

Abstract
Werner syndrome (WS) is a rare human autosomal recessive premature aging disorder characterized by early onset of aging-associated diseases, chromosomal instability, and cancer predisposition. The function of the DNA helicase encoded by WRN, the gene responsible for WS, has been studied extensively. WRN helicase is involved in the maintenance of chromosome integrity through DNA replication, repair, and recombination by interacting with a variety of proteins associated with DNA repair and telomere maintenance. The accelerated aging associated with WS is reportedly caused by telomere dysfunction, and the underlying mechanism of the disease is yet to be elucidated. Although it was reported that the life expectancy for patients with WS has improved over the last two decades, definitive therapy for these patients has not seen much development. Severe symptoms of the disease, such as leg ulcers, cause a significant decline in the quality of life in patients with WS. Therefore, the establishment of new therapeutic strategies for the disease is of utmost importance. Induced pluripotent stem cells (iPSCs) can be established by the introduction of several pluripotency genes, including Oct3/4, Sox2, Klf4, and c-myc into differentiated cells. iPSCs have the potential to differentiate into a variety of cell types that constitute the human body, and possess infinite proliferative capacity. Recent studies have reported the generation of iPSCs from the cells of patients with WS, and they have concluded that reprogramming represses premature senescence phenotypes in these cells. In this review, we summarize the findings of WS patient-specific iPSCs (WS iPSCs) and focus on the roles of telomere and telomerase in the maintenance of these cells. Finally, we discuss the potential use of WS iPSCs for clinical applications.
Keywords: Werner syndrome (WS), accelerated aging, chromosomal instability, telomere dysfunction, induced pluripotent stem cells (iPSCs), reprogramming, telomerase, premature senescence phenotypes
INTRODUCTION
Werner syndrome (WS) is a rare human autosomal recessive disorder characterized by early onset of aging-associated diseases, chromosomal instability, and cancer predisposition (Goto, 1997, 2000). Fibroblasts from patients with WS exhibit premature replicative senescence (Salk et al., 1981b). WRN, the gene responsible for the disease, encodes a RecQ-type DNA helicase (Oshima et al., 1996; Yu et al., 1996; Goto et al., 1997; Matsumoto et al., 1997) that is involved in the maintenance of chromosome integrity during DNA replication, repair, and recombination (Shimamoto et al., 2004; Rossi et al., 2010).
WRN is a member of the RecQ helicase gene family, and other members of the family include BLM and RTS/RECQL4, which are mutated in Bloom syndrome (BS) and Rothmund–Thomson syndrome (RTS), respectively (Ellis et al., 1995; Kitao et al., 1999). BS and RTS, along with WS, are characterized by chromosomal instability, due to which RecQ helicases are considered to be the guardian angels of the genome (Shimamoto et al., 2004; Bohr, 2008). There are five members in the RecQ helicase gene family, including RECQL1 (Seki et al., 1994) and RECQL5 (Kitao et al., 1998; Shimamoto et al., 2000), the mutations of which have yet to be identified in human diseases.
Major clinical symptoms of WS include common age-associated diseases, such as insulin-resistant diabetes mellitus, and atherosclerosis. Recent advances in drug therapy for these diseases are available and are known to increase the lifespan of patients with WS. However, there is no effective therapy for intractable features, such as severe skin ulcers leading to a decrease in quality of life (QOL), which is a serious problem in patients with WS. Thus, there is an urgent need to develop a new treatment strategy for this syndrome. Regenerative medicine, such as autologous cell transplantation, could be considered as one of the therapeutic strategies for WS, and a potential choice is the use of patient-specific iPSCs.
Somatic cell reprogramming follows the introduction of several pluripotency genes, including Oct3/4, Sox2, Klf4, c-myc, Nanog, and Lin-28, into differentiated cells such as dermal fibroblasts, blood cells, and others (Takahashi and Yamanaka, 2006; Takahashi et al., 2007; Yu et al., 2007; Aoi et al., 2008; Stadtfeld and Hochedlinger, 2010; Okita and Yamanaka, 2011). During reprogramming, somatic cell-specific genes are suppressed, while embryonic stem cell (ESC)-specific pluripotency genes are induced, leading to the generation of induced pluripotent stem cells (iPSCs) with undifferentiated states and pluripotency (Stadtfeld et al., 2008). Somatic cell reprogramming generates iPSCs characterized by pluripotency and infinite proliferative potential similar to the ESCs, and this technology opens up new possibilities for tailor-made regenerative medicine (Stadtfeld and Hochedlinger, 2010; Okita and Yamanaka, 2011).
Recently, two groups reported the generation of iPSCs from the cells of patients with WS and came to the similar conclusion that reprogramming repressed premature senescence phenotypes in WS cells (Cheung et al., 2014; Shimamoto et al., 2014). They demonstrated the successful reprogramming of cells from patients with WS into iPSCs with restored telomere function and stable karyotypes, suggesting that the induction of the gene encoding human telomerase reverse transcriptase (hTERT) during reprogramming suppresses telomere dysfunction in WS cells lacking WRN. In this review, we summarize the findings of WS patient-specific iPSCs (WS iPSCs) reported in the literature, and focus on the roles of telomere and telomerase in maintenance of these cells. We also review the recent progress in the clinical management of WS and explore stem cell therapy as a new strategy for WS treatment. WS iPSCs will provide opportunities not only for a better understanding of the pathogenic processes and modeling of the complex features of WS, but also for drug screening as well as the discovery and development of a new strategy for its treatment.
FUNCTION OF WRN HELICASE
Prolonged S-phase and reduction in frequency of DNA replication initiation observed in WS cells have implicated the role of WRN helicase in DNA replication (Hanaoka et al., 1983; Poot et al., 1992). The fact that WRN helicase interacts with several factors involved in DNA replication, including RPA, PCNA, FEN-1, and Topoisomerase I, supports this theory (Figure 1; Shimamoto et al., 2004; Rossi et al., 2010). WS cells are hypersensitive to a Topoisomerase I inhibitor, camptothecin (Okada et al., 1998; Poot et al., 1999), and WRN nuclear foci induced by the DNA damage caused by camptothecin are co-localized with RPA in the S-phase (Sakamoto et al., 2001). In addition, WRN helicase forms or unwinds the Holliday junction intermediate associated with a regressed replication fork (Sharma et al., 2004; Machwe et al., 2007). These observations suggest that the WRN helicase is involved in the re-initiation of a stalled replication fork. WS cells also show hypersensitivity to 4NQO that induces oxidative damage (Gebhart et al., 1988). Since accumulation of oxidative DNA damage is associated with aging, it is suggested that the WRN helicase is associated with one of the oxidative repair mechanisms, base excision repair (BER), and is known to interact with BER factors, polδ, polβ, PCNA, RPA, FEN-1, and PARP-1 (Figure 1; Rossi et al., 2010). Furthermore, the WRN helicase unwinds a BER substrate produced by uracil-DNA glycosylase and AP endonuclease (Ahn et al., 2004). It is also known that the helicase interacts with the double-strand break repair factors Ku, DNA-PKcs, and the Mre11-Rad50-Nbs1 complex, as well as the telomeric DNA protecting proteins, TRF1, TRF2, and POT1 (Figure 1; Shimamoto et al., 2004; Rossi et al., 2010). Additionally, Tahara et al. (1997) reported abnormal telomere dynamics in WS lymphoblastoid cell lines (LCLs) with weak or no telomerase activity. These findings suggest that the WRN helicase is involved in telomere metabolism. WRN helicase is shown to resolve Holliday junctions (Sharma et al., 2004), G-quadruplexes formed in telomere G-rich sequences (Mohaghegh et al., 2001), and higher-ordered DNA structures, such as the D-loop (Opresko et al., 2004). These DNA structures formed at telomere ends must be resolved during DNA replication to be accessible to DNA polymerases and telomerase, therefore, WRN helicase might function in the resolution of higher order structures in telomeric DNA.
FIGURE 1.

Function of WRN helicase. WRN helicase functions through interaction with factors involved in replication (RPA, PCNA, FEN-1, Topoisomerase I), base excision repair (BER; polδ, polβ, PCNA, RPA, FEN-1, PARP-1), double-strand break repair (Ku, DNA-PKcs, Mre11-Rad50-Nbs1 complex), and telomere maintenance, (TRF1, TRF2, POT1).
ROLES OF TELOMERE IN REPLICATIVE LIFESPAN AND IMMORTALITY
Telomeres, the ends of linear chromosomes in eukaryotes, are ribonucleoprotein-containing specialized structures essential for the protection of chromosomes from a sensing mechanism of double-stranded DNA breaks (Chan and Blackburn, 2004). Mammalian telomeres are composed of TTAGGG repeat sequences, while their specific binding protein complex, shelterin, is composed of the six proteins TRF1, TRF2, RAP1, TIN2, POT1, and TPP1. The chromosome ends are capped by t-loop structures formed by the telomeric DNA and shelterin complex to protect them from DNA damage responses (Palm and de Lange, 2008). In normal human cells, progressive telomere shortening occurs with each successive cell division because of the “end replication problem,” wherein regions of RNA primers involved in lagging strand DNA synthesis at most chromosome ends cannot be replaced with DNA during DNA replication (Harley et al., 1990; Levy et al., 1992). Most of the cells in the human body, such as terminally differentiated cells, have no detectable telomerase activity. Further, tissue stem cells such as hematopoietic stem cells (Vaziri et al., 1994; Allsopp et al., 2001, 2003), epidermal stem cells (Flores et al., 2005), and neural stem cells (Ferron et al., 2004) do not exhibit substantial telomerase activity that can add telomeric repeats sufficient to prevent their chromosomal ends from attrition with successive cell division, which is a major cause of human and other organismal aging (Blasco, 2007). On the other hand, germline stem cells and cancer cells express high levels of telomerase that maintains telomere length sufficient for their immortality (Flores et al., 2006). The human telomerase holoenzyme complex consists of a telomerase reverse transcriptase subunit, hTERT, and a template RNA, TERC, which are the basic components required for catalytic activity. (Egan and Collins, 2012) In addition, it also consists of other accessory proteins, including dyskerin, NHP2, NOP10, and NAF1 required for its assembly and stability (Egan and Collins, 2012). Introduction of hTERT is necessary and sufficient for the activation of telomerase in cells, as other components are already expressed in most normal cells and tissues (Nakayama et al., 1998; Chang et al., 2002). hTERT can elongate telomeres, extend the lifespan of normal cells, and immortalize cells such as dermal diploid fibroblasts (Bodnar et al., 1998; Vaziri and Benchimol, 1998; Jiang et al., 1999; Morales et al., 1999). Homologous recombination between telomeres, known as ALT (alternative lengthening of telomeres) is an alternative mechanism for the maintenance of telomere length, and has been observed in subsets of cancer cells, telomerase-deficient ESCs and iPSCs (Dunham et al., 2000; Niida et al., 2000; Wang et al., 2012). These findings indicate that the telomerase-dependent and -independent mechanisms of telomere maintenance are essential for cellular immortality.
WS FIBROBLASTS EXHIBIT PREMATURE REPLICATIVE SENESCENCE
Intrinsic DNA damage caused by the loss of WRN helicase could activate stress responses leading to cellular senescence. Senescence is defined as a state of permanent cell cycle arrest mediated by the p53-p21Cip1/Waf1 and p16INK4A-RB pathways. It is one of the tumor suppressor mechanisms exerted in cells that undergo replicative aging with telomere attrition, generation of reactive oxygen species, abnormal proliferation by oncogene activation, and DNA damage activated by DNA damaging agents such as ionizing radiation (Kuilman et al., 2010; Salama et al., 2014). Stress-associated p38 mitogen-activated protein kinase is constitutively activated in WS fibroblasts (Davis et al., 2005). Activation of p38 is known to mediate cellular senescence in the presence of elevated p21 levels (Haq et al., 2002; Iwasa et al., 2003), and p38 inhibitors can suppress premature senescence phenotypes of WS fibroblasts by reducing p21 expression (Davis et al., 2005). These observations indicate that p38 is a major mediator of the reduced replicative lifespan of WS fibroblasts. Meanwhile, activation of p38 also mediates induction of the senescence-associated secretory phenotype (SASP; Freund et al., 2011) that is the hallmark of aging. It is widely accepted that age-associated inflammatory responses contribute to human aging mechanisms (Goto, 2008). WS fibroblasts express inflammatory cytokines (Kumar et al., 1993), and WS is associated with inflammatory conditions responsible for common age-associated diseases, such as atherosclerosis, diabetes, and osteoporosis (Rubin et al., 1992; Murano et al., 1997; Yokote et al., 2004a; Davis and Kipling, 2006). Taken together, these findings suggest that premature replicative senescence with concomitant induction of p21 and SASP, mediated by the activation of p38, could be pathogenic hallmarks of WS.
TELOMERASE BYPASSES PREMATURE REPLICATIVE SENESCENCE IN WS FIBROBLASTS
As mentioned previously, WRN helicase might play an important role in telomere maintenance. This has been verified by Crabbe et al. (2004) wherein, defects in WRN helicase caused impairment of telomeric lagging-strand synthesis and accelerated telomere loss during DNA replication. Moreover, the telomere loss caused by mutation in the WRN gene involved telomere dysfunction such as chromosome end fusions (Crabbe et al., 2007). It is postulated that the absence of WRN causes stalled replication forks at the sites of unresolved G-quadruplexes at the lagging telomere, which would produce degradable substrates for factors involved in DNA repair and recombination, leading to accelerated telomere shortening (Figures 2A,B; Multani and Chang, 2007). More importantly, telomerase prevented sister telomere loss (STL) caused by defective telomeric lagging-strand synthesis and suppressed chromosome end fusions in WRN-deficient cells (Crabbe et al., 2004, 2007). These results demonstrate that telomerase can provide WS fibroblasts with a complementation effect by adding telomeric DNA “TTAGGG” to lagging telomeres that are lost during replication (Figure 2C). Since telomerase is also known to bypass premature replicative senescence in WS fibroblasts (Wyllie et al., 2000), it is suggested that premature senescence in WS cells might be caused by defects in telomeric lagging-strand synthesis followed by telomere loss during DNA replication (Sugimoto, 2014).
FIGURE 2.

Telomerase bypasses premature replicative senescence in WS fibroblasts. (A) G-quadruplexes at the lagging telomere are normally unwound by WRN helicase, making it possible to complete replication of lagging strand G-rich telomeres. (B) The absence of WRN causes stalled replication forks at the sites of unresolved G-quadruplexes at the lagging telomere, which would produce degradable substrates for factors involved in DNA repair and recombination, leading to accelerated telomere shortening. (C) Telomerase can add telomeric DNA “TTAGGG” to lagging telomeres that are lost during replication in WS cells, which overcomes the lack of WRN, enabling complete replication of lagging strand G-rich telomeres. This figure is based on reference (Multani and Chang, 2007).
PATHOLOGY IN RECENT WS PATIENTS AND THEIR LIFESPAN
Although WS patients usually grow normally until they reach the late teens, they generally exhibit short stature during adulthood due to impaired maturation. In their 20s and 30s, WS patients start to prematurely develop common age-associated diseases, including cataract, graying of hair and hair loss, atrophic skin, skin ulcers, abdominal fat accumulation, osteoporosis, insulin resistant diabetes mellitus, hypogonadism, atherosclerosis, and cancer (Epstein et al., 1966; Goto, 1997; Mori et al., 2001). Recent increase in life expectancy of patients with WS as well as normal individuals suggest that present-day environment, including diet and medical treatment, might have an effect in delaying and/or improving common age-associated diseases. The clinical review of recent WS case reports was updated in a recent study (Goto et al., 2013). In addition, a nation-wide epidemiological survey was conducted in Japan from 2009 to 2011 to elucidate the current clinical picture of WS, as most patients suffering from this disease are of Japanese origin (Takemoto et al., 2013).
It was found that from 2004 to 2008, patients with WS generally survived until their early 50s; this life expectancy is higher than that in 1966 (below the age of 40), with malignancy and cardiac infarction being the major causes of death (Goto et al., 2013). Several symptoms such as short stature with stocky trunk, bilateral cataracts, graying of hair and hair loss, osteoporosis, and atherosclerosis are still the hallmarks of WS. Skin abnormalities including atrophy, sclerosis, ulcers, pigmentation, and subcutaneous calcification have also been observed recently in most WS patients. Endocrine and metabolic diseases including insulin-resistant diabetes mellitus, hypogonadism, and hyperlipidemia are constantly reported, but not observed in all patients with this disease (Goto et al., 2013).
Our recent epidemiological survey revealed that progeroid changes of the hair, bilateral cataracts, soft-tissue calcifications, and skin abnormalities, including atrophy and intractable ulcers, are the most prominent diagnostic clinical features of WS (Takemoto et al., 2013). Bird-like face and abnormal voice are also the discriminating features of WS. The following features are not observed in all WS patients but are critical symptoms: endocrine and metabolic diseases, such as glucose and/or lipid metabolism abnormalities; bone diseases, such as osteoporosis; atherosclerosis; hypogonadism; short stature; and malignancy (Okabe et al., 2012; Onishi et al., 2012; Takemoto et al., 2013). These endocrine and metabolic symptoms are common age-associated diseases in normal individuals, and recent longevity in the general Japanese population could be attributed to recent advances in medicine. In the same way, the use of current medical care for the treatment of the several critical symptoms of patients with WS might increase their life expectancy (Yokote and Saito, 2008; Goto et al., 2013).
CURRENT STRATEGIES FOR TREATMENT OF WS
Recent advances in drug therapy for common age-associated diseases are also available for patients with WS. For example, in most of these patients, insulin-resistant diabetes improved by administration of the PPAR-γ agonist pioglitazone that is generally used for the treatment of type 2 diabetes mellitus. In these cases, pioglitazone ameliorated glycemic irregularities and hyperlipidemia as well as impaired insulin sensitivity (Yokote et al., 2004b; Honjo et al., 2008). Insulin-resistant diabetes is also improved by treatment with the dipeptidyl peptidase-4 inhibitor sitagliptin in patients with WS (Kitamoto et al., 2012; Watanabe et al., 2013). Furthermore, hyperlipidemia is one of the predictors of coronary artery disease in WS, and statins have been shown to address this issue in patients with WS (Kobayashi et al., 2000). Since premature senescence in WS cells seem to be caused by accelerated telomere loss during DNA replication (Crabbe et al., 2007), the relationship between telomere and these drugs should be considered in the light of protection against aging. It was reported that a short telomere length is a risk factor for coronary heart disease, which is attenuated when combined with the intake of statins (Brouilette et al., 2007). This may be corroborated by a previous finding which suggests that statins prevent telomere dysfunction caused by the loss of telomere repeat-binding factor, TRF2, in cultured endothelial progenitor cells (Spyridopoulos et al., 2004). A PPAR-γ agonist was reported to increase the expression of TRF2 and prevent apoptosis of endothelial progenitor cells (Gensch et al., 2007). Thus, it is likely that improvement in the condition of patients with WS is associated with the effects of these drugs.
INCREASED LONGEVITY AND QOL IN WS PATIENTS
As mentioned earlier, recent protocols for drug therapy in patients with WS have led to an improvement in their lifespan. The average life expectancy of patients with WS at the Chiba University hospital has increased by more than 10 years from 1987 to 2007 (Yokote and Saito, 2008), and a most recent record reported that the longest-living patient had survived until the age of 64 (Yokote, Personal communication). This retrospective study revealed that 7 of the 11 living patients with WS after 1997 had a history of taking statins and/or pioglitazone, suggesting that medical procedures, possibly improved by drug development, as well as early detection and early intervention may increase the life expectancy of patients with WS. However, improvement in the QOL of these patients is also imperative, as skin ulcers are known to have a negative effect on it (Goto et al., 2013).
Excruciatingly painful skin ulcers in patients with WS are considered to be caused by multiple factors, including dermal fragility caused by a decrease in connective and fat tissues, a delay in wound healing caused by impaired proliferative ability of dermal cells, and poor circulation associated with diabetes and arteriosclerotic lesions, and are extremely difficult to treat (Yeong and Yang, 2004; Takemoto et al., 2013). Severe ulcers are commonly found in heels, ankles, elbows, and other areas subject to pressure, and can be surgically treated in some cases only (Yeong and Yang, 2004). However, drug therapy including basic fibroblast growth factor spray, hydrocolloid dressing, and PGE1 preparation have little effect on the ulcers in WS, although it is reported that topical platelet-derived growth factor-BB and the endothelin receptor antagonist bosentan has shown some beneficial effects (Wollina et al., 2004; Noda et al., 2011). Most deep and severe leg ulcers with necrosis require amputations (Yeong and Yang, 2004; Goto et al., 2013). In spite of the increase in the average of life expectancy in WS patients due to the recent improvement in drug therapy for common age-associated diseases, the decrease in QOL caused by excruciatingly painful ulcers is still a major problem that needs to be addressed in these patients.
Intractable skin ulcers also include diabetic ulcers, stasis ulcers, arterial ulcers associated with arteriosclerotic obliteration and Buerger’s disease, ulcers associated with connective tissue disease, and radiation-induced ulcers. These ulcers might be treated with debridement ointment under infection control for the enhancement of granulation tissue with vascularization and connective tissue repair (Brem and Lyder, 2004; Sorensen et al., 2004). If the affected area contains necrotic tissue, surgical debridement would be performed followed by skin grafting or flap as required (Sorensen et al., 2004). However, as described above, ulcers in patients with WS heal poorly because of atrophic connective and fat tissues, impaired proliferative ability of dermal fibroblasts, and poor circulation, leading to limited healing of skin grafts and flap as a result of defective granulation tissue formation. At present, there is an urgent need to develop an effective therapeutic strategy for the treatment of severe ulcers in patients with WS.
TELOMERE REJUVENATION IN iPSCs BY REPROGRAMMING
Induced pluripotent stem cells are similar to ESCs, which are generated from individual somatic cells such as dermal fibroblasts, blood cells, and other cell types by the introduction of several pluripotency genes, including Oct3/4, Sox2, Klf4, c-myc, Nanog, and Lin-28 (Takahashi and Yamanaka, 2006; Takahashi et al., 2007; Yu et al., 2007; Aoi et al., 2008; Stadtfeld and Hochedlinger, 2010; Okita and Yamanaka, 2011). Because of their ability to differentiate into various cell types as well as their unlimited proliferative potential, iPSCs, like ESCs, are expected to contribute to regenerative medicine (Takahashi et al., 2007; Stadtfeld and Hochedlinger, 2010). However, unlike ESCs, iPSCs are generated from individual patients, therefore, they can be applied to tailor-made medicine based on syngeneic cell transplantation without allograft rejection (Robinton and Daley, 2012; Lin et al., 2013; Takahashi and Yamanaka, 2013). Moreover, disease-specific iPSCs that can differentiate into multiple cell types can be used to resolve the pathogenic processes of several diseases where cell types available from patients are usually limited to patient-derived lymphocytes and/or fibroblasts.
The reprogramming process includes several key events that define the mechanism of reprogramming of somatic cells into an ES-like state. One proposed idea separates the process into three distinct phases in human and mouse (Figure 3; Samavarchi-Tehrani et al., 2010; Golipour et al., 2012; David and Polo, 2014). In the Oct3/4, Sox2, Klf4, and c-Myc (OSKM)-driven reprogramming of mouse embryonic fibroblasts, changes in the expression of genes related to the mesenchymal-to-epithelial transition (MET) are observed in the initiation phase (Mikkelsen et al., 2008; Samavarchi-Tehrani et al., 2010; David and Polo, 2014), along with the loss of mesenchymal cell surface markers, CD44 and Thy1, and a gain of the pluripotency markers, alkaline phosphatase activity, and ESC markers (Stadtfeld et al., 2008; Samavarchi-Tehrani et al., 2010; O’Malley et al., 2013; David and Polo, 2014). MET is also observed in reprogramming of human fibroblasts. Using Tra-1-60 positive intermediate reprogrammed cells, similar events are observed during reprogramming of human fibroblasts (Figure 3; Takahashi et al., 2014). MET-associated gene expression change occurs in early phage, where induction of epithelial marker genes such as CDH1 and EpCAM, and suppression of mesenchymal genes including SNAI2, ZEB1, and FN1 is observed. In transient stage, intermediate cells transiently express genes related to the primitive streak, including BRACHYURY, MIXL1, CER1, LHX1, and EOMES (Figure 3; Takahashi et al., 2014). These events, along with the later phases, are directly or indirectly regulated though the OSKM transcription network by which chromatin decondensation, loss of suppressive histone modification, DNA demethylation, and gain of active histone modification are directed in the genes to be activated, while a concomitantly opposite regulation is observed in the lineage-specific genes to be inactivated (Apostolou and Hochedlinger, 2013; Buganim et al., 2013; Papp and Plath, 2013). During late stage, activation of the first pluripotency-associated genes, including endogenous Oct3/4, Nanog, Sall4, and Esrrb, followed by subsequent activation of Sox2 and Dppa4 is essentially required to initiate the transformation into pluripotent cells (Figure 3; Stadtfeld et al., 2008; Samavarchi-Tehrani et al., 2010; Buganim et al., 2012; Golipour et al., 2012; David and Polo, 2014; Takahashi et al., 2014). The cells activating the first pluripotency genes successfully shift into late stage from transient stage, leading to the accomplishment of a full reprogramming state that ensures sustained self-renewal ability and differentiation potential. In late stage, successive passages are required to eliminate small differences in gene expression profiles between human iPSCs and hESCs, and to erase epigenetic memory derived from somatic cells used in reprogramming (David and Polo, 2014).
FIGURE 3.
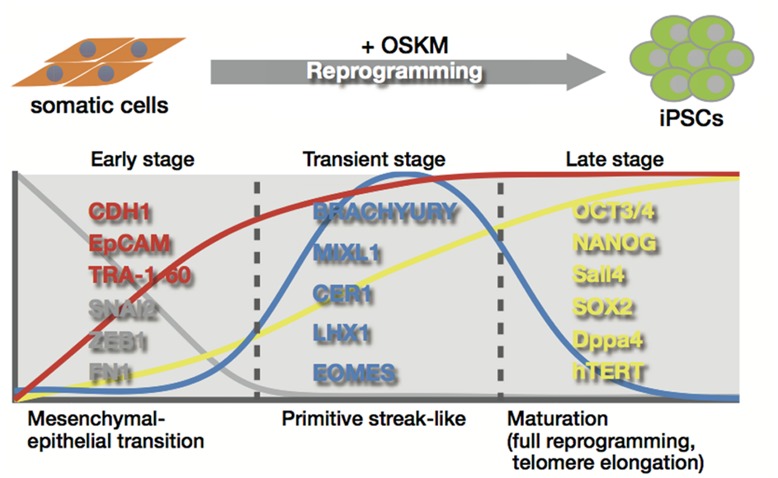
Telomere rejuvenation by reprogramming. The reprogramming process is divided into three distinct stages; early stage, transient stage, and late stage. Telomerase activation and telomere elongation occur in the late stage by induction of telomerase reverse transcriptase subunit and the telomerase RNA (TERC) component, which induce telomere rejuvenation similar in length and their epigenetic state to those of ESCs.
Late stage also involves telomerase activation and telomere elongation that provide somatic cells with infinite proliferative potential (Figure 3; Takahashi et al., 2007; Stadtfeld et al., 2008; Marion et al., 2009b; Wang et al., 2012). During the reprogramming process, telomerase is activated by induction of telomerase reverse transcriptase subunit (hTERT in humans) and the telomerase RNA component (TERC; Takahashi et al., 2007; Agarwal et al., 2010; Ji et al., 2013), which are likely to be regulated in stem cells by Wnt/β-catenin signaling with KLF4 and/or TCF4 and OCT3/4 and NANOG, respectively (Agarwal et al., 2010; Wong et al., 2010; Hoffmeyer et al., 2012; Zhang et al., 2012). Although c-Myc is known to induce telomerase activity through direct activation of the hTERT gene (Wang et al., 1998; Wu et al., 1999), it might be less involved in telomerase activation during reprogramming, because OSK of the Yamanaka 4 factors without c-Myc is shown to generate iPSCs with enough telomerase activity (Marion et al., 2009b). hESCs have much longer telomeres with higher expression levels of hTERT and stronger telomerase activity than the differentiated cells (Thomson et al., 1998), and telomerase-dependent telomere maintenance is critical for the growth of mammalian ESCs (Niida et al., 1998). Telomere elongation accompanied by telomerase activation occurs during reprogramming, leading to acquisition of telomeres similar in length to those of ESCs after reprogramming (Marion et al., 2009b). iPSC generation also involves epigenetic alterations to ESC-like states with reduced histone codes associated with heterochromatin, and enhanced transcription at the telomere loci. Elevated frequencies in telomeric sister chromatid exchanges and telomere elongation were observed even when old cells with shortened telomeres were used (Marion et al., 2009b). These observations indicate that telomeres are rejuvenated toward an ESC-like state during reprogramming (Figure 3).
However, telomerase-deficient cells with critically shortened telomeres fail to be reprogrammed, suggesting that iPSC generation requires a minimum telomere length to be reprogrammed (Marion et al., 2009b). Critically shortened telomeres resulting from the progression of replicative aging in normal human cells lose the protective function of the chromosomal ends and are recognized as endogenous DNA damage. As a result, dysfunctional telomeres induce DNA damage responses including the activation of ataxia telangiectasia mutated (ATM), ATM- and Rad3-related (ATR), and downstream CHK1 and CHK2 kinases, as well as the phosphorylation of p53, inducing cellular senescence via stimulation of the expression of the cyclin-dependent kinase inhibitor (CDKI) p21 (d’Adda di Fagagna et al., 2003; Herbig et al., 2004; Deng et al., 2008). It has been shown that the activation of p53 significantly suppresses reprogramming efficiency, known as the reprogramming barrier (Hong et al., 2009; Kawamura et al., 2009; Li et al., 2009; Utikal et al., 2009), while suppression of p53 improves the reprogramming efficiency in cells with critically shortened telomeres (Marion et al., 2009a). These findings demonstrate that activation of telomerase during reprogramming plays a pivotal role not only in telomere elongation with chromatin state characteristic of ESCs, but also in the restoration and maintenance of the protective functions of the telomere at the chromosomal ends, in order to suppress DNA damage responses.
iPSCs AS A POTENTIAL STRATEGY FOR WS TREATMENT
As described above, skin ulcers in patients with WS heal poorly, and so far no effective therapy has been developed to treat them or the other symptoms associated with WS. Thus, there is an urgent need to develop a new treatment strategy in order to improve the health and QOL of patients with WS. Understanding the molecular basis and development of therapeutics requires an appropriate disease modeling system. Primary cells from affected tissues of these patients are required for better understanding of the pathogenic processes and complex features involved with this disease. However, their use is usually limited to patient-derived lymphocytes and/or fibroblasts, which are difficult to propagate in culture for extended periods of time. Thus, regenerative medicine such as autologous cell transplantation could be used as a therapeutic strategy for WS, which provides cells with high proliferative ability and differentiation potential in large quantities over a long period.
The skin, composed of epidermis and dermis, is one of the main affected tissues in WS. A recent study demonstrated that hESCs can generate a homogeneous population of epithelial cells expressing postnatal keratinocyte markers in squamous epithelia, and these hESC-derived keratinocytes could reconstitute a functional pluri-stratified epithelium (Guenou et al., 2009). On the other hand, human epidermal keratinocytes can reconstitute stratified epithelium in culture, and it is known that expanded culture of epidermal stem cells from a tiny skin biopsy can cover the whole body surface of an individual, because of the high proliferative potential of these cells (De Luca et al., 2006), thus raising the argument as to whether pluripotent cell-derived epithelium can be used for clinical purposes (Pellegrini and Luca, 2009). In the case of WS, as the regenerative potential of adult skin cells is expected to be hampered due to their impaired proliferative ability, the concerns over premature senescence phenotype in cells from these patients might be eliminated by the development of rejuvenated resources. Patient-specific iPSCs might be a potential candidate that can meet these requirements. The epoch-making invention of iPSCs has the potential to bring innovation to regenerative medicine as well as drug discovery, as these cells are known to possess the ability to differentiate into all cell types, including those belonging to the skin, hair root, blood vessel, bone, and pancreatic islets.
GENERATION OF iPSCs FROM WS PATIENT CELLS
During the reprogramming process, both telomere and telomerase play protective roles at chromosomal ends against DNA damage responses, causing a reprogramming barrier (Marion et al., 2009a,b). Fibroblasts from patients with WS exhibit premature senescence caused by accelerated telomere loss during DNA replication (Salk et al., 1981b; Crabbe et al., 2007). Thus, it is interesting to note that forced expression of the telomerase catalytic gene hTERT in WS fibroblasts bypassed the phenotype, raising the question as to whether WS fibroblasts can be reprogrammed into iPSCs. In addition, there are doubts as to whether WS iPSCs, if successfully generated, can maintain hESC-like characteristics during long-term culture. Inconsistent consequences of the generation of patient-specific iPSCs from dyskeratosis congenita (DKC), another disease involving telomere abnormalities, have been reported (Agarwal et al., 2010; Batista et al., 2011). Batista et al. demonstrated that DKC iPSCs presented with progressive telomere shortening and loss of self-renewal ability in long-term culture (Batista et al., 2011). Therefore, it is important to evaluate the properties of iPSCs derived from the cells of the patient with telomere dysfunction over the long term. The findings from a recent study by Cheung et al. demonstrating the successful generation of disease-specific iPSCs from cells of patients with WS were in accordance with one of our current works, leading us to believe that reprogramming repressed premature senescence phenotypes in WS cells (Cheung et al., 2014; Shimamoto et al., 2014).
WS iPSCs were generated from the patient’s fibroblasts and were quite similar to normal iPSCs in their characteristics as pluripotent stem cells, including their hESC-like morphology, expression of pluripotency genes, and hESC-specific surface markers, global gene expression profiles, embryoid body (EB) formation and subsequent differentiation into three embryonic germ layers, and teratoma formation. The WS iPSCs maintained their telomeres with reactivation of endogenous telomerase by induction of hTERT as well as other components of telomerase, such as TERC and DKC1, and were sustained in culture for more than 35 (Cheung et al., 2014) and 150 passages (Shimamoto et al., 2014) without morphological changes and loss of growth capacity. These observations indicate that induction levels of telomerase activity during reprogramming are sufficient for generation and subsequent cloning and maintenance of iPSCs from WS fibroblasts (Figure 4).
FIGURE 4.
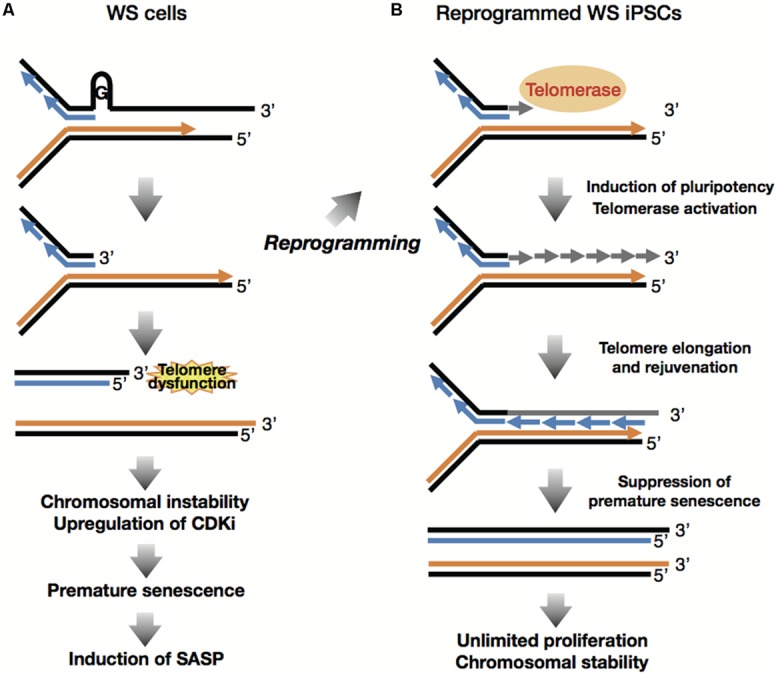
Reprogramming suppresses premature senescence phenotypes. (style="color:gray" A) Accelerated telomere shortening during replication induces telomere dysfunction and DNA damage response at the lagging telomere, leading to induction of premature senescence and SASP following chromosomal instability and upregulation of CDK inhibitors. (B) Reprogramming involves induction of pluripotency and global epigenetic alteration toward ESC-like state, which suppress senescence phenotypes including CDKi and SASP. Reprogramming also overcomes the lack of WRN through activation of telomerase, consequently inducing telomere elongation and cellular immortality.
REPROGRAMMING SUPPRESSES PREMATURE SENESCENCE PHENOTYPES AND GENOMIC INSTABILITY OF WS FIBROBLASTS
Expression levels of senescence-associated genes including the CDKIs as well as the SASP factors, were compared between WS fibroblasts and WS iPSCs, because it is widely accepted that age-associated inflammatory responses, including SASP, contribute to human aging mechanisms (Goto, 2008). The results demonstrated that in addition to the CDKI genes p21Cip1/Waf1 and p16INK4A, the SASP genes such as IL-6, gp130, IGFBP5, IGFBP7, ANGPTL2, and TIMP1 were highly expressed in cells of patients with WS as compared with PDL-matched normal fibroblasts. However, the expression levels of the same genes were suppressed in their iPSC derivatives to the level generally seen in normal iPSCs. These observations revealed that reprogramming suppresses and rejuvenates the premature aging phenotypes of WS fibroblasts (Figure 4; Shimamoto et al., 2014).
WS is characterized by genomic instability and chromosomal aberrations, including translocations, inversions, and deletions that have been observed during culture of patient-derived cells (Salk et al., 1981a). As the generation and subsequent maintenance of iPSCs involve extensive cell division, WS iPSCs may acquire additional chromosomal abnormalities during the process. In one of our recent works, we performed a chromosomal profiling analysis which indicated karyotype stability in WS iPSCs in long-term culture (Shimamoto et al., 2014). We performed G-banding stain and multicolor fluorescence in situ hybridization, and showed that 3 of 6 WS iPSC clones had the same karyotypes as their parental cells after approximately 100 passages, suggesting that karyotypes of WS cells are stabilized following reprogramming (Figure 4; Shimamoto et al., 2014). Normal human iPSCs are known to acquire genomic instability with high incidence of additions, deletions and translocations (Martins-Taylor et al., 2011; Taapken et al., 2011). Thus, given the genomic instability of WS cells, these data reveal the unexpected maintenance of chromosomal profiles in WS iPSC clones during long-term culture, indicating the possibility of its application clinically, although general risk factors, including genetic and epigenetic abnormalities (Gore et al., 2011; Hussein et al., 2011; Lister et al., 2011; Pera, 2011) and the potential for tumorigenicity (Kiuru et al., 2009; Ben-David and Benvenisty, 2011) and immunogenicity (Zhao et al., 2011; Araki et al., 2013) must be taken into consideration.
RECAPITULATION OF PREMATURE SENESCENCE PHENOTYPES IN DIFFERENTIATED CELLS
Differentiated cells including mesenchymal stem cells (MSCs) and other cell types derived from WS iPSCs were examined to determine their roles as models of WS, because the WS iPSCs do not exhibit any of the characteristic features of the syndrome. Cheung et al. demonstrated premature senescence of WS MSCs with elevated expression levels of p53, p21, and p16; accelerated telomere shortening; and impaired telomeric lagging-strand synthesis that causes telomere loss and dysfunction (Cheung et al., 2014). They also showed that WS iPSC-derived neural progenitor cells (NPCs) expressing telomerase activity maintained telomere and proliferative capacity with NPC phenotypes, and treatment with telomerase inhibitor was seen to decrease growth and increase the incidence of γH2AX in WS NPCs. These results, together with the fact that hTERT rescues premature senescence and telomere dysfunction, suggest that premature senescence in WS MSCs is due to insufficient levels of telomerase activity downregulated during differentiation (Figure 5A). Thus, adequate telomerase activity could maintain tissue stem cell function in WS (Cheung et al., 2014).
FIGURE 5.

Healthy stem cells from gene-corrected WS iPSCs. (A) WS iPSC-derived stem cells with insufficient levels of telomerase activity induce premature senescence and stem cell dysfunction. (B) Gene-corrected WS iPSCs can produce healthy stem cells by suppressing premature senescence due to functional WRN helicase.
Differentiated cells derived from WS iPSC EBs were also examined for their growth defects. We found that the cells underwent premature senescence with a higher rate of SA-β-gal positive cells, upregulation of p21 concomitantly with downregulation of hTERT and induction of SASP genes (Figure 5A; Shimamoto et al., 2014). Since EB-derived differentiated cells include a variety of cell types originating from the three germ layers, these results suggest that EB-mediated iPSC differentiation could provide a simple and rapid method for the identification of cell lineages other than the MSCs in WS.
ATM, a causative gene for premature aging syndrome ataxia telangiectasia (AT) is required for the maintenance of stem cells including hematopoietic and spermatogonial stem cells (Ito et al., 2004; Takubo et al., 2008). Furthermore, the pathophysiology of AT might be associated with dysregulation of the reservoir of adult stem cell populations (Wong et al., 2003). Recent findings have shown dysfunction of vascular smooth muscle cells (VSMCs) and their progenitor SMCs in Hutchinson–Gilford Progeria syndrome (HGPS; Liu et al., 2011; Zhang et al., 2011). Taken together, these findings suggest that premature aging syndromes including WS, AT, and HGPS are stem cell dysfunction-associated diseases.
GENE-CORRECTED WS iPSCs AND ITS CLINICAL APPLICATION
Autologous cell transplantation can be selected as one of the therapeutic strategies for treating the intractable symptoms of WS, including skin ulcers. Clinical application of iPSCs requires unlimited proliferative ability and differentiation potential into various cell types with healthy conditions that could replace the affected area. Although WS iPSCs are almost indistinguishable from normal iPSCs in many aspects, differentiated cells from WS iPSCs manifest premature aging phenotypes (Figure 5A; Cheung et al., 2014; Shimamoto et al., 2014). Thus, gene-corrected WS iPSCs could offer a unique treatment strategy for patients with WS. Recent progress in gene therapy and genome engineering technology provides powerful tools for genome editing, including zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and RNA-guided engineered nucleases derived from the bacterial clustered regularly interspaced short palindromic repeat (CRISPR)-Cas system (Kim and Kim, 2014; Li et al., 2014). This technology could be used for correction of the disease-specific mutations in iPSCs by gene targeting (Yusa et al., 2011; Suzuki et al., 2014) and establishment of disease-specific iPSCs from wild-type iPSC lines (Soldner et al., 2011). Because WS is inherited in an autosomal recessive manner, a single gene-targeting event of specific mutations in WRN loci might be sufficient for recovery from WS, which could be confirmed by examining differentiated cells such as MSCs for the restoration of telomere dysfunction and premature growth defects (Figure 5B). In addition to the safety of iPSCs (Okano et al., 2013), evaluation of whole-genome sequencing and epigenomic analysis will be needed before their clinical application because WS patient cells are reported to have chromosomal aberrations including translocations, inversions, and deletions (Salk et al., 1981a). Furthermore, the differentiation potential and corrected phenotypes in gene-corrected WS iPSCs must be warranted for their clinical use. For example, reconstituted epithelium might be clinically applicable for skin ulcers after finding evidence that gene-corrected WS iPSC-derived keratinocytes could form functional pluri-stratified epithelium with fibroblast-containing fibrin dermal matrix in vivo (Guenou et al., 2009).
CONCLUSION
Recent findings including ours, demonstrate that reprogramming bypasses premature senescence and suppresses genomic instability in WS cells, leading to sustained undifferentiated states with the ability to differentiate into three embryonic germ layers over the long term. It is noteworthy that WS iPSCs exhibited stable chromosomal profiles, and this unexpected property might be achieved by the expression of the endogenous telomerase gene induced during reprogramming. As normal iPSCs exhibited higher expression levels of WRN protein than normal fibroblasts, WRN helicase might have a role in chromosomal stability as well as telomere maintenance in iPSCs. Thus, thorough safety tests, especially concerning genetic and epigenetic abnormalities and the potential for tumorigenicity, will be necessary before the clinical application of these cells. The use of WS iPSCs will enhance our understanding of the pathogenic processes and modeling of complex features associated with WS. In addition, it can provide opportunities for drug screening and the discovery and development of new strategies for the treatment this disease. Finally, although challenges and concerns remain regarding the general safety and risk of iPSCs as well as the WS-specific defects, a recent clinical trial using patient-specific iPSCs at the RIKEN Center for Developmental Biology (CDB) will encourage and promote stem cell research toward clinical application (Reardon and Cyranoski, 2014), at the same time, we need to consider the results of the RIKEN CDB clinical trial in a calm manner.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a Grant-in-Aid for Challenging Exploratory Research No. 25670030 (to Akira Shimamoto), for Scientific Research No. 26115002 on Innovative Areas “Stem Cell Aging and Disease” (to Koutaro Yokote ), and for Scientific Research No. 20014015 (to Hidetoshi Tahara) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. This work was also supported by a Health and Labor Sciences Research Grant from the Ministry of Health Labor and Welfare of Japan (to Akira Shimamoto and Koutaro Yokote). The authors would like to thank Enago (www.enago.jp) for the English language review.
REFERENCES
- Agarwal S., Loh Y. H., McLoughlin E. M., Huang J., Park I. H., Miller J. D., et al. (2010). Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature 464 292–296 10.1038/nature08792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn B., Harrigan J. A., Indig F. E., Wilson D. M., III, Bohr V. A. (2004). Regulation of WRN helicase activity in human base excision repair. J. Biol. Chem. 279 53465–53474 10.1074/jbc.M409624200 [DOI] [PubMed] [Google Scholar]
- Allsopp R. C., Cheshier S., Weissman I. L. (2001). Telomere shortening accompanies increased cell cycle activity during serial transplantation of hematopoietic stem cells. J. Exp. Med. 193 917–924 10.1084/jem.193.8.917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allsopp R. C., Morin G. B., DePinho R., Harley C. B., Weissman I. L. (2003). Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood 102 517–520 10.1182/blood-2002-07-2334 [DOI] [PubMed] [Google Scholar]
- Aoi T., Yae K., Nakagawa M., Ichisaka T., Okita K., Takahashi K., et al. (2008). Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 321 699–702 10.1126/science.1154884 [DOI] [PubMed] [Google Scholar]
- Apostolou E., Hochedlinger K. (2013). Chromatin dynamics during cellular reprogramming. Nature 502 462–471 10.1038/nature12749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki R., Uda M., Hoki Y., Sunayama M., Nakamura M., Ando S., et al. (2013). Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nature 494 100–104 10.1038/nature11807 [DOI] [PubMed] [Google Scholar]
- Batista L. F., Pech M. F., Zhong F. L., Nguyen H. N., Xie K. T., Zaug A. J., et al. (2011). Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 474 399–402 10.1038/nature10084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David U., Benvenisty N. (2011). The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat. Rev. Cancer 11 268–277 10.1038/nrc3034 [DOI] [PubMed] [Google Scholar]
- Blasco M. A. (2007). Telomere length, stem cells and aging. Nat. Chem. Biol. 3 640–649 10.1038/nchembio.2007.38 [DOI] [PubMed] [Google Scholar]
- Bodnar A. G., Ouellette M., Frolkis M., Holt S. E., Chiu C. P., Morin G. B., et al. (1998). Extension of life-span by introduction of telomerase into normal human cells. Science 279 349–352 10.1126/science.279.5349.349 [DOI] [PubMed] [Google Scholar]
- Bohr V. A. (2008). Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem. Sci. 33 609–620 10.1016/j.tibs.2008.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem H., Lyder C. (2004). Protocol for the successful treatment of pressure ulcers. Am. J. Surg. 188 9–17 10.1016/S0002-9610(03)00285-X [DOI] [PubMed] [Google Scholar]
- Brouilette S. W., Moore J. S., McMahon A. D., Thompson J. R., Ford I., Shepherd J., et al. (2007). Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. Lancet 369 107–114 10.1016/S0140-6736(07)60071-3 [DOI] [PubMed] [Google Scholar]
- Buganim Y., Faddah D. A., Cheng A. W., Itskovich E., Markoulaki S., Ganz K., et al. (2012). Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 150 1209–1222 10.1016/j.cell.2012.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y., Faddah D. A., Jaenisch R. (2013). Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 14 427–439 10.1038/nrg3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S. R., Blackburn E. H. (2004). Telomeres and telomerase. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359 109–121 10.1098/rstb.2003.1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J. T., Chen Y. L., Yang H. T., Chen C. Y., Cheng A. J. (2002). Differential regulation of telomerase activity by six telomerase subunits. Eur. J. Biochem. 269 3442–3450 10.1046/j.1432-1033.2002.03025.x [DOI] [PubMed] [Google Scholar]
- Cheung H. H., Liu X., Canterel-Thouennon L., Li L., Edmonson C., Rennert O. M. (2014). Telomerase protects werner syndrome lineage-specific stem cells from premature aging. Stem Cell Reports 2 534–546 10.1016/j.stemcr.2014.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe L., Jauch A., Naeger C. M., Holtgreve-Grez H., Karlseder J. (2007). Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc. Natl. Acad. Sci. U.S.A. 104 2205–2210 10.1073/pnas.0609410104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe L., Verdun R. E., Haggblom C. I., Karlseder J. (2004). Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 306 1951–1953 10.1126/science.1103619 [DOI] [PubMed] [Google Scholar]
- d’Adda di Fagagna F., Reaper P. M., Clay-Farrace L., Fiegler H., Carr P., Von Zglinicki T., et al. (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature 426 194–198 10.1038/nature02118 [DOI] [PubMed] [Google Scholar]
- David L., Polo J. M. (2014). Phases of reprogramming. Stem Cell Res. 12 754–761 10.1016/j.scr.2014.03.007 [DOI] [PubMed] [Google Scholar]
- Davis T., Baird D. M., Haughton M. F., Jones C. J., Kipling D. (2005). Prevention of accelerated cell aging in Werner syndrome using a p38 mitogen-activated protein kinase inhibitor. J. Gerontol. A Biol. Sci. Med. Sci. 60 1386–1393 10.1093/gerona/60.11.1386 [DOI] [PubMed] [Google Scholar]
- Davis T., Kipling D. (2006). Werner Syndrome as an example of inflamm-aging: possible therapeutic opportunities for a progeroid syndrome? Rejuvenation Res. 9 402–407 10.1089/rej.2006.9.402 [DOI] [PubMed] [Google Scholar]
- De Luca M., Pellegrini G., Green H. (2006). Regeneration of squamous epithelia from stem cells of cultured grafts. Regen. Med. 1 45–57 10.2217/17460751.1.1.45 [DOI] [PubMed] [Google Scholar]
- Deng Y., Chan S. S., Chang S. (2008). Telomere dysfunction and tumour suppression: the senescence connection. Nat. Rev. Cancer 8 450–458 10.1038/nrc2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham M. A., Neumann A. A., Fasching C. L., Reddel R. R. (2000). Telomere maintenance by recombination in human cells. Nat. Genet. 26 447–450 10.1038/82586 [DOI] [PubMed] [Google Scholar]
- Egan E. D., Collins K. (2012). Biogenesis of telomerase ribonucleoproteins. RNA 18 1747–1759 10.1261/rna.034629.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis N. A., Groden J., Ye T. Z., Straughen J., Lennon D. J., Ciocci S., et al. (1995). The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 83 655–666 10.1016/0092-8674(95)90105-1 [DOI] [PubMed] [Google Scholar]
- Epstein C. J., Martin G. M., Schultz A. L., Motulsky A. G. (1966). Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 45 177–221 10.1097/00005792-196605000-00001 [DOI] [PubMed] [Google Scholar]
- Ferron S., Mira H., Franco S., Cano-Jaimez M., Bellmunt E., Ramirez C., et al. (2004). Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development 131 4059–4070 10.1242/dev.01215 [DOI] [PubMed] [Google Scholar]
- Flores I., Benetti R., Blasco M. A. (2006). Telomerase regulation and stem cell behaviour. Curr. Opin. Cell Biol 18 254–260 10.1016/j.ceb.2006.03.003 [DOI] [PubMed] [Google Scholar]
- Flores I., Cayuela M. L., Blasco M. A. (2005). Effects of telomerase and telomere length on epidermal stem cell behavior. Science 309 1253–1256 10.1126/science.1115025 [DOI] [PubMed] [Google Scholar]
- Freund A., Patil C. K., Campisi J. (2011). p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30 1536–1548 10.1038/emboj.2011.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhart E., Bauer R., Raub U., Schinzel M., Ruprecht K. W., Jonas J. B. (1988). Spontaneous and induced chromosomal instability in Werner syndrome. Hum. Genet. 80 135–139 10.1007/BF00702855 [DOI] [PubMed] [Google Scholar]
- Gensch C., Clever Y. P., Werner C., Hanhoun M., Bohm M., Laufs U. (2007). The PPAR-gamma agonist pioglitazone increases neoangiogenesis and prevents apoptosis of endothelial progenitor cells. Atherosclerosis 192 67–74 10.1016/j.atherosclerosis.2006.06.026 [DOI] [PubMed] [Google Scholar]
- Golipour A., David L., Liu Y., Jayakumaran G., Hirsch C. L., Trcka D., et al. (2012). A late transition in somatic cell reprogramming requires regulators distinct from the pluripotency network. Cell Stem Cell 11 769–782 10.1016/j.stem.2012.11.008 [DOI] [PubMed] [Google Scholar]
- Gore A., Li Z., Fung H. L., Young J. E., Agarwal S., Antosiewicz-Bourget J., et al. (2011). Somatic coding mutations in human induced pluripotent stem cells. Nature 471 63–67 10.1038/nature09805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M. (1997). Hierarchical deterioration of body systems in Werner’s syndrome: implications for normal ageing. Mech. Ageing Dev. 98 239–254 10.1016/S0047-6374(97)00111-5 [DOI] [PubMed] [Google Scholar]
- Goto M. (2000). Werner’s syndrome: from clinics to genetics. Clin. Exp. Rheumatol. 18 760–766. [PubMed] [Google Scholar]
- Goto M. (2008). Inflammaging (inflammation + aging): A driving force for human aging based on an evolutionarily antagonistic pleiotropy theory? Biosci Trends 2 218–230. [PubMed] [Google Scholar]
- Goto M., Imamura O., Kuromitsu J., Matsumoto T., Yamabe Y., Tokutake Y., et al. (1997). Analysis of helicase gene mutations in Japanese Werner’s syndrome patients. Hum. Genet. 99 191–193 10.1007/s004390050336 [DOI] [PubMed] [Google Scholar]
- Goto M., Ishikawa Y., Sugimoto M., Furuichi Y. (2013). Werner syndrome: a changing pattern of clinical manifestations in Japan (1917∼2008). Biosci. Trends 7 13–22. [PubMed] [Google Scholar]
- Guenou H., Nissan X., Larcher F., Feteira J., Lemaitre G., Saidani M., et al. (2009). Human embryonic stem-cell derivatives for full reconstruction of the pluristratified epidermis: a preclinical study. Lancet 374 1745–1753 10.1016/S0140-6736(09)61496-3 [DOI] [PubMed] [Google Scholar]
- Hanaoka F., Takeuchi F., Matsumura T., Goto M., Miyamoto T., Yamada M. (1983). Decrease in the average size of replicons in a Werner syndrome cell line by Simian virus 40 infection. Exp. Cell Res. 144 463–467 10.1016/0014-4827(83)90425-1 [DOI] [PubMed] [Google Scholar]
- Haq R., Brenton J. D., Takahashi M., Finan D., Finkielsztein A., Damaraju S., et al. (2002). Constitutive p38HOG mitogen-activated protein kinase activation induces permanent cell cycle arrest and senescence. Cancer Res. 62 5076–5082. [PubMed] [Google Scholar]
- Harley C. B., Futcher A. B., Greider C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature 345 458–460 10.1038/345458a0 [DOI] [PubMed] [Google Scholar]
- Herbig U., Jobling W. A., Chen B. P., Chen D. J., Sedivy J. M. (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell. 14 501–513 10.1016/S1097-2765(04)00256-4 [DOI] [PubMed] [Google Scholar]
- Hoffmeyer K., Raggioli A., Rudloff S., Anton R., Hierholzer A., Del Valle I., et al. (2012). Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science 336 1549–1554 10.1126/science.1218370 [DOI] [PubMed] [Google Scholar]
- Hong H., Takahashi K., Ichisaka T., Aoi T., Kanagawa O., Nakagawa M., et al. (2009). Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 460 1132–1135 10.1038/nature08235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo S., Yokote K., Fujishiro T., Maezawa Y., Sato S., Koshizaka M., et al. (2008). Early amelioration of insulin resistance and reduction of interleukin-6 in Werner syndrome using pioglitazone. J. Am. Geriatr. Soc. 56 173–174 10.1111/j.1532-5415.2007.01484.x [DOI] [PubMed] [Google Scholar]
- Hussein S. M., Batada N. N., Vuoristo S., Ching R. W., Autio R., Narva E., et al. (2011). Copy number variation and selection during reprogramming to pluripotency. Nature 471 58–62 10.1038/nature09871 [DOI] [PubMed] [Google Scholar]
- Ito K., Hirao A., Arai F., Matsuoka S., Takubo K., Hamaguchi I., et al. (2004). Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431 997–1002 10.1038/nature02989 [DOI] [PubMed] [Google Scholar]
- Iwasa H., Han J., Ishikawa F. (2003). Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 8 131–144 10.1046/j.1365-2443.2003.00620.x [DOI] [PubMed] [Google Scholar]
- Ji G., Ruan W., Liu K., Wang F., Sakellariou D., Chen J., et al. (2013). Telomere reprogramming and maintenance in porcine iPS cells. PLoS ONE 8:e74202 10.1371/journal.pone.0074202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X. R., Jimenez G., Chang E., Frolkis M., Kusler B., Sage M., et al. (1999). Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat. Genet. 21 111–114 10.1038/5056 [DOI] [PubMed] [Google Scholar]
- Kawamura T., Suzuki J., Wang Y. V., Menendez S., Morera L. B., Raya A., et al. (2009). Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460 1140–1144 10.1038/nature08311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Kim J. S. (2014). A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 15 321–334 10.1038/nrg3686 [DOI] [PubMed] [Google Scholar]
- Kitamoto T., Takemoto M., Fujimoto M., Ishikawa T., Onishi S., Okabe E., et al. (2012). Sitagliptin successfully ameliorates glycemic control in Werner syndrome with diabetes. Diabetes Care 35 e83. 10.2337/dc12-1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao S., Ohsugi I., Ichikawa K., Goto M., Furuichi Y., Shimamoto A. (1998). Cloning of two new human helicase genes of the RecQ family: biological significance of multiple species in higher eukaryotes. Genomics 54 443–452 10.1006/geno.1998.5595 [DOI] [PubMed] [Google Scholar]
- Kitao S., Shimamoto A., Goto M., Miller R. W., Smithson W. A., Lindor N. M., et al. (1999). Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat. Genet. 22 82–84 10.1038/8788 [DOI] [PubMed] [Google Scholar]
- Kiuru M., Boyer J. L., O’Connor T. P., Crystal R. G. (2009). Genetic control of wayward pluripotent stem cells and their progeny after transplantation. Cell Stem Cell 4 289–300 10.1016/j.stem.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi J., Murano S., Yokote K., Mori S., Matsunaga A., Sasaki J., et al. (2000). Marked decrease in plasma apolipoprotein A-I and high density lipoprotein-cholesterol in a case with Werner syndrome. Clin. Chim. Acta 293 63–73 10.1016/S0009-8981(99)00219-3 [DOI] [PubMed] [Google Scholar]
- Kuilman T., Michaloglou C., Mooi W. J., Peeper D. S. (2010). The essence of senescence. Genes Dev. 24 2463–2479 10.1101/gad.1971610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Vinci J. M., Millis A. J., Baglioni C. (1993). Expression of interleukin-1 alpha and beta in early passage fibroblasts from aging individuals. Exp. Gerontol. 28 505–513 10.1016/0531-5565(93)90039-G [DOI] [PubMed] [Google Scholar]
- Levy M. Z., Allsopp R. C., Futcher A. B., Greider C. W., Harley C. B. (1992). Telomere end-replication problem and cell aging. J. Mol. Biol. 225 951–960 10.1016/0022-2836(92)90096-3 [DOI] [PubMed] [Google Scholar]
- Li H., Collado M., Villasante A., Strati K., Ortega S., Canamero M., et al. (2009). The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460 1136–1139 10.1038/nature08290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Suzuki K., Kim N. Y., Liu G. H., Izpisua Belmonte J. C. (2014). A cut above the rest: targeted genome editing technologies in human pluripotent stem cells. J. Biol. Chem. 289 4594–4599 10.1074/jbc.R113.488247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. T., Otsu M., Nakauchi H. (2013). Stem cell therapy: an exercise in patience and prudence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368 20110334 10.1098/rstb.2011.0334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R., Pelizzola M., Kida Y. S., Hawkins R. D., Nery J. R., Hon G., et al. (2011). Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471 68–73 10.1038/nature09798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. H., Barkho B. Z., Ruiz S., Diep D., Qu J., Yang S. L., et al. (2011). Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 472 221–225 10.1038/nature09879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machwe A., Xiao L., Lloyd R. G., Bolt E., Orren D. K. (2007). Replication fork regression in vitro by the Werner syndrome protein (WRN): holliday junction formation, the effect of leading arm structure and a potential role for WRN exonuclease activity. Nucleic Acids Res. 35 5729–5747 10.1093/nar/gkm561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion R. M., Strati K., Li H., Murga M., Blanco R., Ortega S., et al. (2009a). A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460 1149–1153 10.1038/nature08287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion R. M., Strati K., Li H., Tejera A., Schoeftner S., Ortega S., et al. (2009b). Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 4 141–154 10.1016/j.stem.2008.12.010 [DOI] [PubMed] [Google Scholar]
- Martins-Taylor K., Nisler B. S., Taapken S. M., Compton T., Crandall L., Montgomery K. D., et al. (2011). Recurrent copy number variations in human induced pluripotent stem cells. Nat. Biotechnol. 29 488–491 10.1038/nbt.1890 [DOI] [PubMed] [Google Scholar]
- Matsumoto T., Imamura O., Yamabe Y., Kuromitsu J., Tokutake Y., Shimamoto A., et al. (1997). Mutation and haplotype analyses of the Werner’s syndrome gene based on its genomic structure: genetic epidemiology in the Japanese population. Hum. Genet. 100 123–130 10.1007/s004390050477 [DOI] [PubMed] [Google Scholar]
- Mikkelsen T. S., Hanna J., Zhang X., Ku M., Wernig M., Schorderet P., et al. (2008). Dissecting direct reprogramming through integrative genomic analysis. Nature 454 49–55 10.1038/nature07056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohaghegh P., Karow J. K., Brosh R. M., Jr., Bohr V. A., Hickson I. D. (2001). The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 29 2843–2849 10.1093/nar/29.13.2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales C. P., Holt S. E., Ouellette M., Kaur K. J., Yan Y., Wilson K. S., et al. (1999). Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat. Genet. 21 115–118 10.1038/5063 [DOI] [PubMed] [Google Scholar]
- Mori S., Murano S., Yokote K., Takemoto M., Asaumi S., Take A., et al. (2001). Enhanced intra-abdominal visceral fat accumulation in patients with Werner’s syndrome. Int. J. Obes. Relat. Metab. Disord. 25 292–295 10.1038/sj.ijo.0801529 [DOI] [PubMed] [Google Scholar]
- Multani A. S., Chang S. (2007). WRN at telomeres: implications for aging and cancer. J. Cell Sci. 120 713–721 10.1242/jcs.03397 [DOI] [PubMed] [Google Scholar]
- Murano S., Nakazawa A., Saito I., Masuda M., Morisaki N., Akikusa B., et al. (1997). Increased blood plasminogen activator inhibitor-1 and intercellular adhesion molecule-1 as possible risk factors of atherosclerosis in Werner syndrome. Gerontology 43(Suppl. 1), 43–52 10.1159/000213885 [DOI] [PubMed] [Google Scholar]
- Nakayama J., Tahara H., Tahara E., Saito M., Ito K., Nakamura H., et al. (1998). Telomerase activation by hTRT in human normal fibroblasts and hepatocellular carcinomas. Nat. Genet. 18 65–68 10.1038/ng0198-65 [DOI] [PubMed] [Google Scholar]
- Niida H., Matsumoto T., Satoh H., Shiwa M., Tokutake Y., Furuichi Y., et al. (1998). Severe growth defect in mouse cells lacking the telomerase RNA component. Nat. Genet. 19 203–206 10.1038/580 [DOI] [PubMed] [Google Scholar]
- Niida H., Shinkai Y., Hande M. P., Matsumoto T., Takehara S., Tachibana M., et al. (2000). Telomere maintenance in telomerase-deficient mouse embryonic stem cells: characterization of an amplified telomeric DNA. Mol. Cell. Biol. 20 4115–4127 10.1128/MCB.20.11.4115-4127.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda S., Asano Y., Masuda S., Miyagawa T., Sugita M., Yamamoto M., et al. (2011). Bosentan: a novel therapy for leg ulcers in Werner syndrome. J. Am. Acad. Dermatol. 65 e54–e55 10.1016/j.jaad.2011.03.013 [DOI] [PubMed] [Google Scholar]
- O’Malley J., Skylaki S., Iwabuchi K. A., Chantzoura E., Ruetz T., Johnsson A., et al. (2013). High-resolution analysis with novel cell-surface markers identifies routes to iPS cells. Nature 499 88–91 10.1038/nature12243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe E., Takemoto M., Onishi S., Ishikawa T., Ishibashi R., He P., et al. (2012). Incidence and characteristics of metabolic disorders and vascular complications in individuals with Werner syndrome in Japan. J. Am. Geriatr. Soc. 60 997–998 10.1111/j.1532-5415.2012.03944.x [DOI] [PubMed] [Google Scholar]
- Okada M., Goto M., Furuichi Y., Sugimoto M. (1998). Differential effects of cytotoxic drugs on mortal and immortalized B-lymphoblastoid cell lines from normal and Werner’s syndrome patients. Biol. Pharm. Bull. 21 235–239 10.1248/bpb.21.235 [DOI] [PubMed] [Google Scholar]
- Okano H., Nakamura M., Yoshida K., Okada Y., Tsuji O., Nori S., et al. (2013). Steps toward safe cell therapy using induced pluripotent stem cells. Circ. Res. 112 523–533 10.1161/CIRCRESAHA.111.256149 [DOI] [PubMed] [Google Scholar]
- Okita K., Yamanaka S. (2011). Induced pluripotent stem cells: opportunities and challenges. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366 2198–2207 10.1098/rstb.2011.0016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi S., Takemoto M., Ishikawa T., Okabe E., Ishibashi R., He P., et al. (2012). Japanese diabetic patients with Werner syndrome exhibit high incidence of cancer. Acta Diabetol. 49(Suppl. 1), S259–S260 10.1007/s00592-012-0424-z [DOI] [PubMed] [Google Scholar]
- Opresko P. L., Otterlei M., Graakjaer J., Bruheim P., Dawut L., Kolvraa S., et al. (2004). The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol. Cell 14 763–774 10.1016/j.molcel.2004.05.023 [DOI] [PubMed] [Google Scholar]
- Oshima J., Yu C. E., Piussan C., Klein G., Jabkowski J., Balci S., et al. (1996). Homozygous and compound heterozygous mutations at the Werner syndrome locus. Hum. Mol. Genet. 5 1909–1913 10.1093/hmg/5.12.1909 [DOI] [PubMed] [Google Scholar]
- Palm W., de Lange T. (2008). How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42 301–334 10.1146/annurev.genet.41.110306.130350 [DOI] [PubMed] [Google Scholar]
- Papp B., Plath K. (2013). Epigenetics of reprogramming to induced pluripotency. Cell 152 1324–1343 10.1016/j.cell.2013.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini G., Luca M. D. (2009). Human Embryonic Stem Cell-Derived Keratinocytes: how Close to Clinics? Cell Stem Cell 6 8–9 10.1016/j.stem.2009.12.002 [DOI] [PubMed] [Google Scholar]
- Pera M. F. (2011). Stem cells: the dark side of induced pluripotency. Nature 471 46–47 10.1038/471046a [DOI] [PubMed] [Google Scholar]
- Poot M., Gollahon K. A., Rabinovitch P. S. (1999). Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Hum. Genet. 104 10–14 10.1007/s004390050903 [DOI] [PubMed] [Google Scholar]
- Poot M., Hoehn H., Runger T. M., Martin G. M. (1992). Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp. Cell Res. 202 267–273 10.1016/0014-4827(92)90074-I [DOI] [PubMed] [Google Scholar]
- Reardon S., Cyranoski D. (2014). Japan stem-cell trial stirs envy. Nature 513 287–288 10.1038/513287a [DOI] [PubMed] [Google Scholar]
- Robinton D. A., Daley G. Q. (2012). The promise of induced pluripotent stem cells in research and therapy. Nature 481 295–305 10.1038/nature10761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M. L., Ghosh A. K., Bohr V. A. (2010). Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst.) 9 331–344 10.1016/j.dnarep.2009.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin C. D., Zerwekh J. E., Reed-Gitomer B. Y., Pak C. Y. (1992). Characterization of osteoporosis in a patient with Werner’s syndrome. J. Am. Geriatr. Soc. 40 1161–1163. [DOI] [PubMed] [Google Scholar]
- Sakamoto S., Nishikawa K., Heo S. J., Goto M., Furuichi Y., Shimamoto A. (2001). Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes Cells 6 421–430 10.1046/j.1365-2443.2001.00433.x [DOI] [PubMed] [Google Scholar]
- Salama R., Sadaie M., Hoare M., Narita M. (2014). Cellular senescence and its effector programs. Genes Dev. 28 99–114 10.1101/gad.235184.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salk D., Au K., Hoehn H., Martin G. M. (1981a). Effects of radical-scavenging enzymes and reduced oxygen exposure on growth and chromosome abnormalities of Werner syndrome cultured skin fibroblasts. Hum. Genet. 57 269–275 10.1007/BF00278942 [DOI] [PubMed] [Google Scholar]
- Salk D., Au K., Hoehn H., Stenchever M. R., Martin G. M. (1981b). Evidence of clonal attenuation, clonal succession, and clonal expansion in mass cultures of aging Werner’s syndrome skin fibroblasts. Cytogenet. Cell Genet. 30 108–117 10.1159/000131597 [DOI] [PubMed] [Google Scholar]
- Samavarchi-Tehrani P., Golipour A., David L., Sung H. K., Beyer T. A., Datti A., et al. (2010). Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7 64–77 10.1016/j.stem.2010.04.015 [DOI] [PubMed] [Google Scholar]
- Seki M., Miyazawa H., Tada S., Yanagisawa J., Yamaoka T., Hoshino S., et al. (1994). Molecular cloning of cDNA encoding human DNA helicase Q1 which has homology to Escherichia coli Rec Q helicase and localization of the gene at chromosome 12p12. Nucleic Acids Res. 22 4566–4573 10.1093/nar/22.22.4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S., Otterlei M., Sommers J. A., Driscoll H. C., Dianov G. L., Kao H. I., et al. (2004). WRN helicase and FEN-1 form a complex upon replication arrest and together process branchmigrating DNA structures associated with the replication fork. Mol. Biol. Cell 15 734–750 10.1091/mbc.E03-08-0567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto A., Kagawa H., Zensho K., Sera Y., Kazuki Y., Osaki M., et al. (2014). Reprogramming Suppresses Premature Senescence Phenotypes of Werner Syndrome Cells and maintains chromosomal stability over Long-Term Culture. PLoS ONE 9:e112900 10.1371/journal.pone.0112900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto A., Nishikawa K., Kitao S., Furuichi Y. (2000). Human RecQ5beta, a large isomer of RecQ5 DNA helicase, localizes in the nucleoplasm and interacts with topoisomerases 3alpha and 3beta. Nucleic Acids Res. 28 1647–1655 10.1093/nar/28.7.1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto A., Sugimoto M., Furuichi Y. (2004). Molecular biology of Werner syndrome. Int. J. Clin. Oncol. 9 288–298 10.1007/s10147-004-0426-0 [DOI] [PubMed] [Google Scholar]
- Soldner F., Laganiere J., Cheng A. W., Hockemeyer D., Gao Q., Alagappan R., et al. (2011). Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146 318–331 10.1016/j.cell.2011.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen J. L., Jorgensen B., Gottrup F. (2004). Surgical treatment of pressure ulcers. Am. J. Surg. 188 42–51 10.1016/S0002-9610(03)00290-3 [DOI] [PubMed] [Google Scholar]
- Spyridopoulos I., Haendeler J., Urbich C., Brummendorf T. H., Oh H., Schneider M. D., et al. (2004). Statins enhance migratory capacity by upregulation of the telomere repeat-binding factor TRF2 in endothelial progenitor cells. Circulation 110 3136–3142 10.1161/01.CIR.0000142866.50300.EB [DOI] [PubMed] [Google Scholar]
- Stadtfeld M., Hochedlinger K. (2010). Induced pluripotency: history, mechanisms, and applications. Genes Dev. 24 2239–2263 10.1101/gad.1963910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M., Maherali N., Breault D. T., Hochedlinger K. (2008). Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell 2 230–240 10.1016/j.stem.2008.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M. (2014). A cascade leading to premature aging phenotypes including abnormal tumor profiles in Werner syndrome (review). Int. J. Mol. Med. 33 247–253 10.3892/ijmm.2013.1592 [DOI] [PubMed] [Google Scholar]
- Suzuki K., Yu C., Qu J., Li M., Yao X., Yuan T., et al. (2014). Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell 15 31–36 10.1016/j.stem.2014.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taapken S. M., Nisler B. S., Newton M. A., Sampsell-Barron T. L., Leonhard K. A., McIntire E. M., et al. (2011). Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol. 29 313–314 10.1038/nbt.1835 [DOI] [PubMed] [Google Scholar]
- Tahara H., Tokutake Y., Maeda S., Kataoka H., Watanabe T., Satoh M., et al. (1997). Abnormal telomere dynamics of B-lymphoblastoid cell strains from Werner’s syndrome patients transformed by Epstein-Barr virus. Oncogene 15 1911–1920 10.1038/sj.onc.1201377 [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131 861–872 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Sasaki A., Yamamoto M., et al. (2014). Induction of pluripotency in human somatic cells via a transient state resembling primitive streak-like mesendoderm. Nat. Commun. 5 3678 10.1038/ncomms4678 [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126 663–676 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. (2013). Induced pluripotent stem cells in medicine and biology. Development 140 2457–2461 10.1242/dev.092551 [DOI] [PubMed] [Google Scholar]
- Takemoto M., Mori S., Kuzuya M., Yoshimoto S., Shimamoto A., Igarashi M., et al. (2013). Diagnostic criteria for Werner syndrome based on Japanese nationwide epidemiological survey. Geriatr. Gerontol. Int. 13 475–481 10.1111/j.1447-0594.2012.00913.x [DOI] [PubMed] [Google Scholar]
- Takubo K., Ohmura M., Azuma M., Nagamatsu G., Yamada W., Arai F., et al. (2008). Stem cell defects in ATM-deficient undifferentiated spermatogonia through DNA damage-induced cell-cycle arrest. Cell Stem Cell 2 170–182 10.1016/j.stem.2007.10.023 [DOI] [PubMed] [Google Scholar]
- Thomson J. A., Itskovitz-Eldor J., Shapiro S. S., Waknitz M. A., Swiergiel J. J., Marshall V. S., et al. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282 1145–1147 10.1126/science.282.5391.1145 [DOI] [PubMed] [Google Scholar]
- Utikal J., Polo J. M., Stadtfeld M., Maherali N., Kulalert W., Walsh R. M., et al. (2009). Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 460 1145–1148 10.1038/nature08285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri H., Benchimol S. (1998). Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr. Biol. 8 279–282 10.1016/S0960-9822(98)70109-5 [DOI] [PubMed] [Google Scholar]
- Vaziri H., Dragowska W., Allsopp R. C., Thomas T. E., Harley C. B., Lansdorp P. M. (1994). Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proc. Natl. Acad. Sci. U.S.A. 91 9857–9860 10.1073/pnas.91.21.9857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Yin Y., Ye X., Liu K., Zhu H., Wang L., et al. (2012). Molecular insights into the heterogeneity of telomere reprogramming in induced pluripotent stem cells. Cell Res. 22 757–768 10.1038/cr.2011.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Xie L. Y., Allan S., Beach D., Hannon G. J. (1998). Myc activates telomerase. Genes Dev. 12 1769–1774 10.1101/gad.12.12.1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K., Kobayashi K., Takemoto M., Ishibashi R., Yamaga M., Kawamura H., et al. (2013). Sitagliptin improves postprandial hyperglycemia by inhibiting glucagon secretion in Werner syndrome with diabetes. Diabetes Care 36 e119. 10.2337/dc13-0709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollina U., Gruner M., Koch A., Kostler E., Hubl W., Hanson N. B., et al. (2004). Topical PDGF-BB results in limited healing in a patient with Werner’s syndrome and chronic leg ulcers. J. Wound Care 13 415–416. [DOI] [PubMed] [Google Scholar]
- Wong C. W., Hou P. S., Tseng S. F., Chien C. L., Wu K. J., Chen H. F., et al. (2010). Kruppel-like transcription factor 4 contributes to maintenance of telomerase activity in stem cells. Stem Cells 28 1510–1517 10.1002/stem.477 [DOI] [PubMed] [Google Scholar]
- Wong K. K., Maser R. S., Bachoo R. M., Menon J., Carrasco D. R., Gu Y., et al. (2003). Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature 421 643–648 10.1038/nature01385 [DOI] [PubMed] [Google Scholar]
- Wu K. J., Grandori C., Amacker M., Simon-Vermot N., Polack A., Lingner J., et al. (1999). Direct activation of TERT transcription by c-MYC. Nat. Genet. 21 220–224 10.1038/6010 [DOI] [PubMed] [Google Scholar]
- Wyllie F. S., Jones C. J., Skinner J. W., Haughton M. F., Wallis C., Wynford-Thomas D., et al. (2000). Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nat. Genet. 24 16–17 10.1038/71630 [DOI] [PubMed] [Google Scholar]
- Yeong E. K., Yang C. C. (2004). Chronic leg ulcers in Werner’s syndrome. Br. J. Plast. Surg. 57 86–88 10.1016/j.bjps.2003.10.011 [DOI] [PubMed] [Google Scholar]
- Yokote K., Hara K., Mori S., Kadowaki T., Saito Y., Goto M. (2004a). Dysadipocytokinemia in werner syndrome and its recovery by treatment with pioglitazone. Diabetes Care 27 2562–2563 10.2337/diacare.27.10.2562 [DOI] [PubMed] [Google Scholar]
- Yokote K., Honjo S., Kobayashi K., Fujimoto M., Kawamura H., Mori S., et al. (2004b). Metabolic improvement and abdominal fat redistribution in Werner syndrome by pioglitazone. J. Am. Geriatr. Soc. 52 1582–1583 10.1111/j.1532-5415.2004.52430_4.x [DOI] [PubMed] [Google Scholar]
- Yokote K., Saito Y. (2008). Extension of the life span in patients with Werner syndrome. J. Am. Geriatr. Soc. 56 1770–1771 10.1111/j.1532-5415.2008.01817.x [DOI] [PubMed] [Google Scholar]
- Yu C. E., Oshima J., Fu Y. H., Wijsman E. M., Hisama F., Alisch R., et al. (1996). Positional cloning of the Werner’s syndrome gene. Science 272 258–262 10.1126/science.272.5259.258 [DOI] [PubMed] [Google Scholar]
- Yu J., Vodyanik M. A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J. L., Tian S., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318 1917–1920 10.1126/science.1151526 [DOI] [PubMed] [Google Scholar]
- Yusa K., Rashid S. T., Strick-Marchand H., Varela I., Liu P. Q., Paschon D. E., et al. (2011). Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature 478 391–394 10.1038/nature10424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Lian Q., Zhu G., Zhou F., Sui L., Tan C., et al. (2011). A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 8 31–45 10.1016/j.stem.2010.12.002 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Toh L., Lau P., Wang X. (2012). Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/beta-catenin pathway in human cancer. J. Biol. Chem. 287 32494–32511 10.1074/jbc.M112.368282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T., Zhang Z. N., Rong Z., Xu Y. (2011). Immunogenicity of induced pluripotent stem cells. Nature 474 212–215 10.1038/nature10135 [DOI] [PubMed] [Google Scholar]