacin for WJ66 is more than 32 μg/mL, corresponding to susceptibility results: sensitive, moderately sensitive, and resistant, respectively. Sequence alignments show that the resistant WJ66 strain contained a point mutation (Q83L) in its GyrA (Figure 5). While no mutations in GyrB were identified, WJ66 contained mutations in ParC (S86I and E421D) and ParE (M227L and F519C).
Figure 5.

Alignment of the gyrA gene among S. maltophilia WJ66, K279a, and D457. Alignment of gyrA sequences of S. maltophilia WJ66, K279a, and D457 using vector NTI suite 9 software.
2.8. Efflux Pumps in WJ66 Genome
Efflux as a mechanism for antibiotic resistance was first described in 1980 [28]. In Gram-negative nosocomial pathogens, MDR is usually mediated by the overproduction of resistance-nodulation-division (RND) type efflux pumps. These pumps tend to have broad substrate profiles, including organic solvents, disinfectants, and antimicrobial drugs from a number of different classes. In the case of Gram-negative bacteria, efflux pumps consist of cytoplasmic, periplasmic, and outer membrane proteins (OMP) that associate to form multicomponent efflux systems [29]. A periplasm spanning membrane-fusion protein (MFP) is usually specific to each RND efflux protein, and it is common to find the pair encoded as part of an operon. A third component, the outer membrane protein, can be encoded in the same operon, but there tends to be fewer OMPs than RND/MFP pairs in a cell, meaning that the OMPs are often promiscuous [10]. The K279a sequence encodes nine RND-type efflux pump genes that fall into the drug resistance type based on sequence homology. WJ66 also possessed these efflux pumps (Table 3).
Table 3.
Characteristics of Sme efflux transporters in S. maltophilia WJ66.
| Name | Systematic ID | Known or putative regulation mechanism | Systematic ID |
|---|---|---|---|
| SmeABC | WJ66_02013, WJ66_02012, WJ66_02011 | Two component regulator smeSR | WJ66_02014, WJ66_02015 |
|
| |||
| SmeDEF | WJ66_02297, WJ66_02296, WJ66_02294 | TetR type transcriptional regulator, smeT | WJ66_02298 |
|
| |||
| SmeVWX | WJ66_01718, WJ66_01720, WJ66_01721 | HTH-type transcriptional regulator | WJ66_01724 |
|
| |||
| SmeYZ | WJ66_04223, WJ66_04224 | Two component regulator | WJ66_02878, WJ66_02879 |
|
| |||
| SmeGH | WJ66_01237, WJ66_01238 | TetR type transcriptional regulator | WJ66_01236 |
|
| |||
| SmeMN | WJ66_02576, WJ66_02575 | ||
|
| |||
| SmeOP | WJ66_00263, WJ66_00264 | TetR type transcriptional regulator | WJ66_00262 |
|
| |||
| SmeIJK | WJ66_03576, WJ66_03575, WJ66_03574 | ||
2.9. The Pathogen-Host Interactions Database (PHI-base)
The PHI-base (http://www.phi-base.org/) contains expertly curated molecular and biological information on genes proven to affect the outcome of pathogen-host interactions. Information is also given on the target sites of some anti-infective molecules.
Little is known about pathogenesis of S. maltophilia and the genomic diversity exhibited by clinical isolates complicates the study of pathogenicity and virulence factors. A recent study about relative virulence in adult-zebrafish model of S. maltophilia infection revealed that abundance of the quorum-sensing factor Ax21 in four strains of S. maltophilia correlates with mortality [30]. ORFs obtained from the GeneMarks annotation were subjected to BLAST analysis against the PHI-base, resulting in the identification of 252 homologous genes. Two genes, ax21 and avrXa21, were found to be homologous to Xanthomonas oryzae genes that are related to plant virulence. The ax21 and avrXa21 genes also have homologs in Metarhizium anisopliae and Saccharomyces cerevisiae. Seven genes were annotated as lethal to host (plant); the first two have homologs in Gibberella zeae, and the other five have homologs in Aspergillus fumigatus. Seven genes were annotated to be resistant to chemical reagents including disinfectants and chemotherapy. Seven genes were annotated with mixed outcomes. A majority of genes, approximately 26, were annotated as partial loss of Ax21 signalling in this opportunistic pathogen strain, these genes include raxB, raxC, raxH, raxR, raxH 2 /phoQ, and raxH 2 /phoP, which has a homolog in Xanthomon oryzae. Thirty-three genes were annotated as genes that do not affect pathogenicity, while 42 genes and 125 genes were annotated as genes that lead to pathogenic loss and virulence attenuation, respectively. While a majority of pathogenic genes were avirulent, foreign outbreaks are examples of pathogenicity and virulence still worthy of our attention.
The rice XA21 receptor binds the AxYS22 peptide corresponding to the N-terminal region of Ax21, a type I-secreted protein that is highly conserved in all Xanthomonas species as well as in Xylella fastidiosa and S. maltophilia [31].
Ax21 is a 194-amino-acid protein and an activator of innate immunity mediated by the XA21 pattern recognition receptor [32]. S. maltophilia displayed Ax21 (Smlt0378 in K279a) functions in the signal between cells. An Ax21 knockout leads to decreased virulence but a lack of raxST sulphated species modifications that do not affect the signal. These findings suggest that sulfation is not required for the regulatory action of Ax21 within S. maltophilia K279a [33]. Ax21 associated genes in S. maltophilia WJ66 are shown in Table 4. Homology analysis showed that WJ66 lacks raxST and raxA, which is consistent with K279a. WJ66_03588 is homologous to the raxB gene, a 169-amino-acid protein that functions in Colicin V secretion and had 97.78% homology to an ABC transporter ATP-binding protein. EPM1 predicted that Smlt2001 from K279a is a putative 590-amino-acid ABC transporter ATP-binding protein that has high homology to PHI_1141 (719 amino acids). Ax21 secretion depends on the RaxB homologous protein Smlt2001. Because of the size difference, confirming WJ66_03588 functions as Smlt2001 will need further verification. Additionally, raxP is missing in WJ66, and raxQ has only 37.04% homology.
Table 4.
Ax21 associated genes in S. maltophilia WJ66.
| Systematic ID | S. name | % identity 1 | Gene name | Phenotype | % identy 2 | Product-NR |
|---|---|---|---|---|---|---|
| WJ66_02052 | PHI_1146 | 59.32 | Ax21 | Effector (plant avirulence determinant) | 100 | Putative uncharacterized protein (precursor) (strain K279a) |
|
| ||||||
| WJ66_01677 | PHI_1146 | 53.67 | Ax21 | Effector (plant avirulence determinant) | 95.81 | Putative uncharacterized protein (precursor) (strain K279a) |
|
| ||||||
| WJ66_03588 | PHI_1141 | 54.55 | raxB | Partially lost Ax21 (synonym: AvrXa21) activity | 97.78 | Colicin V secretion ABC transporter ATP-binding protein (strain EPM1) |
|
| ||||||
| WJ66_00260 | PHI_1142 | 71.26 | raxC | Partially lost Ax21 (synonym: AvrXa21) activity | 100 | Putative outer membrane protein (precursor) (strain K279a) |
|
| ||||||
| WJ66_03632 | PHI_1136 | 80.79 | raxR | Partially lost Ax21 (synonym: AvrXa21) activity | 100 | Putative two-component system response regulator (strain K279a) |
|
| ||||||
| WJ66_03633 | PHI_1137 | 58.11 | raxH | Partially lost Ax21 (synonym: AvrXa21) activity | 99.53 | Putative two-component system sensor histidine (strain K279a) |
|
| ||||||
| WJ66_03508 | PHI_1144 | 98.21 | raxR2/phoP | Partially lost Ax21 (synonym: AvrXa21) activity/affected bacterial pathogenicity | 100 | Putative two-component system response regulator (strain K279a) |
|
| ||||||
| WJ66_03509 | PHI_1145 | 77.07 | raxH2/phoQ | Partially lost Ax21 (synonym: AvrXa21) activity | 100 | Putative two-component regulator sensor histidine kinase transmembrane transcriptional regulatory protein (precursor) (strain K279a) |
|
| ||||||
| WJ66_04429 | PHI_1139 | 37.04 | raxQ | Partially lost Ax21 (synonym: AvrXa21) activity | 98.86 | Elongation factor Tu (strain K279a) |
% identity 1 and % identity 2 values were results from the identity when subjected to BLAST analysis against PHI database and NR database, respectively.
3. Conclusions
In summary, the genomic sequence of the resistant bacteraemia-associated S. maltophilia isolate WJ66 is highly homologous with K279a. Tiny differences in nucleic acid levels lead to different in vitro antibiotic susceptibility phenotypes. Comparative genomic analyses revealed a considerable diversity among S. maltophilia strains, providing new insights into the differences and similarities that may explain the diverse nature of these strains. Drug-resistant gene mutations, drug-resistant enzymes, and horizontal drug resistance gene transfer in bacteria contribute to drug resistance. We obtained useful information about the unexplored aspects of S. maltophilia. The work will improve our understanding of the correlation between pathogenic traits and specific genes or gene clusters of S. maltophilia.
4. Material and Methods
4.1. Bacterial Strains, In Vitro Antimicrobial Sensitivity Assays, and Genomic DNA Extraction
S. maltophilia WJ66 was originally isolated in a blood sample from an elder male patient undergoing chemotherapy. The patient has diabetic nephropathy, chronic renal insufficiency, uraemia, renal anaemia, lung infection, hypertension, cerebral infarction (old), urinary tract infection, and hyperuricemia and did not respond to therapy with piperacillin-tazobactam, ceftazidime, or imipenem. The bacterium was performed the antimicrobial susceptibility assay and the MLST sequencing as previously described [34].
Susceptibility assays for the different antibiotics were performed in Mueller Hinton (MH) broth, using the twofold dilution method in 96-well microporous plates. The results were recorded after 20 h incubation at 37°C [35]. MICs were determined in MH medium by Etest (AB BIOMERIEUX, Arizona City, Sweden), according to the manufacturer's instructions. To ensure that the observed changes were consistent, all MICs were determined in three independent assays, using different bacterial cultures on different days. In most cases, there were no interassay differences in the MIC values. In a few cases, there were one dilution differences. For the latter, the assay was repeated one more time to further assure assay reliability. Control strains were Escherichia coli (ATCC25922) and Pseudomonas aeruginosa (ATCC27853). Genomic DNA was extracted using Invitrogen PureLink Genomic DNA Mini Kit according to the manufacturer's instructions.
4.2. Sequence Assembly, Prediction, and Annotation
After the DNA was prepared, a whole-genome shotgun (WGS) sequencing strategy and Illumina HiSeq 2000 sequencing technology with a paired-end protocol was used to determine the genome sequence of S. maltophilia WJ66. The low-quality reads were filtered, and the remaining reads were assembled into contigs with Velvet by the de novo assembly method. ORFs were predicted by the GeneMark analysis. The rRNA was predicted by using RNAmmer and the tRNA was identified with tRNA scan-SE. Then, the genomic sequence was annotated automatically using blast software for searching the SwissPort, UniPort, and the NR database; the cutoff of E-value was 1e − 5.
4.3. Nucleotide Sequence Accession Number
This entire genome shotgun project has been deposited in the DDBJ/EMBL/GenBank under the accession AZRF00000000. The version described in this paper is the first version, accession AZRF00000000.
4.4. Phylogenetic Analysis
After a maximum likelihood phylogenetic analysis, we construct a neighbour-joining phylogenetic tree with one single copy gene family. Sequences from Xanthomonas campestris pv. campestris strain 8004 were used as an outgroup.
4.5. Genome Alignments of S. maltophilia WJ66 and K279a
Annotated genome sequences and statistics of strain K279a from GenBank can be accessed through genome. Line figures depict the whole genome alignment results using MUMmer [19, 36]. The query genomic sequence was the strain WJ66 and the subject genome was K279a. The red and blue lines represent direct and reverse alignments, respectively.
Supplementary Material
Table S1 Class 1 integron within S. maltophilia WJ66 genome.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grants 81071399 and 81271887 to Y. Li) and National Science and Technology Major Project funded projects (Grant 2013ZX10004202002).
Conflict of Interests
The authors declare that they have no conflict of interests.
Authors' Contribution
Y. Li, J. Yue, and S. Lu conceived and designed the experiments. Y. Zhao, W. Niu, Y. Sun, and H. Hao performed the experiments. D. Yu, G. Xu, X. Shang, and X. Tang contributed with reagents/materials/analysis tools. Y. Zhao and J. Yue wrote the paper. All authors read and approved the final paper. Y. Zhao, W. Niu, and Y. Sun contributed equally to this work.
References
- 1.Ryan R. P., Monchy S., Cardinale M., et al. The versatility and adaptation of bacteria from the genus Stenotrophomonas . Nature Reviews Microbiology. 2009;7(7):514–525. doi: 10.1038/nrmicro2163. [DOI] [PubMed] [Google Scholar]
- 2.Brooke J. S. Stenotrophomonas maltophilia: an emerging global opportunistic pathogen. Clinical Microbiology Reviews. 2012;25(1):2–41. doi: 10.1128/cmr.00019-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calza L., Manfredi R., Chiodo F. Stenotrophomonas (Xanthomonas) maltophilia as an emerging opportunistic pathogen in association with HIV infection: a 10-year surveillance study. Infection. 2003;31(3):155–161. doi: 10.1007/s15010-003-3113-6. [DOI] [PubMed] [Google Scholar]
- 4.Naidu P., Smith S. A review of 11 years of Stenotrophomonas maltophilia blood isolates at a tertiary care institute in Canada. Canadian Journal of Infectious Diseases and Medical Microbiology. 2012;23(4):165–169. doi: 10.1155/2012/762571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waters V. J., Gómez M. I., Soong G., Amin S., Ernst R. K., Prince A. Immunostimulatory properties of the emerging pathogen Stenotrophomonas maltophilia . Infection and Immunity. 2007;75(4):1698–1703. doi: 10.1128/iai.01469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nseir S., Di Pompeo C., Brisson H., et al. Intensive care unit-acquired Stenotrophomonas maltophilia: incidence, risk factors, and outcome. Critical Care. 2006;10(5, article R143) doi: 10.1186/cc5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samonis G., Karageorgopoulos D. E., Maraki S., et al. Stenotrophomonas maltophilia infections in a general hospital: patient characteristics, antimicrobial susceptibility, and treatment outcome. PLoS ONE. 2012;7(5) doi: 10.1371/journal.pone.0037375.e37375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jang T.-N., Wang F.-D., Wang L.-S., Liu C.-H., Liu I.-M. Xanthomonas maltophilia bacteremia: an analysis of 32 cases. Journal of the Formosan Medical Association. 1992;91(12):1170–1176. [PubMed] [Google Scholar]
- 9.Victor M. A., Arpi M., Bruun B., Jonsson V., Hansen M. M. Xanthomonas maltophilia bacteremia in immunocompromised hematological patients. Scandinavian Journal of Infectious Diseases. 1994;26(2):163–170. doi: 10.3109/00365549409011780. [DOI] [PubMed] [Google Scholar]
- 10.Crossman L. C., Gould V. C., Dow J. M., et al. The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome Biology. 2008;9(4, article R74) doi: 10.1186/gb-2008-9-4-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tseng C.-C., Fang W.-F., Huang K.-T., et al. Risk factors for mortality in patients with nosocomial Stenotrophomonas maltophilia pneumonia. Infection Control and Hospital Epidemiology. 2009;30(12):1193–1202. doi: 10.1086/648455. [DOI] [PubMed] [Google Scholar]
- 12.Gould V. C., Okazaki A., Avison M. B. Coordinate hyperproduction of SmeZ and SmeJK efflux pumps extends drug resistance in Stenotrophomonas maltophilia. Antimicrobial Agents and Chemotherapy. 2013;57(1):655–657. doi: 10.1128/aac.01020-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sassera D., Leardini I., Villa L., et al. Draft genome sequence of Stenotrophomonas maltophilia strain EPM1, found in association with a culture of the human parasite Giardia duodenalis . Genome Announcements. 2013;1(2) doi: 10.1128/genomea.00182-13.e00182-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chauhan A., Green S., Pathak A., Thomas J., Venkatramanan R. Whole-genome sequences of five oyster-associated bacteria show potential for crude oil hydrocarbon degradation. Genome Announcements. 2013;1(5) doi: 10.1128/genomeA.00802-13.e00802-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lira F., Hernández A., Belda E., et al. Whole-genome sequence of Stenotrophomonas maltophilia D457, a clinical isolate and a model strain. Journal of Bacteriology. 2012;194(13):3563–3564. doi: 10.1128/jb.00602-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L., Morrison M., Cuív P. O., Evans P., Rickard C. M. Genome sequence of Stenotrophomonas maltophilia strain AU12-09, isolated from an intravascular catheter. Genome Announcements. 2013;1(3) doi: 10.1128/genomeA.00195-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banerjee R., Roy D. Codon usage and gene expression pattern of Stenotrophomonas maltophilia R551-3 for pathogenic mode of living. Biochemical and Biophysical Research Communications. 2009;390(2):177–181. doi: 10.1016/j.bbrc.2009.09.062. [DOI] [PubMed] [Google Scholar]
- 18.Adamek M., Overhage J., Bathe S., Winter J., Fischer R., Schwartz T. Genotyping of environmental and clinical Stenotrophomonas maltophilia isolates and their pathogenic potentia. PLoS ONE. 2011;6(11) doi: 10.1371/journal.pone.0027615.e27615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delcher A. L., Phillippy A., Carlton J., Salzberg S. L. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Research. 2002;30(11):2478–2483. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McArthur A. G., Waglechner N., Nizam F., et al. The comprehensive antibiotic resistance database. Antimicrobial Agents and Chemotherapy. 2013;57(7):3348–3357. doi: 10.1128/aac.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang L. L., Chen H. F., Chang C. Y., Lee T. M., Wu W. J. Contribution of integrons, and SmeABC and SmeDEF efflux pumps to multidrug resistance in clinical isolates of Stenotrophomonas maltophilia . Journal of Antimicrobial Chemotherapy. 2004;53(3):518–521. doi: 10.1093/jac/dkh094. [DOI] [PubMed] [Google Scholar]
- 22.Stokes H. W., Hall R. M. A novel family of potentially mobile DNA elements encoding site-specific gene-integration functions: integrons. Molecular Microbiology. 1989;3(12):1669–1683. doi: 10.1111/j.1365-2958.1989.tb00153.x. [DOI] [PubMed] [Google Scholar]
- 23.Fluit A. C., Schmitz F. J. Class 1 integrons, gene cassettes, mobility, and epidemiology. European Journal of Clinical Microbiology & Infectious Diseases. 1999;18(11):761–770. doi: 10.1007/s100960050398. [DOI] [PubMed] [Google Scholar]
- 24.Barbolla R., Catalano M., Orman B. E., et al. Class 1 integrons increase trimethoprim-sulfamethoxazole MICs against epidemiologically unrelated Stenotrophomonas maltophilia isolates. Antimicrobial Agents and Chemotherapy. 2004;48(2):666–669. doi: 10.1128/aac.48.2.666-669.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toleman M. A., Bennett P. M., Bennett D. M. C., Jones R. N., Walsh T. R. Global emergence of trimethoprim/sulfamethoxazole resistance in Stenotrophomonas maltophilia mediated by acquisition of sul genes. Emerging Infectious Diseases. 2007;13(4):559–565. doi: 10.3201/eid1304.061378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacoby G. A. Mechanisms of resistance to quinolones. Clinical Infectious Diseases. 2005;41(supplement 2):S120–S126. doi: 10.1086/428052. [DOI] [PubMed] [Google Scholar]
- 27.Alonso A., Martínez J. L. Multiple antibiotic resistance in Stenotrophomonas maltophilia . Antimicrobial Agents and Chemotherapy. 1997;41(5):1140–1142. doi: 10.1128/aac.41.5.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McMurry L., Petrucci R. E., Jr., Levy S. B. Active efflux of tetracycline encoded by four genetically different tetracyline resistance determinants in Escherichia coli . Proceedings of the National Academy of Sciences of the United States of America. 1980;77(7):3974–3977. doi: 10.1073/pnas.77.7.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez J. L., Sánchez M. B., Martínez-Solano L., et al. Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiology Reviews. 2009;33(2):430–449. doi: 10.1111/j.1574-6976.2008.00157.x. [DOI] [PubMed] [Google Scholar]
- 30.Ferrer-Navarro M., Planell R., Yero D., et al. Abundance of the quorum-sensing factor Ax21 in four strains of correlates with mortality rate in a New Zebrafish model of infection. PLoS ONE. 2013;8(6) doi: 10.1371/journal.pone.0067207.e67207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han S. W., Lee S. W., Ronald P. C. Secretion, modification, and regulation of Ax21. Current Opinion in Microbiology. 2011;14(1):62–67. doi: 10.1016/j.mib.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 32.Lee SW, Han S. W., Sririyanum M., Park C. J., Seo Y. S., Ronald P. C. A type I-secreted, sulfated peptide triggers XA21-mediated innate immunity. Science. 2009;326(5954):850–853. doi: 10.1126/science.1173438. [DOI] [PubMed] [Google Scholar]
- 33.McCarthy Y., Dow J. M., Ryan R. P. The Ax21 protein is a cell-cell signal that regulates virulence in the nosocomial pathogen Stenotrophomonas maltophilia . Journal of Bacteriology. 2011;193(22):6375–6378. doi: 10.1128/jb.05949-11. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Kaiser S., Biehler K., Jonas D. A Stenotrophomonas maltophilia multilocus sequence typing scheme for inferring population structured. Journal of Bacteriology. 2009;191(9):2934–2943. doi: 10.1128/jb.00892-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sánchez M. B., Hernández A., Rodríguez-Martínez J. M., Martínez-Martínez L., Martínez J. L. Predictive analysis of transmissible quinolone resistance indicates Stenotrophomonas maltophilia as a potential source of a novel family of Qnr determinants. BMC Microbiology. 2008;8, article 148 doi: 10.1186/1471-2180-8-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ng V., Lin W.-J. Comparison of assembled Clostridium botulinum A1 genomes revealed their evolutionary relationship. Genomics. 2014;103(1):94–106. doi: 10.1016/j.ygeno.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Class 1 integron within S. maltophilia WJ66 genome.