

Abstract
Background
Placental insufficiency is the leading cause of intrauterine growth restriction in the developed world and results in chronic hypoxemia in the fetus. Oxygen is essential for fetal heart development, but a hypoxemic environment in utero can permanently alter development of cardiomyocytes. The present study aimed to investigate the effect of placental restriction and chronic hypoxemia on total number of cardiomyocytes, cardiomyocyte apoptosis, total length of coronary capillaries, and expression of genes regulated by hypoxia.
Methods and Results
We induced experimental placental restriction from conception, which resulted in fetal growth restriction and chronic hypoxemia. Fetal hearts in the placental restriction group had fewer cardiomyocytes, but interestingly, there was no difference in the percentage of apoptotic cardiomyocytes; the abundance of the transcription factor that mediates hypoxia‐induced apoptosis, p53; or expression of apoptotic genes Bax and Bcl2. Likewise, there was no difference in the abundance of autophagy regulator beclin 1 or expression of autophagic genes BECN1, BNIP3, LAMP1, and MAP1LC3B. Furthermore, fetuses exposed to normoxemia (control) or chronic hypoxemia (placental restriction) had similar mRNA expression of a suite of hypoxia‐inducible factor target genes, which are essential for angiogenesis (VEGF, Flt1, Ang1, Ang2, and Tie2), vasodilation (iNOS and Adm), and glycolysis (GLUT1 and GLUT3). In addition, there was no change in the expression of PKC‐ε, a cardioprotective gene with transcription regulated by hypoxia in a manner independent of hypoxia‐inducible factors. There was an increased capillary length density but no difference in the total length of capillaries in the hearts of the chronically hypoxemic fetuses.
Conclusion
The lack of upregulation of hypoxia target genes in response to chronic hypoxemia in the fetal heart in late gestation may be due to a decrease in the number of cardiomyocytes (decreased oxygen demand) and the maintenance of the total length of capillaries. Consequently, these adaptive responses in the fetal heart may maintain a normal oxygen tension within the cardiomyocyte of the chronically hypoxemic fetus in late gestation.
Keywords: angiogenesis, apoptosis, hypoxia, myocytes, pregnancy
Introduction
Hypoxia during early fetal life is essential for normal heart growth, especially for embryonic outflow track remodeling1 and coronary vessel growth.2 Oxygen homeostasis is tightly regulated by hypoxia‐inducible factors (HIFs) within fetal tissues. During acute hypoxia, HIFs recruit mechanisms to increase oxygen supply (erythropoesis, angiogenesis, and vasodilation), decrease oxygen demand (increased glycolysis coupled with decreased oxidative metabolism), and regulate the cell cycle, apoptosis, and autophagy (for review, see Semenza3). HIFs function in a heterodimeric complex consisting of an oxygen‐regulated α isoform (HIF‐1α, HIF‐2α or HIF‐3α) and a constitutively expressed β isoform. Together, HIFs act as a transcription factor that binds to a hypoxia response element upstream of the promoter of genes required for the response to cellular hypoxia. The degradation of HIF‐α subunits is oxygen dependent because prolyl hydroxylases (prolyl hydroxylase domains [PHDs] 1 to 3) require oxygen as a cosubstrate. During acute hypoxia, PHDs cannot hydroxylate HIF‐α, leading to increased HIF‐α protein stability and translocation to the nucleus, whereupon they form a heterodimer with HIF‐1β and recruit coactivators CREB binding protein and p300 to induce gene transcription (for review, see Kaelin and Ratcliffe4). In addition, the stabilization of HIF‐1α protein also directly stabilizes and causes the accumulation of tumor suppressor p53 protein,5 a transcription factor that regulates cell cycle activity and apoptosis, thus mediating hypoxia‐induced apoptosis.
A lower‐than‐normal oxygen supply in utero can result in lifelong consequences.6 Exposure to chronic hypoxemia in utero results in intrauterine growth restriction and low birth weight.7 A series of worldwide epidemiological studies have demonstrated that low birth weight is a predictor of ischemic heart disease and heart failure in adulthood, and this association is supported by studies in a variety of animal models of fetal hypoxemia (for review, see McMillen and Robinson8). The cardiomyocytes present at birth constitute the majority of the cardiomyocytes that an individual will have for a lifetime9; therefore, considerable focus has been placed on understanding the impact of reduced oxygen in utero on cardiomyocyte development.
Chronic hypoxemia in the fetus can have different effects on cardiomyocyte development depending on the timing in relation to cardiac development and duration and degree of the insult.10 In this context, “chronic” has been used to define periods of exposure to hypoxia for periods ranging from 24 hours to several weeks. Interpretation of experimental studies on the impact of fetal hypoxemia is further complicated by the fact that exposure to hypoxemia for 1 week constitutes one‐third of gestation in rats but only 5% of gestation in sheep and less in humans. Maternal hypoxia during the last week of gestation in rats increases cardiomyocyte apoptosis and accelerates cardiomyocyte maturation.11 In adulthood, these rats have an increased susceptibility to ischemia‐reperfusion injury,12 which is partly due to decreased expression of cardioprotective protein kinase C‐ε (PKCε).13 Interestingly, the hypoxia induced decrease in PKCε mRNA expression is independent of HIFs; instead, it is mediated by intracellular reactive oxygen species.14 Unlike humans, however, rats are born with an immature cardiovascular system, with all cardiomyocytes capable of proliferating.15 Terminally differentiated human cardiomyocytes, in the form of binucleated cardiomyocytes, have been observed at 0.8 of gestation, which is similar to sheep, where binucleation begins at 0.7 of gestation (for review, see Botting et al16). Studies of placental insufficiency in sheep, which results in chronic fetal hypoxemia, hypoglycemia, and low birth weight, report altered fetal heart growth in late gestation.17–19 Placental insufficiency, induced by the removal of maternal endometrial caruncles prior to conception (long‐term model of placental restriction [PR])19–20 or for 20 days in late gestation due to umbilicoplacental embolization,17–18 causes a delay in the transition of proliferating mononucleated cardiomyocytes to terminally differentiated binucleated cardiomyocytes. In the PR model, chronic hypoxemia occurs for at least the last 40 days of gestation (earliest recording of fetal PaO2 in the PR model is 102 days of gestation;20 term: 150 days), and despite both PR and umbilicoplacental embolization fetuses being exposed to chronic hypoxemia in late gestation, only umbilicoplacental embolization fetuses, which are exposed to hypoxemia for 20 days, have a decreased percentage of mononucleated cardiomyocytes undergoing proliferation.18–19 Interestingly, the reverse is true for cardiomyocyte size, for which chronic hypoxemia for the last 40 days of gestation results in cardiomyocytes that are larger than expected for the size of the heart,19 but chronic hypoxemia for up to 20 days in late gestation does not.18 This hypertrophy persists into postnatal life and is associated with an increased abundance of cardiac growth factor IGF2R.21
We hypothesize that the increase in relative cardiomyocyte size reflects a decrease in the endowment of cardiomyocytes in late gestation. Because PR fetuses have an equivalent percentage of proliferative cardiomyoctes in late gestation,19 we further hypothesis that PR fetuses will have greater cardiomyocyte apoptosis mediated by hypoxia in addition to a greater length of coronary capillaries and increased expression of genes that are unregulated in these processes in response to acute hypoxia.
Methods
Animals and Surgical Procedures
All procedures were approved by the University of South Australia and the University of Adelaide animal ethics committees. In total, 32 control and 22 PR fetuses were used in this study. PR was induced by performing a carunclectomy in nonpregnant ewes, whereby the majority of the endometrial caruncles were removed from the uterus prior to conception.19 All surgery was performed under aseptic conditions with general anesthesia induced by sodium thiopentone (1.25 g/mL, intravenous; Boehringer Ingelheim) and maintained with 3% to 4% halothane in oxygen. At surgery, antibiotics were administered to the ewe (153.5 mg procaine penicillin, 393 mg benzathine penicillin, 500 mg dihydrostreptomycin; Lyppards). Ewes recovered from surgery for >12 weeks prior to entering a mating program.
Vascular surgery was performed at 119±1 days of gestation with general anesthesia induced by sodium thiopentone (Pentothal; 1.25 g; Rhone Merieux) and maintained by inhalation of halothane (2.5% to 4%) in oxygen, as described previously.19 Briefly, vascular catheters (Critchley Electrical Products) were inserted in the maternal jugular vein, the fetal carotid artery and jugular vein, and the amniotic cavity. Fetal catheters were exteriorized through a small incision in the ewe's flank. Ewes were administered antibiotics during surgery and for 3 days after surgery (as above, intramuscularly). Fetuses were administered antibiotics during surgery (intramuscularly; 150 mg procaine penicillin, 112.5 mg benzathine penicillin, 250 mg dihydrostreptomycin; Lyppards) and for 4 days after surgery (intra‐amniotically; 500 mg ibimicyn; GenePharm).
Arterial Blood Gas Measurements
Fetal carotid artery blood samples were collected daily to monitor fetal health by measuring blood gases (PaO2, PaCO2, oxygen saturation, pH, hemoglobin, and arterial oxygen content19) at 39°C with an ABL 520 analyzer (Radiometer). Animals recovered for 4 days postoperatively before blood samples were recorded for experimental comparisons.
Tissue Collection
At 140±1 days of gestation, ewes and fetuses were humanely killed with an overdose of sodium pentobarbital (8.2 g; Vibrac Aus), and fetuses were delivered by hysterotomy, weighed, and exsanguinated. The fetal heart was dissected and weighed. Because the experimental aims of this study required the heart to be processed in different ways, the right ventricle (RV) was dissected, weighed, and stored in 4% formaldehyde to determine the total number of cardiomyocytes and capillary length density (control, n=8; PR, n=5); frozen for gene analysis (control, n=9; PR n=8); or enzymatically digested using a reverse Langendorff apparatus, as previously described,19 to determine the average number of nuclei per cardiomyocyte (control, n=14; PR, n=7) and the percentage of apoptotic cardiomyocytes (control, n=6; PR, n=6). The left ventricle (LV) was dissected and weighed, and a piece was frozen for gene and protein analysis (control, n=15; PR, n=12).
Total Number of Cardiomyocytes and Capillary Length Density
The estimation of total cardiomyocyte number and capillary length were performed using design‐unbiased stereological techniques.22
Tissue sampling
RV samples were fixed in 4% formaldehyde and serially sectioned into 2‐mm slices. Using the smooth fractionator principle,23 5 to 6 slices were selected and further cut to create cubes of ≤2 mm3. Using the same principle, tissue cubes were divided into groups of 8 to 12, with 1 group embedded in glycolmethacrylate (Technovit 7100; Axlab) for cardiomyocyte‐number estimation and the other group becoming isotropic with the isector24 and embedded in paraffin for capillary length analysis.
Cardiomyocyte number estimation
The average number of nuclei per cardiomyocyte (NN(nuclei/cm)) was determined from isolated cardiomyocytes from the RV using Equation 1. Briefly, isolated cardiomyocytes were stained with methylene blue to visualize cardiomyocyte nuclei. The number of mononucleated (ΣQ−(mono)) and binucleated (ΣQ−(bi)) cardiomyocytes in a total of 300 cardiomyocytes was determined.
| 1 |
The numerical density of cardiomyocyte nuclei was determined using the optical disector technique on glycol methacrylate–embedded sections.25 From the center of each glycol methacrylate block, a 30‐μm‐thick section was cut and mounted on Superfrost Plus slides (Menzel‐Gläser). To visualize cardiomyocyte nuclei, sections were stained with Mayer's hematoxylin and 0.15% basic fuchsin. Point counting was performed in about 20 two‐dimensional unbiased counting frames of surface area 500 μm2, which were systematic, uniformly, randomly assigned by newCAST software (Visiopharm) to each ventricle piece. A disector height 10 μm in the center of each section determined after a z‐axis analysis was used to determine the numerical density of nuclei in a minimum of 6 ventricle pieces per animal. The numerical density of nuclei in the RV (NV(nuclei/rv)) was determined using Equation 2, in which ΣQ−(nuc) is the number of nuclei, h is the Z height analyzed, a/p is the area of ventricle that each point represents, and ΣP(rv) is the sum of points that hit ventricle tissue.
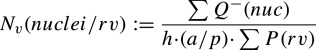 |
2 |
The number of cardiomyocytes in the RV (N(cm,rv)) was determined by dividing the NV(nuclei/rv) by the average number of nuclei per cardiomyocyte and multiplying by the volume of the RV (volume of RV equals postmortem wet weight divided by 1.06 g/cm3).26
Length density and total length of capillaries
The length density of coronary capillaries in the RV (Lv(cap/rv)) was determined in 5‐μm‐thick paraffin sections on Superfrost Plus slides. Nonspecific antibody binding was blocked by 1% bovine serum albumin (0.2% gelatin, 0.05% saponin in phosphate‐buffered saline). The primary antibody anti‐α‐smooth muscle actin (1:4000; A2547; Sigma‐Aldrich) was used to identify pericytes that wrap around the endothelial cells of capillaries. Primary and secondary antibody anti‐mouse‐HRP (1:200; 7076; Cell Signaling) were diluted in phosphate‐buffered saline containing 0.1% bovine serum albumin and 0.3% Triton X. Antigen location was visualized with 3,3′‐diaminobenzidine tetrahydrochloride (4170; Kem‐en‐tec Diagnostics), and nuclei were visualized with Mayer's hematoxylin.
Capillaries were visualized at a magnification of ×600 with a BX53 microscope (Olympus) equipped with a motorized stage (ProScanIII Motorized Stage Systems; Prior) and digital camera (DP72; Olympus). Point counting was performed in about 20 two‐dimensional unbiased counting frames of surface area 10 000 μm2, which were uniformly randomly assigned by newCAST software (Visiopharm) to each ventricle piece. Lv(cap/rv) was calculated using Equation 1,26 in which (ΣQ−(cap)) was the number of capillary profiles within the counting frame.
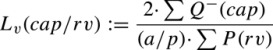 |
3 |
The total length of capillaries in the RV (L(cap,rv)) was calculated by multiplying the Lv(cap/rv) by the volume of the RV.
Transferase dUTP Nick‐End Labeling
Apoptosis was measured by the presence of DNA fragmentation with terminal deoxynucleotidyl transferase dUTP nick‐end labeling (Invitrogen) and visualized with 3,3′‐diaminobenzidine tetrahydrochloride (metal‐enhanced DAB substrate kit; Thermo‐Fischer Scientific). Isolated, paraformaldehyde‐fixed cardiomyocytes from the RV were dried onto polylysine‐coated Superfrost Plus slides and fixed with acetone. Positive control slides were generated by inducing DNA nicks with DNAse I (AMPD1; Sigma‐Aldrich), and negative control slides were generated by the absence of either terminal deoxynucleotidyl transferase or nucleotides. The percentage of apoptotic cardiomyocytes was determined by the presence of positive terminal deoxynucleotidyl transferase dUTP nick‐end labeling staining in at least 1 nuclei of 200 mononucleated and 200 binucleated cardiomyocytes.
Measurement of mRNA Expression
RNA was isolated from the LV and RV (≈100 mg) of each fetus, and cDNA was synthesized as described previously.21 Controls containing no Superscript III and no RNA transcript were used to test for genomic DNA and reagent contamination, respectively. The reference genes tyrosine 3‐monooxygenase (YWHAZ), glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH),27 and phosphoglycerate kinase 1 (PGK1)27 were chosen from a suite of reference genes based on expression analysis using the geNorm component of the qBase relative quantification analysis software28 because their expression was stable across samples.29 The expression of target and reference mRNA transcripts were measured by quantitative reverse‐transcription polymerase chain reaction using Fast SYBR Green Master Mix (Applied Biosystems) in a final volume of 6 μL on a ViiA 7 Fast Real‐Time PCR system (Applied Biosystems), as described previously.29
Primers were validated to generate a single transcript, as confirmed by the presence of one double‐stranded DNA product of the correct size and sequence (Table 1). Controls containing no cDNA were included for each primer set on each plate to test for reagent contamination. Melt curve and dissociation curves were also run to check for nonspecific product formation. Amplification efficiency reactions were performed on 5 triplicate serial dilutions of cDNA template for each primer set. Amplification efficiencies were determined from the slope of a plot of Ct (defined as the threshold cycle with the lowest significant increase in fluorescence) against the log of the cDNA template concentration (1 to 100 ng). The amplification efficiency was close to 100%. Each sample was run in triplicate for target and reference genes. The reactions were quantified by setting the threshold within the exponential growth phase of the amplification curve and obtaining corresponding Ct values. The abundance of each transcript relative to the abundance of the 3 stable reference genes was calculated using DataAssist software version 3.0 (Applied Biosystems) and expressed as mean normalized expression.28
Table 1.
Primer Sequences Used in Quantitative Reverse Transcription Polymerase Chain Reaction to Measure Genes of Interest
| Gene | Primers | Accession Number |
|---|---|---|
| Tyrosine 3‐monooxygenase (YWAHZ; reference gene) | Fwd 5′‐CCTGGAGAAACCTGCCAAGT‐3′ | AY970970 |
| Rev 5′‐GCCAAATTCATTGTCGTACCA‐3′ | ||
| Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH; reference gene) | Fwd 5′‐TGTAGGAGCCCGTAGGTCATCT‐3′ | DQ152956.1 |
| Rev 5′‐ TTCTCTCTGTATTCTCGAGCCATCT‐3′ | ||
| Phosphoglycerate kinase 1 (PGK1; reference gene) | Fwd 5′‐ACTCCTTGCAGCCAGTTGCT‐3′ | NM_001034299 |
| Rev 5′‐AGCACAAGCCTTCTCCACTTCT‐3′ | ||
| Beclin 1 (BECN1) | Fwd 5′‐ GAACCTCAGCCGAAGACTAAAG‐3′ | XM_004012945.1 |
| Rev 5′‐CTAAGAGGGTGTCTGTGCATTC‐3′ | ||
| BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) | Fwd 5′‐GTTCCCGACTCTGCTTCTATTT‐3′ | XM_004020372.1 |
| Rev 5′‐GTCACAGTGGGAGCTCTTG‐3′ | ||
| Lysosomal‐associated membrane protein 1 (LAMP1) | Fwd 5′‐CTTGAGAGCTGGCACTTAGAA‐3′ | XM_004012369.1 |
| Rev 5′‐CTCCAACAGGAAACGGAGAA ‐3′ | ||
| Microtubule‐associated protein 1 light chain 3β (MAP1LC3B) | Fwd 5′‐ACGCTCCTTGAGAGGATCTA‐3′ | NM_022818 |
| Rev 5′‐CAGCAGCATGGTTTCCTTATTT‐3′ | ||
| Hypoxia‐inducible factor 1α (HIF‐1α) | Fwd 5′‐TGAGCTTGCTCATCAGTTGCCA‐3′ | AY485676.1 |
| Rev 5′‐ACGCAAATAGCTGATGGTGAGCCT‐3′ | ||
| Hypoxia‐inducible factor 2α (HIF‐2α) | Fwd 5′‐TACAGGTTCCTCCCCGTCAC‐3′ | NM_174725.2 |
| Rev 5′‐CTTGTCAGCTGTCATTGTCGC‐3′ | ||
| Hypoxia‐inducible factor 3α (HIF‐3α) | Fwd 5′‐GTGGAGTTCCTGGGCATCAG‐3′ | EU340262.1 |
| Rev 5′‐CCCGTCAGAAGGAAGCTCAG‐3′ | ||
| Hypoxia‐inducible factor 1β (HIF‐1β) | Fwd 5′‐AGGTGTGGCAATAGCTCTGTGGAT‐3′ | NM_173993.1 |
| Rev 5′‐AGGCCTTGATATAGCCTGTGCAGT‐3′ | ||
| Vascular endothelial growth factor A (VEGFA) | Fwd 5′‐TGTAATGACGAAAGTCTGGAG‐3′ | AF071015.1 |
| Rev 5′‐TCACCGCCTCGGCTTGTCACA‐3′ | ||
| VEGF receptor 1 (Flt1) | Fwd 5′‐CCGAAGGGAAGAAGGTGGTC‐3′ | NM_001191132.2 |
| Rev 5′‐GACTGTTGTCTCGCAGGTCA‐3′ | ||
| Angiopoietin 1 (ANGPT1; Ang1) | Fwd 5′‐TGCAAATGTGCCCTCATGCT‐3′ | AY881028.1 |
| Rev 5′‐TTCCATGGTTCTGTCCCGCT‐3′ | ||
| Angiopoietin 2 (ANGPT2; Ang2) | Fwd 5′‐AGAACCAGACCGCTGTGATG‐3′ | AY881029.1 |
| Rev 5′‐TGCAGTTTGCTTATTTCACTGGT‐3′ | ||
| Tyrosine‐protein kinase receptor (TEK/Tie2) | Fwd 5′‐CAGTTTACCAGGTGGACATC‐3′ | AY288926.1 |
| Rev 5′‐ACATTTTGGAAGGCTTGGGC‐3′ | ||
| Solute carrier family 2 (facilitated glucose transporter), member 1 (SLC2A1/GLUT1) | Fwd 5′‐ATCGTGGCCATCTTTGGCTTTGTG‐3′ | U89029.1 |
| Rev 5′‐CTGGAAGCACATGCCCACAATGAA‐3′ | ||
| Solute carrier family 2 (facilitated glucose transporter), member 3 (SLC2A3/GLUT3) | Fwd 5′‐AGAGTATGCGGATGTCGCAG‐3′ | NM_001009770.1 |
| Rev 5′‐CACCGATAGTGGCGTAGACC‐3′ | ||
| Inducible nitric oxide synthase (iNOS) | Fwd 5′‐AAGGCAGCCTGTGAGACATT‐3′ | AF223942.1 |
| Rev 5′‐CAGATTCTGCTGCGATTTGA‐3′ | ||
| Adrenomedullin (Adm) | Fwd 5′‐GGGGTGCAAGCCTCACTATT‐3′ | NM_173888.3 |
| Rev 5′‐ CACATTCCACGCAGCAAACA‐3′ | ||
| B‐cell CLL/lymphoma 2 (Bcl2) | Fwd 5′‐GTGGAGGAGCTCTTCAGGGA‐3′ | HM630309.1 |
| Rev 5′‐GTTGACGCTCTCCACACACA‐3′ | ||
| Bcl‐2 associated protein (Bax) | Fwd 5′‐CAGGATGCATCCACCAAGAAGC‐3′ | AF163774.1 |
| Rev 5′‐TTGAAGTTGCCGTCGGAAAACATT‐3′ | ||
| Protein kinase C‐ε (PKCε) | Fwd 5′‐AGCACCCGTTCTTCAAGGAG‐3′ | XM_004005978.1 |
| Rev 5′‐TGCTTGCAGCATCACCAAAC‐3′ | ||
| Egl nine homolog 2 (EGLN2/PHD‐1) | Fwd 5′‐ATGGTGGCATGTTACCCAGG‐3′ | NM_001102193.1 |
| Rev 5′‐AGGGGCTCAATGTTGGCTAC‐3′ | ||
| Egl nine homolog 1 (EGLN1/PHD2) | Fwd 5′‐TGGAGATGGAAGATGTGTGA‐3′ | NM_001206046.2 |
| Rev 5′‐TTGGGTTCAATGTCAGCAAA‐3′ | ||
| Egl nine homolog 3 (EGLN3/PHD3) | Fwd 5′‐TGCTACCCAGGAAATGGAACAGGT‐3′ | NM_001101164.1 |
| Rev 5′‐GCTTGGCATCCCAGTTCTTGTTCA‐3′ |
Quantification of Protein Abundance by Western Blot
Proteins were extracted from the LV by sonication in lysis buffer (50 mmol/L Tris‐HCL [pH 8], 150 mmol/L NaCl, 1% NP‐40, 1 mmol/L Na orthovanadate, 30 mmol/L Na fluoride, 10 mmol/L Na pyrophosphate, 10 mmol/L EDTA, and a protease inhibitor tablet [cOmplete Mini; Roche]). Total protein concentration was determined by Micro BCA assay (Thermo‐Fisher Scientific). Protein was diluted to a concentration of 5 mg/mL in 1× sodium dodecyl sulfate sample buffer (containing 75 mmol/L DL‐Dithiothreitol) and Coomassie blue stain used to confirm equal protein loading on sodium dodecyl sulfate polyacrylamide gel electrophoresis before diluted protein was used for experimental blots. Proteins were transferred to a nitrocellulose membrane (Amersham Hybond‐C extra; GE Healthcare Life Sciences) using boric acid transfer buffer. Nonspecific antibody binding was blocked with 5% skim milk in Tris‐buffered saline with 1% Tween‐20 or 5% bovine serum albumin in Tris‐buffered saline with 1% Tween‐20. Primary antibodies anti‐p53 (1:200, mouse monoclonal antibody, OP104L; Merck), anti‐PHD1 (1:500, rabbit polyclonal antibody, NB100‐310; Novus Biological) and anti‐PHD2 (1:500, rabbit polyclonal antibody, NB100‐137; Novus Biological) were incubated overnight. Secondary antibodies mouse‐HRP (1:2000, 7075; Cell Signaling) and rabbit‐HRP (1:2000, 7076; Cell Signaling) were incubated for 1 hour at room temperature. SuperSignal West Pico chemiluminescent substrate (Thermo‐Fisher Scientific) and an ImageQuant LAS 4000 (GE Healthcare) were used to detect and image antigens of interest. ImageQuantTL Analysis Toolbox (GE Healthcare) was used to quantify the protein bands, and the ratio of band density from 50% and 100% of the loading control constituted from an equal amount of protein extracts from each animal was used to ensure linearity of density measurement. Each antibody was repeated on new blots to ensure reproducibility of results.
Statistical Analyses
Fetuses were included in the control group if the ewe did not undergo carunclectomy surgery and if they had a mean gestational PaO2 >17 mm Hg; fetuses were included in the PR group if they were chronically hypoxemic, defined as a mean gestational PaO2 <17 mm Hg.19,30 Because some twin pregnancies (11 of 22 pregnancies in the control group; 8 of 19 pregnancies in the PR group) were included in this study, a 2‐way analysis of variance for the effect of treatment group and fetal number, nested for litter, was used, and there was no statistical interaction between treatment and fetal number for any parameters measured in this study. Pearson correlation and linear regression analysis were used to determine the relationship between the total number of cardiomyocytes in the RV and either fetal weight or the length of coronary capillaries. A probability level of 5% (P<0.05) was considered significant. Box plots were used instead of bar graphs to describe the distribution of data.
Results
Arterial Blood Gas and Fetal Weight Measurements
PR resulted in reduced PaO2, oxygen saturation, and arterial oxygen content but did not alter the concentration of hemoglobin compared with controls (Table 2). PR and control fetuses had equivalent PaCO2 and base excess, but PR fetuses had lower pH, although they were not clinically acidotic (Table 2). Fetuses exposed to chronic hypoxemia had reduced body and asymmetric growth, represented by increased brain weight relative to body weight. Chronically hypoxemic fetuses had smaller hearts but maintained heart weight relative to body weight in late gestation compared with controls (Table 3).
Table 2.
Fetal Arterial Blood Gas Measurements
| Control | PR | |
|---|---|---|
| PaO2 on day of postmortem, mm Hg | 20.8±0.7 | 12.7±0.5* |
| Mean gestational PaO2, mm Hg | 21.9±0.5 | 13.7±0.4* |
| Mean gestational O2 saturation, % | 65.8±1.7 | 37.5±1.7* |
| Mean gestational hemoglobin, g/dL | 10.5±0.22 | 11.3±0.61 |
| Mean gestational O2 content, mL/dL | 9.6±0.2 | 5.8±0.3* |
| Mean gestational PaCO2, mm Hg | 49.2±0.6 | 50.4±1.2 |
| Mean gestational pH | 7.384±0.005 | 7.369±0.005* |
| Mean gestational base excess, mEq/L | 2.8±0.3 | 3.5±0.5 |
Values are mean±SEM (SD). PR indicates placental restriction.
P<0.05.
Table 3.
Fetal Body and Heart Weight Measurements
| Control | PR | |
|---|---|---|
| Fetal weight, kg | 4.68±0.12 | 2.52±0.16* |
| Heart weight, g | 32.17±0.79 | 18.77±0.81* |
| Relative heart weight, g/kg | 6.71±0.09 | 7.12±0.19 |
| Relative brain weight, g/kg | 12.75±0.37 | 19.77±0.88* |
Values are mean±SEM (SD). PR indicates placental restriction.
P<0.05.
Cardiomyocyte Endowment, Apoptosis, and Autophagy
Fetuses exposed to chronic hypoxemia had a decreased total number of cardiomyocytes and binucleated cardiomyocytes in the RV (Figure 1A through 1C). The total number of cardiomyocytes in the RV was positively associated with fetal body weight (regression analysis: P<0.001; R2=0.968; y=1.95−0.70x+0.17x2; Figure 1D). Despite reduction in the total number of cardiomyocytes, there was no effect of exposure to chronic hypoxemia on the percentage of apoptotic cardiomyocytes or the mRNA expression of the anti‐apoptotic gene B‐cell CLL/lymphoma 2 (Bcl2) or the pro‐apoptotic gene Bcl2 associated protein (Bax). RV (Figure 2A through 2C). Furthermore, fetuses exposed to chronic hypoxemia had an equivalent abundance of p53 protein in the LV (Figure 2D), which is responsible for hypoxia‐induced apoptosis. Paradoxically, fetuses exposed to chronic hypoxemia had a decrease in mRNA expression of pro‐apoptotic Bax; however, there was also a decrease in the mRNA expression of antiapoptotic Bcl‐2 in the LV (Figure 2E and 2F). Furthermore, exposure to chronic hypoxemia did not alter the expression of autophagic genes Beclin‐1 (BECN1), BCL2/adenovirus E1B 19kDa interacting protein 3 (BNIP3), Lysosomal‐associated membrane protein 1 (LAMP1) or Microtubule‐associated protein 1 light chain 3 beta (MAP1LC3B) or the abundance of Beclin‐1 protein (Table 4).
Figure 1.
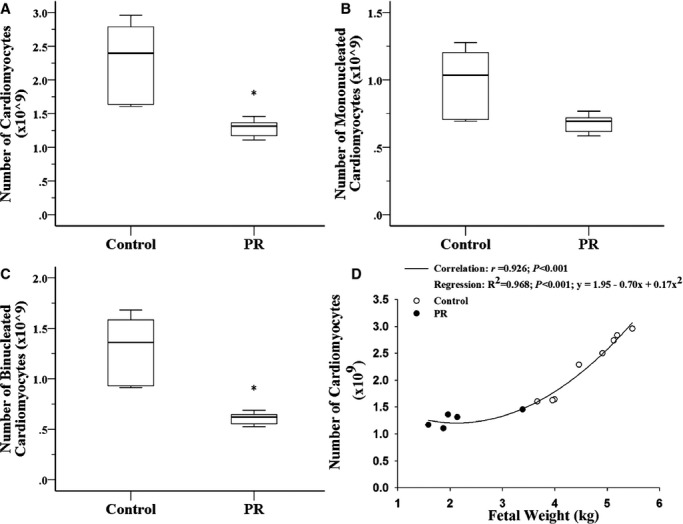
Placental restriction (PR) resulting in chronic hypoxemia reduced the total number of cardiomyocytes (A) and binucleated cardiomyocytes (C) but not mononucleated cardiomyocytes (B) in the right ventricle. The total number of cardiomyocytes is positively correlated with fetal weight (D). *P<0.05; Control, ○; PR, ●.
Figure 2.
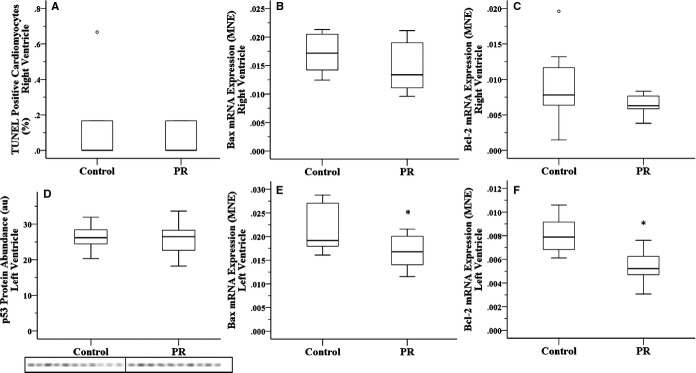
Placental restriction (PR) resulting in chronic hypoxemia did not alter the percentage of apoptotic cardiomyocytes (A) or the mRNA expression of pro‐apoptotic gene Bax (B) or antiapoptotic gene Bcl2 (C) in the right ventricle. PR did not affect the protein abundance of hypoxia‐mediated apoptosis regulator p53 (D) but resulted in decreased mRNA expression of both pro‐apoptotic gene Bax (E) and antiapoptotic gene Bcl2 (F) in the left ventricle. Open circle represents an outlier, defined as being >1.5×IQR; *P<0.05. MNE indicates mean normalized expression; TUNEL, terminal deoxynucleotidyl transferase dUTP nick‐end labeling.
Table 4.
Expression of Markers of Autophagy
| Left Ventricle | Right Ventricle | |||
|---|---|---|---|---|
| Control | PR | Control | PR | |
| Gene | ||||
| BECN1 | 0.077±0.005 | 0.071±0.006 | 0.081±0.013 | 0.05±0.008 |
| BNIP3 | 0.438±0.040 | 0.493±0.049 | 0.541±0.115 | 0.397±0.064 |
| LAMP1 | 0.154±0.012 | 0.180±0.023 | 0.172±0.039 | 0.095±0.011 |
| MAP1LC3B | 0.097±0.004 | 0.103±0.009 | 0.129±0.051 | 0.067±0.010 |
| Protein | ||||
| Beclin 1 | 34.3±8.3 | 55.6±12.8 | ||
Values are mean±SEM (SD). PR indicates placental restriction.
Expression of HIFs and Hypoxia Mediated Genes
There was no difference in the mRNA expression of HIF‐1α, HIF‐2α, HIF‐3α, and HIF‐1β and genes with hypoxia response elements that are involved in angiogenesis (Vascular endothelial growth factor [VEGF], VEGF Receptor 1 [Flt1], Angiopoietin‐2 [Ang2] and Tyrosine‐protein kinase receptor [Tie2]), vascular tone (Inducible nitric oxide synthase [iNOS]), and glysolysis (Solute carrier family 2 (facilitated glucose transporter), member 1 [GLUT1] and member 3 [GLUT3]). or in the hypoxia‐regulated cardioprotective gene Protein kinase C‐e (PKCε) in either the LV or the RV of fetuses exposed to chronic hypoxemia compared with controls (Table 5). Fetuses exposed to chronic hypoxemia had a decrease in the mRNA expression of the angiogenic gene Ang1 and the vasoactive gene Adrenomedullin (Adm) in the LV, but there was no change in the RV.
Table 5.
mRNA Expression of HIFs, Genes With Hypoxia Response Elements, and Genes Involved in Cardioprotection and HIF‐α Stability
| Left Ventricle | Right Ventricle | |||
|---|---|---|---|---|
| Control | PR | Control | PR | |
| Gene | ||||
| Oxygen sensing | ||||
| Hif‐1α | 0.390±0.035 | 0.343±0.036 | 0.348±0.022 | 0.285±0.018 |
| Hif‐2α | 0.389±0.026 | 0.499±0.083 | 0.487±0.090 | 0.387±0.047 |
| Hif‐3α | 0.073±0.010 | 0.061±0.013 | 0.065±0.006 | 0.093±0.013 |
| Hif‐1β | 0.060±0.010 | 0.049±0.008 | 0.056±0.005 | 0.076±0.010 |
| Angiogenesis | ||||
| VEGF | 0.611±0.054 | 0.637±0.122 | 0.436±0.076 | 0.603±0.065 |
| Flt‐1 | 0.102±0.009 | 0.108±0.011 | 0.115±0.006 | 0.140±0.012 |
| Ang‐1 | 0.018±0.002 | 0.009±0.002* | 0.047±0.016 | 0.019±0.004 |
| Ang‐2 | 0.016±0.001 | 0.015±0.003 | 0.018±0.003 | 0.021±0.002 |
| Tie‐2 | 0.075±0.009 | 0.055±0.011 | 0.054±0.005 | 0.085±0.014 |
| Vasodilation | ||||
| iNOS | 0.010±0.002 | 0.006±0.001 | 0.006±0.001 | 0.007±0.001 |
| Adm | 0.013±0.001 | 0.009±0.001* | 0.007±0.001 | 0.006±0.001 |
| Glucose metabolism | ||||
| GLUT1 | 0.050±0.006 | 0.056±0.010 | 0.042±0.010 | 0.053±0.009 |
| GLUT3 | 1.020±0.158 | 0.931±0.072 | 1.384±0.327 | 1.729±0.271 |
| Cardio‐protection | ||||
| PKCε | 0.053±0.006 | 0.048±0.007 | 0.072±0.011 | 0.050±0.008 |
| HIF‐α stability | ||||
| PHD1 | 0.081±0.007 | 0.077±0.006 | 0.071±0.007 | 0.072±0.009 |
| PHD2 | 0.391±0.026 | 0.344±0.048 | 0.379±0.029 | 0.418±0.055 |
| PHD3 | 0.284±0.020 | 0.450±0.047* | 0.340±0.036 | 0.436±0.058 |
Values are mean±SEM (SD). Adm indicates adrenomedullin; Ang, angiopoietin; GLUT, glucose transporter; HIF, hypoxia‐inducible factor; iNOS, inducible nitric oxide synthase; PHD, prolyl hydroxylases; PKCε, protein kinase C‐epsilon; PR, placental restriction; VEGF, vascular endothelial growth factor.
P<0.05.
Length of Coronary Capillaries
Fetuses exposed to chronic hypoxemia had an increased capillary length density (Figure 3A) but a similar total length of capillaries in the RV compared with controls (Figure 3B). Interestingly, control fetuses maintained a positive relationship between the number of cardiomyocytes and the length of capillaries (y=−4.55+3.68x; Figure 3C), but PR fetuses did not. A similar length of capillaries with a reduction in the number of cardiomyocytes would indicate that there was a greater length of capillaries per cardiomyocyte in the RV of the PR fetus (Figure 3D).
Figure 3.
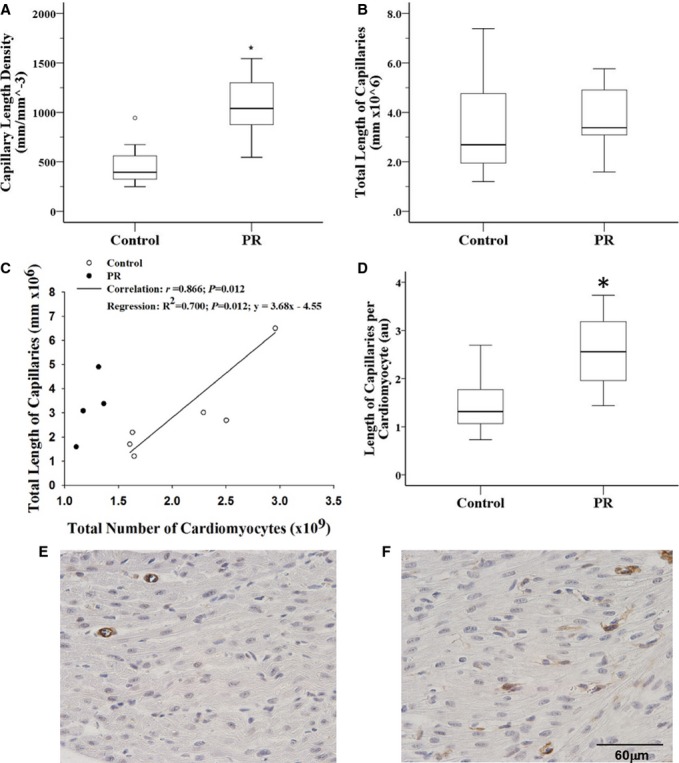
Placental restriction (PR; A and F) resulting in chronic hypoxemia increased capillary length density in the right ventricle compared with controls (A and E); however, there was a similar total length of capillaries (B). In the control right ventricle, there was a significant positive correlation between the total number of cardiomyocytes and the total length of capillaries; however, this correlation was not present in the PR group (C). Note, analysis of combined data from the control and PR groups demonstrated a significant correlation between the total length of capillaries and cardiomyocyte number (r=0.620, P=0.042); however, simple/linear regression analysis failed to reach significance. There is an increase in the length of capillaries per cardiomyocyte in the PR group compared with the control group (D; presented as arbitrary units (au) due to the 2 analyses being performed in sections embedded in different compounds; control, n=7; PR, n=4; these numbers are reduced because not every animal had both components analyzed). Coronary capillaries were identified with immunohistochemistry for α‐smooth muscle actin in the pericytes that surround the capillaries (brown) and counterstained with Mayer's hematoxylin. An open circle represents an outlier, defined as being >1.5×IQR; *P<0.05.
Expression and Abundance of HIF Regulators, PHDs
Exposure to chronic hypoxemia resulted in increased mRNA expression of the HIF destabilizing gene PHD3 but only in the LV (Table 5), coupled with an increased abundance of PHD2 protein (Figure 4). There was no change in the mRNA expression of PHD1 or PHD2 in either ventricle (Table 5). Similarly, there was no change in the protein abundance of PHD1 (Figure 4).
Figure 4.
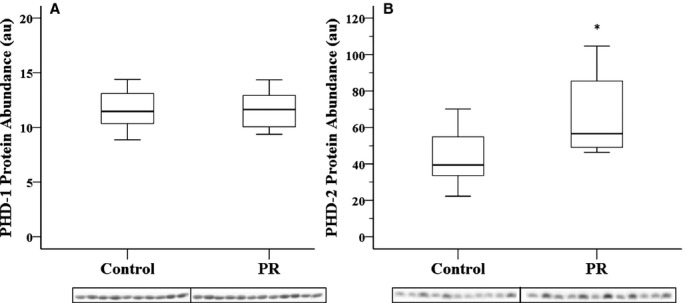
PR resulting in chronic hypoxemia does not change the protein abundance of PHD1 (A) but increases the protein abundance of PHD2 (B) in the left ventricle. Treatment groups were alternated across the Western blot to minimize transfer bias; *P<0.05. PHD indicates prolyl hydroxylase domain; PR, placental restriction.
Discussion
In the present study, experimental induction of PR in the sheep from conception resulted in chronic fetal hypoxemia, reduced fetal growth, and fewer cardiomyocytes in the fetal heart. This study is the first to demonstrate that there is a reduction in the total number of cardiomyocytes in a large animal model of intrauterine growth restriction in which the timing of cardiomyocyte maturation is similar to humans. Consistent with the current findings, maternal protein restriction in rats reduces fetal growth, heart weight, and total number of cardiomyocytes at birth.31 Furthermore, rats exposed to maternal hypoxia during the last week of pregnancy may also have a lower number of cardiomyocytes because adult offspring have the same heart weight as controls but larger individual cardiomyocytes.32 A reduction in the total number of cardiomyocytes may have critical consequences for heart health in later life because at birth, the hearts of both sheep and human contain the majority of the cardiomyocytes they will have for life.9,33 Consequently, the remaining cardiomyocytes will be required to increase in size to increase their capacity for contractile force generation, rendering the individual vulnerable to heart disease.34–35 This premise is supported by studies in the hypertrophic heart rat model, which has a smaller body from 2 days of age and a heart that contains smaller and fewer cardiomyocytes that have prematurely exited the cell cycle,36 and these rats develop cardiac hypertrophy in the absence of hypertension by 2 months of age.37 Interestingly, in the present study, the linear relationship between the number of cardiomyocytes and fetal weight, as previously reported in sheep with naturally occurring variations in birth weight,38 only holds true for fetuses with a body weight >3 kg. It appears that there may be a critical threshold for the number of cardiomyocytes required to maintain function in fetuses <3 kg. A reduction in cardiomyocyte endowment, as observed in the present model of chronic hypoxemia and intrauterine growth restriction, provides a potential link for the epidemiological association between intrauterine growth restriction and increased incidence of ischemic heart disease and heart failure in adult life.
Maternal hypoxia from 15 to 21 days of gestation in rats results in an increase in HIF‐1α protein abundance in the fetal heart at 21 days of gestation.11 Furthermore, fetal rats exposed to maternal hypoxia had a greater percentage of binucleated cardiomyocytes, which were also larger in size. This is in contrast to previous studies in fetal sheep in which hypoxemia due to umbilicoplacental embolization18 or PR19 not only reduced the percentage of binucleated cardiomyocytes, PR also reduced their size.19 Furthermore, in contrast to the present study, fetal rats exposed to maternal hypoxia in the last week of gestation had a greater percentage of terminal deoxynucleotidyl transferase dUTP nick‐end labeling–positive or apoptotic cardiomyocytes, possibly due to a decrease in antiapoptotic Bcl2 protein abundance and an increase in the pro‐apoptotic protein Fas.11 In the present study, chronic hypoxemia did not alter the mRNA expression of the antiapoptotic gene Bcl2 or the pro‐apoptotic gene Bax in the RV, whereas in the LV, exposure to chronic hypoxemia decreased the mRNA expression of Bcl2 and Bax. A decrease in both anti‐ and pro‐apoptotic factors may suggest that there is a similar Bcl2:Bax ratio, an index of mitochondrial‐mediated apoptosis, that is trigged when the ratio favors Bax.39 Although HIFs do not transcribe Bax, its expression can be regulated by hypoxia due to an HIF‐1α–dependent interaction with the transcription factor p53.5,40 Tumor suppressor p53 protein is stabilized in response to cellular stressors, including hypoxia,41 to arrest the cell cycle, induce apoptosis, and regulate autophagy.42 Another important p53‐43 and hypoxia‐induced44 regulator of cardiac cell death is BNIP3, which encodes a pro‐apoptotic Bcl2 family protein and can induce apoptosis44 and necrosis45 and is linked to the induction of autophagy in cardiac ischemia–reperfusion injury.46 Autophagy is utilized to maintain cellular homeostasis both at baseline and in response to stress, such as hypoxia. Autophagy is also promoted by Beclin‐1, which forms the autophagasome and is encoded by the BECN1 gene. During hypoxia, increased autophagy has been associated with increased transcription of autophagy‐related genes such as BNIP3,47–49 BECN1,50 MAP1LC3B,49 and LAMP1.51
In the present study, chronic hypoxemia for at least the last third of gestation in fetal sheep did not lead to increased terminal deoxynucleotidyl transferase dUTP nick‐end labeling–positive cardiomyocytes; an accumulation of p53 protein; an increase in Bax, BNIP3, BECN1, LAMP‐1, or MAP1LC3B transcription; or an increase in Beclin‐1 abundance. This suggests that in late gestation, the heart is not experiencing greater apoptosis or autophagy and thus may not be hypoxic.
Despite PR fetuses being hypoxemic in late gestation, we did not observe an increase in the cardiac mRNA expression of genes with hypoxia response elements, which are crucial for a cell's response to hypoxia to increase oxygen supply by angiogenesis (VEGF, Flt1, Ang2, and Tie2) and vasodilatation (iNOS and Adm) or to decrease oxygen demand by increasing anaerobic metabolism (GLUT1 and GLUT3). It is possible that this lack of change is due to the small sample size and a lack of power or that the heart is not hypoxic at the time point we studied. An alternative interpretation of the results presented in this study is that chronic hypoxia is unable to upregulate hypoxia‐responsive processes due to the “desensitization” and destabilization of HIF‐α subunits. Ginouves and colleagues suggested that reduction in mitochondrial respiration during hypoxia results in an increase in intracellular oxygen, enabling PHDs to be active despite hypoxia.52 Mice exposed to chronic hypoxia (24 hours; 8% oxygen in air) do not have stabilized HIF‐1α protein in the kidney, brain, and thymus, despite HIF‐1α being present after 6 hour of hypoxia (acute).52 After acute hypoxia, there was a decrease in PHD activity, which was reactivated after 24 hours of hypoxia. Furthermore, silencing all PHDs using small interfering RNA after 24 hours of hypoxia resulted in stabilization of HIF‐1α protein, providing evidence that it is PHD mediated. The theory of HIF‐1α desensitization is supported by studies in the skeletal muscle of exercising mice exposed to 1 day of hypobaric hypoxia (acute) during which HIF‐1α protein was stabilized versus 1 week of hypobaric hypoxia (chronic) during which no HIF‐1α protein was measured.53 In the present study, chronic hypoxemia resulted in an increase in the mRNA expression of PHD3 but only in the LV, as well as an increase in the abundance of PHD‐2 protein. The transcription of PHD2 and PHD3 is regulated by HIFs, possibly to promote rapid degradation of HIF‐α once normoxia is achieved. Consequently, the increase in PHDs and the absence of increased transcription of hypoxia‐responsive genes in our study present different interpretations, either that the heart is chronically hypoxic and HIF‐1α has been desensitized or that the heart is not hypoxic. Considering that we did not see a difference in the mRNA expression of PKCε, for which transcription due to prenatal hypoxia in cardiomyocytes is inhibited by intracellular reactive oxygen species and is independent of HIFs, the evidence suggests that the heart is not hypoxic.13–14 This lack of transcriptional activation in the heart of PR fetuses suggests that, despite exposure to chronic hypoxemia, the heart is not experiencing cellular hypoxia, potentially due to a lower demand for oxygen because it contains fewer cardiomyocytes and they are smaller.19 Considering that there is also no difference reported in the percentage of cardiomyocytes in the cell cycle,19 we suggest that, earlier in gestation, there has been either a reduction in the rate of proliferation (as observed in fetal sheep studies in which hypoxemia has been present for up to 20 days)18 or an increase in cardiomyocyte apoptosis (as observed in rat offspring of maternal hypoxia in the last week of gestation)11 that has resulted in the reduced number of cardiomyocytes observed in late gestation in the present study.
Considering that we did not observe a change in the mRNA expression of the angiogenic genes VEGF, Flt1, Ang1, Ang2, or Tie2 in the RV, it was surprising to observe an almost doubling of capillary length density in the RV of the chronically hypoxemic fetus. An increase in capillary density (angiogenesis) in response to acute hypoxia has been well documented and is essential to increased oxygen supply (for review, see Pugh and Ratcliffe54). Interestingly, there was no difference in the total length of capillaries in the RV of fetuses exposed to chronic hypoxemia compared with controls, suggesting an increased length of capillaries per cardiomyocyte. We speculate that cardiomyocytes and capillaries are differentially sensitive to the early environment in the PR fetus, and this ensures that each cardiomyocyte may have normal oxygen tension.
Conclusion
In the present study, chronic hypoxemia for at least the last third of gestation,20 resulted in growth‐restricted fetuses with smaller hearts that contained fewer cardiomyocytes. Despite a reduction in the number of cardiomyocytes, there is no difference in the percentage of terminal deoxynucleotidyl transferase dUTP nick‐end labeling–positive or apoptotic cardiomyocytes or in the protein abundance of the hypoxia‐mediated apoptosis regulator p53. Furthermore, there was neither a difference nor a decrease in the mRNA expression of the pro‐apoptotic gene Bax, the transcription of which is typically upregulated during hypoxia by p53.40 In addition, there was no change in the expression of autophagic genes that have been previously associated with hypoxia‐mediated autophagy or in the protein abundance of a key regulator of autophagy, Beclin‐1, compared with controls. Interestingly, there was no difference in the mRNA expression of HIFs (HIF‐1α, HIF‐2α, HIF‐3α, and HIF‐1β) or target genes with hypoxia response elements that are central to hypoxia‐mediated angiogenesis (VEGF, Flt1, Ang2, or Tie2), glycolysis (GLUT1 or GLUT3), and vasodilation (iNOS and Adm). Furthermore, we did not observe a decrease in the mRNA expression of PKCε, for which transcriptional regulation due to prenatal hypoxia is independent of HIFs. Despite chronic hypoxemia, PR fetuses had a similar length of capillaries compared with controls, suggesting an increased length of capillaries per cardiomyocyte. These data suggest that in late gestation, the heart of the chronically hypoxemic fetus is not experiencing cellular hypoxia potentially due to a decrease in oxygen demand (fewer and smaller cardiomyocytes) and an appropriate oxygen supply (maintenance of the total length of capillaries despite a smaller heart). This study suggests that the adaptation to hypoxia occurs earlier in gestation.
Sources of Funding
This study was supported by an National Health and Medical Research Council (NHMRC) Project grant to C.M. and J.L.M. J.L.M. was supported by a Career Development Award from the National Heart Foundation (NHF) of Australia (CR07A3328) and the NHMRC (511341) and a South Australian Cardiovascular Research Network Fellowship (CR10A4988). The Centre for Stochastic Geometry and Advanced Bioimaging was supported by Villum Foundation.
Disclosures
None.
Acknowledgments
We are grateful to Jayne Skinner for her expert assistance during sheep surgery and postoperative care of the ewe and her fetus. We also thank Stacey Dunn and Dr Sheridan Gentili for their technical assistance with Western blotting; Darran Tosh, Robb Muirhead, and Bernard Chuang for their technical assistance with real‐time polymerase chain reaction; and Jonnie Andersen and Maj‐Britt Lundorf for their technical assistance in determining total cardiomyocyte number.
References
- 1.Sugishita Y, Leifer DW, Agani F, Watanabe M, Fisher SA. Hypoxia‐responsive signaling regulates the apoptosis‐dependent remodeling of the embryonic avian cardiac outflow tract. Dev Biol. 2004; 273:285-296. [DOI] [PubMed] [Google Scholar]
- 2.Yue X, Tomanek RJ. Stimulation of coronary vasculogenesis/angiogenesis by hypoxia in cultured embryonic hearts. Dev Dyn. 1999; 216:28-36. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL. HIF‐1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000; 88:1474-1480. [DOI] [PubMed] [Google Scholar]
- 4.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008; 30:393-402. [DOI] [PubMed] [Google Scholar]
- 5.An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild‐type p53 by hypoxia‐inducible factor 1alpha. Nature. 1998; 392:405-408. [DOI] [PubMed] [Google Scholar]
- 6.Patterson AJ, Zhang L. Hypoxia and fetal heart development. Curr Mol Med. 2010; 10:653-666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gagnon R. Placental insufficiency and its consequences. Eur J Obstet Gynecol Reprod Biol. 2003; 110suppl 1:S99-S107. [DOI] [PubMed] [Google Scholar]
- 8.McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev. 2005; 85:571-633. [DOI] [PubMed] [Google Scholar]
- 9.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe‐Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009; 324:98-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrison JL. Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin Exp Pharmacol Physiol. 2008; 35:730-743. [DOI] [PubMed] [Google Scholar]
- 11.Bae S, Xiao Y, Li G, Casiano CA, Zhang L. Effect of maternal chronic hypoxic exposure during gestation on apoptosis in fetal rat heart. Am J Physiol Heart Circ Physiol. 2003; 285:H983-H990. [DOI] [PubMed] [Google Scholar]
- 12.Li G, Xiao Y, Estrella JL, Ducsay CA, Gilbert RD, Zhang L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J Soc Gynecol Investig. 2003; 10:265-274. [DOI] [PubMed] [Google Scholar]
- 13.Patterson AJ, Chen M, Xue Q, Xiao D, Zhang L. Chronic prenatal hypoxia induces epigenetic programming of PKC{epsilon} gene repression in rat hearts. Circ Res. 2010; 107:365-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patterson AJ, Xiao D, Xiong F, Dixon B, Zhang L. Hypoxia‐derived oxidative stress mediates epigenetic repression of PKCε gene in foetal rat hearts. Cardiovasc Res. 2012; 93:302-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996; 28:1737-1746. [DOI] [PubMed] [Google Scholar]
- 16.Botting KJ, Wang KC, Padhee M, McMillen IC, Summers‐Pearce B, Rattanatray L, Cutri N, Posterino GS, Brooks DA, Morrison JL. Early origins of heart disease: low birth weight and determinants of cardiomyocyte endowment. Clin Exp Pharmacol Physiol. 2012; 39:814-823. [DOI] [PubMed] [Google Scholar]
- 17.Bubb KJ, Cock ML, Black MJ, Dodic M, Boon WM, Parkington HC, Harding R, Tare M. Intrauterine growth restriction delays cardiomyocyte maturation and alters coronary artery function in the fetal sheep. J Physiol. 2007; 578:871-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louey S, Jonker SS, Giraud GD, Thornburg KL. Placental insufficiency decreases cell cycle activity and terminal maturation in fetal sheep cardiomyocytes. J Physiol. 2007; 580:639-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrison JL, Botting KJ, Dyer JL, Williams SJ, Thornburg KL, McMillen IC. Restriction of placental function alters heart development in the sheep fetus. Am J Physiol Regul Integr Comp Physiol. 2007; 293:R306-R313. [DOI] [PubMed] [Google Scholar]
- 20.Simonetta G, Rourke AK, Owens JA, Robinson JS, McMillen IC. Impact of placental restriction on the development of the sympathoadrenal system. Pediatr Res. 1997; 42:805-811. [DOI] [PubMed] [Google Scholar]
- 21.Wang KC, Zhang L, McMillen IC, Botting KJ, Duffield JA, Zhang S, Suter CM, Brooks DA, Morrison JL. Fetal growth restriction and the programming of heart growth and cardiac insulin‐like growth factor 2 expression in the lamb. J Physiol. 2011; 589:4709-4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muhlfeld C, Nyengaard JR, Mayhew TM. A review of state‐of‐the‐art stereology for better quantitative 3D morphology in cardiac research. Cardiovasc Pathol. 2010; 19:65-82. [DOI] [PubMed] [Google Scholar]
- 23.Gundersen HJ. The smooth fractionator. J Microsc. 2002; 207:191-210. [DOI] [PubMed] [Google Scholar]
- 24.Nyengaard JR, Gundersen JG. The isector: a simple and direct method for generating isotropic, uniform random sections from small specimen. J Microsc. 1992; 165:427-431. [Google Scholar]
- 25.Gundersen HJ. Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J Microsc. 1986; 143:3-45. [PubMed] [Google Scholar]
- 26.Bruel A, Oxlund H, Nyengaard JR. The total length of myocytes and capillaries, and total number of myocyte nuclei in the rat heart are time‐dependently increased by growth hormone. Growth Horm IGF Res. 2005; 15:256-264. [DOI] [PubMed] [Google Scholar]
- 27.Passmore M, Nataatmadja M, Fraser JF. Selection of reference genes for normalisation of real‐time RT‐PCR in brain‐stem death injury in Ovis aries. BMC Mol Biol. 2009; 10:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real‐time quantitative PCR data. Genome Biol. 2007; 8:R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soo PS, Hiscock J, Botting KJ, Roberts CT, Davey AK, Morrison JL. Maternal undernutrition reduces P‐glycoprotein in guinea pig placenta and developing brain in late gestation. Reprod Toxicol. 2012; 33:374-381. [DOI] [PubMed] [Google Scholar]
- 30.Gentili S, Morrison JL, McMillen IC. Intrauterine growth restriction and differential patterns of hepatic growth and expression of IGF1, PCK2, and HSDL1 mRNA in the sheep fetus in late gestation. Biol Reprod. 2009; 80:1121-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corstius HB, Zimanyi MA, Maka N, Herath T, Thomas W, van der Laarse A, Wreford NG, Black MJ. Effect of intrauterine growth restriction on the number of cardiomyocytes in rat hearts. Pediatr Res. 2005; 57:796-800. [DOI] [PubMed] [Google Scholar]
- 32.Li G, Bae S, Zhang L. Effect of prenatal hypoxia on heat stress‐mediated cardioprotection in adult rat heart. Am J Physiol Heart Circ Physiol. 2004; 286:H1712-H1719. [DOI] [PubMed] [Google Scholar]
- 33.Burrell JH, Boyn AM, Kumarasamy V, Hsieh A, Head SI, Lumbers ER. Growth and maturation of cardiac myocytes in fetal sheep in the second half of gestation. Anat Rec A Discov Mol Cell Evol Biol. 2003; 274:952-961. [DOI] [PubMed] [Google Scholar]
- 34.Thornburg KL, Louey S, Giraud GD. The role of growth in heart development. Nestle Nutr Workshop Ser Pediatr Program. 2008; 61:39-51. [DOI] [PubMed] [Google Scholar]
- 35.Porrello ER, Widdop RE, Delbridge LM. Early origins of cardiac hypertrophy: does cardiomyocyte attrition programme for pathological ‘catch‐up’ growth of the heart? Clin Exp Pharmacol Physiol. 2008; 35:1358-1364. [DOI] [PubMed] [Google Scholar]
- 36.Porrello ER, Bell JR, Schertzer JD, Curl CL, McMullen JR, Mellor KM, Ritchie RH, Lynch GS, Harrap SB, Thomas WG, Delbridge LM. Heritable pathologic cardiac hypertrophy in adulthood is preceded by neonatal cardiac growth restriction. Am J Physiol Regul Integr Comp Physiol. 2009; 296:R672-R680. [DOI] [PubMed] [Google Scholar]
- 37.Harrap SB, Danes VR, Ellis JA, Griffiths CD, Jones EF, Delbridge LM. The hypertrophic heart rat: a new normotensive model of genetic cardiac and cardiomyocyte hypertrophy. Physiol Genomics. 2002; 9:43-48. [DOI] [PubMed] [Google Scholar]
- 38.Stacy V, De Matteo R, Brew N, Sozo F, Probyn ME, Harding R, Black MJ. The influence of naturally occurring differences in birthweight on ventricular cardiomyocyte number in sheep. Anat Rec (Hoboken). 2009; 292:29-37. [DOI] [PubMed] [Google Scholar]
- 39.Takagi‐Morishita Y, Yamada N, Sugihara A, Iwasaki T, Tsujimura T, Terada N. Mouse uterine epithelial apoptosis is associated with expression of mitochondrial voltage‐dependent anion channels, release of cytochrome C from mitochondria, and the ratio of Bax to Bcl‐2 or Bcl‐X. Biol Reprod. 2003; 68:1178-1184. [DOI] [PubMed] [Google Scholar]
- 40.Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011; 2:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia‐mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996; 379:88-91. [DOI] [PubMed] [Google Scholar]
- 42.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53‐regulated genes. Nat Rev Mol Cell Biol. 2008; 9:402-412. [DOI] [PubMed] [Google Scholar]
- 43.Wang EY, Gang H, Aviv Y, Dhingra R, Margulets V, Kirshenbaum LA. p53 Mediates autophagy and cell death by a mechanism contingent on BNIP3. Hypertension. 2013; 62:70-77. [DOI] [PubMed] [Google Scholar]
- 44.Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA. Hypoxia and acidosis activate cardiac myocyte death through the Bcl‐2 family protein BNIP3. Proc Natl Acad Sci USA. 2002; 99:12825-12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis‐like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000; 20:5454-5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamacher‐Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves BNIP3 and autophagy. Cell Death Differ. 2007; 14:146-157. [DOI] [PubMed] [Google Scholar]
- 47.Masschelein E, Van Thienen R, D'Hulst G, Hespel P, Thomis M, Deldicque L. Acute environmental hypoxia induces LC3 lipidation in a genotype‐dependent manner. FASEB J. 2014; 28:1022-1034. [DOI] [PubMed] [Google Scholar]
- 48.Ishihara M, Urushido M, Hamada K, Matsumoto T, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Horino T, Fujieda M, Fujimoto S, Terada Y. Sestrin‐2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol. 2013; 305:F495-F509. [DOI] [PubMed] [Google Scholar]
- 49.de Theije CC, Langen RC, Lamers WH, Schols AM, Kohler SE. Distinct responses of protein turnover regulatory pathways in hypoxia‐ and semistarvation‐induced muscle atrophy. Am J Physiol Lung Cell Mol Physiol. 2013; 305:L82-L91. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Pan XL, Ding LJ, Liu DY, Da‐Peng L, Jin T. Aberrant expression of Beclin‐1 and LC3 correlates with poor prognosis of human hypopharyngeal squamous cell carcinoma. PLoS One. 2013; 8:e69038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Escobar ML, Echeverria OM, Sanchez‐Sanchez L, Mendez C, Pedernera E, Vazquez‐Nin GH. Analysis of different cell death processes of prepubertal rat oocytes in vitro. Apoptosis. 2010; 15:511-526. [DOI] [PubMed] [Google Scholar]
- 52.Ginouves A, Ilc K, Macias N, Pouyssegur J, Berra E. PHDs overactivation during chronic hypoxia “desensitizes” HIFalpha and protects cells from necrosis. Proc Natl Acad Sci USA. 2008; 105:4745-4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Le Moine CM, Morash AJ, McClelland GB. Changes in HIF‐1alpha protein, pyruvate dehydrogenase phosphorylation, and activity with exercise in acute and chronic hypoxia. Am J Physiol Regul Integr Comp Physiol. 2011; 301:R1098-R1104. [DOI] [PubMed] [Google Scholar]
- 54.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003; 9:677-684. [DOI] [PubMed] [Google Scholar]