

Abstract
Background
The expression of a novel cardiac glucose transporter, SGLT1, is increased in glycogen storage cardiomyopathy secondary to mutations in PRKAG2. We sought to determine the role of SGLT1 in the pathogenesis of PRKAG2 cardiomyopathy and its role in cardiac structure and function.
Methods and Results
Transgenic mice with cardiomyocyte‐specific overexpression of human T400N mutant PRKAG2 cDNA (TGT400N) and transgenic mice with cardiomyocyte‐specific RNA interference knockdown of SGLT1 (TGSGLT1‐DOWN) were crossed to produce double‐transgenic mice (TGT400N/TGSGLT1‐DOWN). Tet‐off transgenic mice conditionally overexpressing cardiac SGLT1 in the absence of doxycycline were also constructed (TGSGLT‐ON). Relative to TGT400N mice, TGT400N/TGSGLT1‐DOWN mice exhibited decreases in cardiac SGLT1 expression (63% decrease, P<0.05), heart/body weight ratio, markers of cardiac hypertrophy, and cardiac glycogen content. TGT400N/TGSGLT1‐DOWN mice had less left ventricular dilation at age 12 weeks compared to TGT400N mice. Relative to wildtype (WT) mice, TGSGLT1‐ON mice exhibited increases in heart/body weight ratio, glycogen content, and markers of cardiac hypertrophy at ages 10 and 20 weeks. TGSGLT1‐ON mice had increased myocyte size and interstitial fibrosis, and progressive left ventricular dysfunction. When SGLT1 was suppressed after 10 weeks of overexpression (TGSGLT1‐ON/OFF), there was a reduction in cardiac hypertrophy and improvement in left ventricular failure.
Conclusions
Cardiac knockdown of SGLT1 in a murine model of PRKAG2 cardiomyopathy attenuates the disease phenotype, implicating SGLT1 in the pathogenesis. Overexpression of SGLT1 causes pathologic cardiac hypertrophy and left ventricular failure that is reversible. This is the first report of cardiomyocyte‐specific transgenic knockdown of a target gene.
Keywords: cardiomyopathy, glucose, heart failure, hypertrophy
Introduction
AMP‐activated protein kinase (AMPK), a heterotrimeric protein composed of a catalytic α subunit and regulatory β and γ subunits, is activated under conditions of energy depletion manifested by increased cellular AMP levels.1–2 Mutations in the gene PRKAG2, encoding the cardiac γ2 regulatory subunit of AMPK, have been demonstrated to produce a distinct glycogen storage cardiomyopathy in human families.3 We and others have shown that inappropriate activation of AMPK by PRKAG2 mutations4–7 leads to increased cardiac glucose uptake and glycogen synthesis8 and activation of cardiac hypertrophic signaling pathways.9–10
Classically, it has been thought that only the GLUT isoforms GLUT1 and GLUT4 are responsible for glucose uptake in cardiomyocytes.11 However, we have recently reported that the sodium‐dependent glucose co‐transporter (SGLT) isoform SGLT1 is present at the protein level in cardiomyocytes, and appears to be localized to the sarcolemma.12 SGLTs transport glucose by a secondary active transport mechanism that uses the sodium concentration gradient established by the Na+/K+‐ATPase pump. We recently showed that cardiac SGLT1 expression is increased in PRKAG2 cardiomyopathy, and that the increased cardiac glucose uptake appears to be mediated partly by SGLT1.13 Specifically, the pharmacological SGLT1 inhibitor phlorizin reduces glycogen storage in PRKAG2 cardiomyopathy. However, whether long‐term specific inhibition of SGLT1 in cardiomyocytes by genetic means results in a reduction in cardiac hypertrophy or improvement in cardiac function is unknown. Furthermore, the effect of overexpression of SGLT1 on the heart is also unknown. Therefore, the objective of the present study was to investigate whether transgenic knockdown of cardiac SGLT1 (TGSGLT1‐DOWN) in mice with the PRKAG2 Thr400Asn mutation (TGT400N) attenuates the cardiomyopathy phenotype, and whether transgenic mice with cardiac SGLT1 overexpression (TGSGLT1‐ON) replicate phenotypic features of the cardiomyopathy.
Methods
Construction of SGLT1 Knockdown (TGSGLT1‐DOWN) and Double‐Transgenic (TGT400N/TGSGLT1‐DOWN) Mice
All studies involving mice conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85‐23, revised 1996) and were approved by the University of Pittsburgh and The University of Iowa Institutional Animal Care and Use Committees. We tested several siRNAs (Invitrogen) for RNA interference of SGLT1 mRNA in isolated adult mouse cardiomyocytes and in the HL‐1 cardiomyocyte line14 relative to negative control scrambled siRNA (Figure 1). We used the sequence of siRNA no. MSS277112 (Invitrogen), which resulted in the greatest RNA interference and reduction of protein levels of SGLT1 in isolated cells, to construct a transgenic mouse model (TGSGLT1‐DOWN) expressing short hairpin RNA for RNA interference of SGLT1. The transgene construct places the short hairpin RNA within a pC126 expression vector (a generous gift of Dr Jeffrey Robbins, University of Cincinnati), containing a highly active cardiomyocyte‐specific α‐myosin heavy‐chain promoter.4,15–16 Prior to insertion into this vector, the siRNA was cloned into a segment of murine genomic DNA comprising the first 1000 bp of the third intron of the murine α‐myosin heavy‐chain gene, at a Bam HI site at a position 595 bp 3′ to the start of the intronic segment. This construct was linearized, size‐fractionated, purified, and micro‐injected into fertilized FVB mouse oocytes at the University of Pittsburgh Transgenic and Chimeric Mouse Facility. The construction of the transgenic mouse with the human T400N mutation in PRKAG2 (TGT400N) has been previously described.4 Double‐transgenic mice (TGT400N/TGSGLT1‐DOWN) were obtained by crossbreeding.
Figure 1.
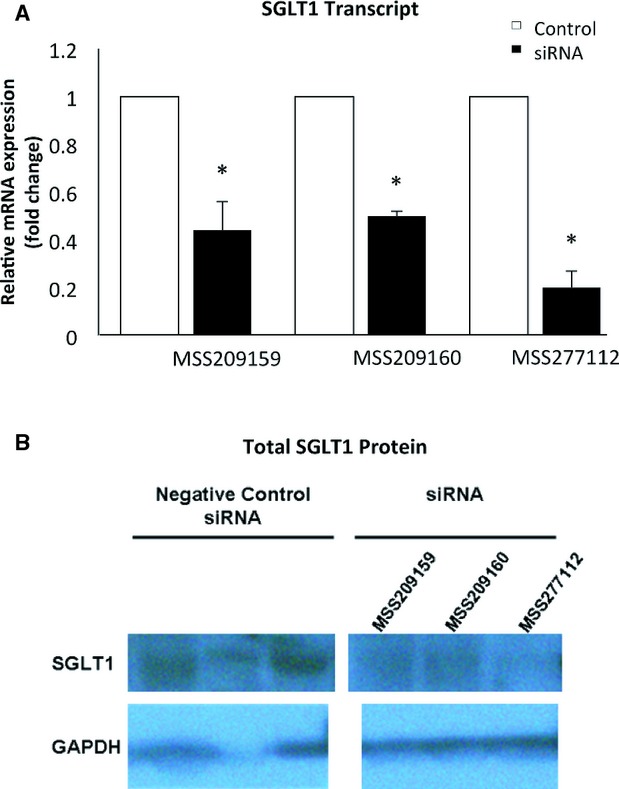
Real‐time quantitative PCR (A) and immunoblots (B) of SGLT1 in HL‐1 cardiomyocytes treated with different siRNAs (Invitrogen). *P<0.01 vs negative control siRNA. PCR indicates polymerase chain reaction; SGLT1, sodium‐dependent glucose co‐transporter 1.
Construction of the SGLT1 Conditionally Overexpressing (TGSGLT1‐ON) Mouse
Constitutive cardiac transgenic overexpression of SGLT1 was lethal at the embryonic or early neonatal stages (data not shown). Therefore, to be able to regulate cardiac transgene expression, a tetracycline‐suppressible binary α‐MHC–driven system was used.17 The transactivator protein, tTA, is required for robust expression of the target responder transgene (SGLT1). Competitive binding of doxycycline to tTA suppresses SGLT1 transactivation, which enables control of transgene expression by oral doxycycline administration. The responder transgene construct was delivered by pronuclear injection into fertilized FVB mouse oocytes at the University of Pittsburgh Transgenic and Chimeric Mouse Facility. Responder and transactivator transgenic mice were crossed to generate conditionally overexpressing SGLT1 mice (TGSGLT1) under the inhibitory control of doxycycline.
Genotyping
Tail snips were collected, DNA was extracted, and genotyping was performed by multiplex polymerase chain reaction with primers specific for each transgene (Table 1). Internal controls included primers for essential myosin light chain and control mouse genomic DNA.
Table 1.
Genotyping Primers
| Transgene | Direction | Primer Sequence |
|---|---|---|
| TGT400N | Forward | 5′‐CGGCACTCTTAGCAAACCTC‐3′ |
| Reverse | 5′‐TTCTGGCTGGCATTTTTCTT‐3′ | |
| TGSGLT1‐DOWN | Forward | 5′‐GACGCTGTCTTCCTCCTCAG |
| Reverse | 5′‐TACTGGTGAGCGGTGGGTGGTT‐3′ | |
| tTA transactivator | Forward | 5′‐AGCGCATTAGAGCTGCTTAATGAGGTC‐3′ |
| Reverse | 5′‐GTCGTAATAATGGCGGCATACTATC‐3′ | |
| SGLT1 responder | Forward | 5′‐TTCTGAGAAGCCTTTGTGGAG‐3′ |
| Reverse | 5′‐CCTAGTCAGACAAAATGATGCAA‐3′ |
SGLT1 indicates sodium‐dependent glucose co‐transporter; tTA, transactivator protein.
Study Protocol for the Conditional Overexpression of SGLT1
The expression of SGLT1 was suppressed in all transgenic mice during conception to age 4 weeks postnatally by administering doxycycline‐containing food (625 mg/kg; Harlan Laboratories) to pregnant and nursing mothers and to weaned offspring. To study the effects of SGLT1 overexpression on cardiac pathology and function, TGSGLT1 male and female mice >4 weeks of age were placed in 2 experimental groups, SGLT1 suppression (TGSGLT1‐OFF) and SGLT1 overexpression (TGSGLT1‐ON), and analyzed at either 10 or 20 weeks of age. In addition, to study the reversibility of the effects of cardiac SGLT1 overexpression, mice were exposed to SGLT1 overexpression at 4 to 10 weeks of age, followed by SGLT1 suppression at 10 to 20 weeks of age (TGSGLT1‐ON/OFF) (Figure 2).
Figure 2.
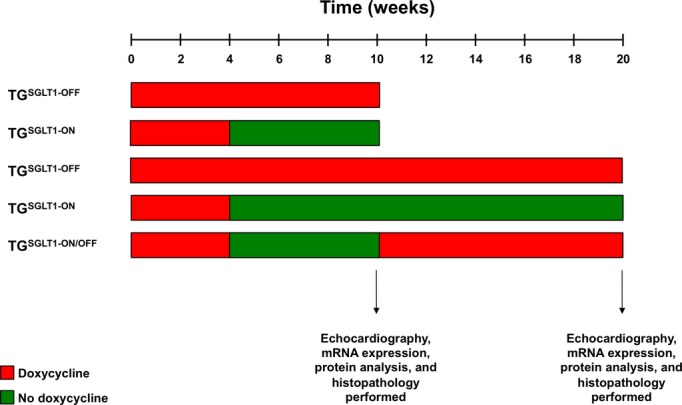
Timeline of SGLT1 overexpressing mouse groups. SGLT1 indicates sodium‐dependent glucose co‐transporter 1.
RNA Analyses
Total RNA was isolated from whole hearts and real‐time quantitative polymerase chain reaction analysis performed as described.16 Primers for quantitative polymerase chain reaction analysis were designed using published sequence information, avoiding regions of homology with other genes (Table 2). Fold‐changes were calculated with normalization to cyclophilin transcript levels.
Table 2.
Real‐Time Quantitative Polymerase Chain Reaction Primers Used to Quantify mRNA Expression
| Transcript | Direction | Primer Sequence |
|---|---|---|
| Glut 1 | Sense | 5′‐GAGTGACGATCTGAGCTACGG‐3′ |
| Antisense | 5′‐CGTTACTCACCTTGCTGCTG‐3′ | |
| Glut 4 | Sense | 5′‐GTAACTTCATTGTCGGCATGG‐3′ |
| Antisense | 5′‐CCTGCTCTAAAAGGGAAGGTG‐3′ | |
| Nppa | Sense | 5′‐GAAAAGCAAACTGAGGGCTCTG‐3′ |
| Antisense | 5′‐CCTACCCCCGAAGCAGCT‐3′ | |
| Nppb | Sense | 5′‐AGGCGAGACAAGGGAGAACA‐3′ |
| Antisense | 5′‐GGAGATCCATGCCGCAGA‐3′ | |
| Procollagen‐1 | Sense | 5′‐GTAAACACCCCAGCGAAGAACTC‐3′ |
| Antisense | 5′‐TCAAACTGGCTGCCACCAT‐3′ | |
| SGLT1 | Sense | 5′‐GTGCACCTGTACCGTTTGTG‐3′ |
| Antisense | 5′‐TTGGAGTCCTCTGGGATGTC‐3′ | |
| Cyclophilin | Sense | 5′‐TGTGCCAGGGTGGTGACTT‐3′ |
| Antisense | 5′‐TCAAATTTCTCTCCGTAGATGGACTT‐3′ |
SGLT1 indicates sodium‐dependent glucose co‐transporter 1.
Protein Analyses
Extraction of total cardiac protein was performed as described previously.9 The extraction of membrane proteins from cardiac tissue was performed using a commercially available kit (no. k268‐50; BioVision) according to the manufacturer's instructions. For membrane protein extraction, 5 to 8 pooled hearts of each genotype totaling at least 1 g in mass were homogenized and processed. The α1 subunit of the Na+/K+‐ATPase, a sarcolemmal membrane marker, was quantified by immunoblot to document adequate enrichment and equal loading of membrane proteins. Immunoblots were performed with antibodies specific for SGLT1 (sc‐20582; Santa Cruz), GLUT1 (ab40084; Abcam), GLUT4 (2213s; Cell Signaling), Na+/K+ ATPase (3010s; Cell Signaling), AMPKα (2532; Cell Signaling), phospho‐Thr172 AMPKα (2531; Cell Signaling), and GAPDH (ab8245; Abcam).
Analysis of Glycogen Content
Glycogen content was determined by the amyloglucosidase digestion method.18
Histopathology
Sections were stained with hematoxylin and eosin to assess myofiber architecture, Masson's trichrome to assess interstitial fibrosis, and periodic acid–Schiff to assess glycogen content.5 For quantitative histomorphometry, hematoxylin and eosin–stained samples were used. Five separate regions per slide were photographed at 40× with myocytes predominantly oriented in cross section. Micrographs were digitized and nucleated myofibers were traced using Image J 1.44p software (National Institutes of Health, Bethesda, MD) in a blinded fashion. Twenty‐five myofibers per heart were examined.19
Echocardiography
Transthoracic echocardiography was performed on mice after sedation with tribromoethanol (125 mg/kg IP) using a VisualSonics Vevo 770 machine with a 45‐MHz transducer.18 Left ventricular end diastolic (LVEDD) and end systolic (LVESD) chamber dimensions and anterior wall thickness (LVAWT) were obtained from M‐mode tracings. LV fractional shortening was calculated as (LVEDD−LVESD)/LVEDD×100%.
Statistical Analysis
Data are expressed as the mean±SE. Statistical analysis was performed using the R program 3.1.0 (R Foundation for Statistical Computing) with P<0.05 considered statistically significant. Prior to testing for significance, the data distributions were examined for normality with the Shapiro‐Wilk test. Comparisons between 2 groups were made using the Student t test or the Wilcoxon–Mann–Whitney U test, as appropriate. For comparisons among 3 or more groups, we used 1‐way ANOVA followed by Tukey's post hoc tests or the Kruskal–Wallis test followed by post hoc pairwise Wilcoxon–Mann–Whitney U tests, as appropriate.
Results
Cardiomyocyte‐Specific Transgenic Knockdown of SGLT1
TGT400N/TGSGLT1‐DOWN mice exhibited a 63% reduction in cardiac SGLT1 transcript levels relative to TGT400N mice at 5 weeks of age (Figure 3). Similarly, immunoblots of membrane protein from 5‐week‐old male and female mice showed decreased expression of SGLT1 in TGSGLT1‐DOWN mice relative to wildtype (WT) mice and in TGT400N/TGSGLT1‐DOWN mice relative TGT400N mice. Expression of SGLT1 in noncardiac tissue, including kidney, intestinal, and liver tissue, was unchanged across groups (data not shown).
Figure 3.
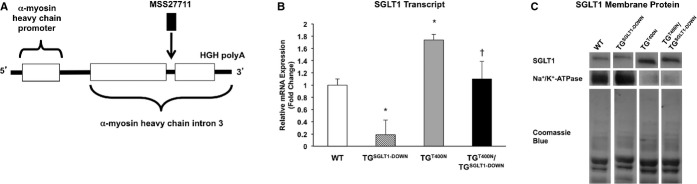
Construction of a cardiomyocyte‐specific SGLT1 knockdown transgenic mouse model (TGSGLT1‐DOWN). A, The sequence of siRNA no. MSS277112 (Invitrogen), which resulted in the greatest RNA interference and reduction of protein levels of SGLT1 in isolated cells (Figure 1), was used to construct a transgenic mouse model (TGSGLT1‐DOWN) expressing hairpin shRNA for RNA interference of SGLT1. The transgene construct places the shRNA within a pC126 expression vector, containing a highly active cardiomyocyte‐specific αMHC promoter. Prior to insertion into this vector, the siRNA was cloned into a segment of murine genomic DNA comprising the first 1000 bp of the third intron of the murine αMHC gene, at a Bam HI site at a position 595 bp 3′ to the start of the intronic segment. B, Relative total cardiac SGLT1 mRNA expression and (C) membrane SGLT1 protein expression in WT, TGSGLT1‐DOWN, TGT400N, and TGT400N/TGSGLT1‐DOWNmice. TGSGLT1‐DOWN mice exhibited decreased expression of SGLT1 transcript and protein relative to WT mice. TGT400N/TGSGLT1‐DOWN mice exhibited decreased expression of SGLT1 transcript and protein relative to TGT400N mice. Lanes depicted are taken from the same immunoblot. QPCR: WT, N=5; TGSGLT1‐DOWN, N=3; TGT400N, N=5; TGT400N/TGSGLT1‐DOWN, N=3. Immunoblot: protein from multiple mice was pooled into 1 lane; WT, N=8; TGSGLT1‐DOWN, N=8; TGT400N, N=5; TGT400N/TGSGLT1‐DOWN, N=5. *P<0.005 vs WT; †P<0.05 vs TGT400N. Data are expressed as mean±SE. MHC indicates myosin heavy‐chain promoter; QPCR, quantitative polymerase chain reaction; SGLT1, sodium‐dependent glucose co‐transporter 1; WT, wildtype.
Targeted Cardiac SGLT1 Knockdown Does Not Impair Murine Cardiac Function
Relative to WT mice, TGSGLT1‐DOWN mice showed no significant differences in heart to body weight ratios (WT 4.15±0.15 versus TGSGLT1‐DOWN 3.93±0.08) and expression levels of Nppa and Nppb (Figure 4A through 4C). In addition, echocardiographic measurements were similar between TGSGLT1‐DOWN and WT mice (Table 3). TGSGLT1‐DOWN mice had a small but significant decrease in cardiac glycogen content relative to WT mice (TGSGLT1‐DOWN 0.54±0.07 versus WT 0.84±0.10 μg/mg cardiac mass, P<0.05) at 5 weeks of age (Figure 4D). There was no significant qualitative difference in the levels of membrane‐associated GLUT4 between TGSGLT1‐DOWN and WT mice, although there appeared to be a reduction in membrane‐associated GLUT1 protein (Figure 5). There were no morphologic abnormalities in myocyte architecture evident in TGSGLT1‐DOWN hearts stained with hematoxylin and eosin (Figure 4E). Finally, TGSGLT1‐DOWN hearts exhibited no differences in AMPK activity relative to WT mice, as reflected by expression of phospho‐Thr172 AMPKα (Figure 6).
Figure 4.

At 5 to 7 weeks of age, relative to TGT400N mice, TGT400N/TGSGLT1‐DOWN mice exhibited decreased heart‐to‐body‐weight ratios (A), downregulation of hypertrophic markers Nppa (B) and Nppb (C), and decreased cardiac glycogen content (D). The decreased cardiac glycogen content was reflected on cardiac histopathology with hematoxylin and eosin (H&E) staining showing a decrease in glycogen‐containing vacuoles in TGT400N/TGSGLT1‐DOWN relative to TGT400N mice (E). QPCR: WT, N=4; TGSGLT1‐DOWN, N=3; TGT400N, N=5; TGT400N/TGSGLT1‐DOWN, N=7. Heart‐to‐body‐weight ratio: WT, N=5; TGSGLT1‐DOWN, N=6; TGT400N, N=7; TGT400N/TGSGLT1‐DOWN, N=7. Glycogen content: WT, N=11; TGSGLT1‐DOWN, N=17; TGT400N, N=12; TGT400N/TGSGLT1‐DOWN, N=17. *P<0.05 vs WT; †P<0.005 vs WT; ‡P<0.05 vs TGT400N; §P<0.01 vs TGT400N. Data are expressed as mean±SE. Bar=2 μm. QPCR indicates quantitative polymerase chain reaction; SGLT1, sodium‐dependent glucose co‐transporter 1; WT, wildtype.
Table 3.
Echocardiography of WT, TGSGLT1‐DOWN, TGT400N, TGT400N/TGSGLT1‐DOWN Mice
| WT | TGSGLT1‐DOWN | TGT400N | TGT400N/TGSGLT1‐DOWN | |
|---|---|---|---|---|
| 7 weeks | ||||
| N | 3 | 3 | 3 | 5 |
| LVAWT, mm | 0.79±0.13 | 0.81±0.09 | 1.99±0.23† | 2.01±0.16† |
| LVEDD, mm | 2.69±0.23 | 2.99±0.08 | 2.56±0.24 | 1.95±0.15* |
| FS, % | 54±10 | 45±3 | 35±6 | 81±5*‡§ |
| HR, bpm | 455±69 | 430±30 | 425±25 | 438±21 |
| 12 weeks | ||||
| N | 5 | 6 | 3 | 5 |
| LVAWT, mm | 0.93±0.03 | 0.96±0.05 | 1.59±0.20† | 1.56±0.10† |
| LVEDD, mm | 2.35±0.16 | 2.39±0.15 | 4.52±0.39† | 3.15±0.21‡ |
| FS, % | 66±6 | 65±5 | 13±4† | 32±3* |
| HR, bpm | 552±21 | 538±26 | 440±10* | 417±19† |
FS indicates fractional shortening; HR, heart rate; LVAWT, left ventricular anterior wall thickness; LVEDD, left ventricular end‐diastolic diameter; SGLT, sodium‐dependent glucose co‐transporter 1; WT, wildtype.
*P<0.05 vs WT; †P<0.005 vs WT; ‡P<0.005 vs TGT400N; §P<0.005 vs TGSGLT1‐DOWN.
Figure 5.
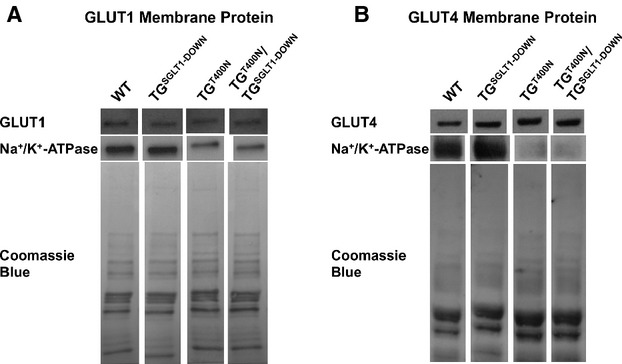
Cardiac membrane protein expression of the facilitated diffusion glucose transporters GLUT1 (A) and GLUT4 (B) in WT, TGSGLT1‐DOWN, TGT400N, and TGT400N/TGSGLT1‐DOWN mice. A Na+/K+‐ATPase immunoblot was used to document loading of sarcolemmal protein, and Coomassie blue staining was used to document loading of total membrane protein. Lanes depicted are taken from the same immunoblot. Protein from multiple mice was pooled into 1 lane; WT, N=8; TGSGLT1‐DOWN, N=8; TGT400N, N=5; TGT400N/TGSGLT1‐DOWN, N=5. SGLT1 indicates sodium‐dependent glucose co‐transporter 1; WT, wildtype.
Figure 6.
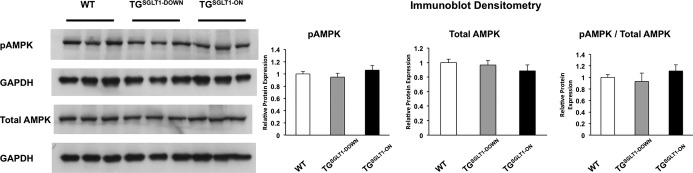
Total cardiac protein immunoblots showed that levels of phospho‐Thr172 AMPKα (pAMPK), reflecting AMPK activity, were not significantly different in WT, TGSGLT1‐DOWN, and TGSGLT1‐ON mice at the age of 10 weeks. Densitometry plots were normalized to GAPDH signal. AMPK indicates AMP‐activated protein kinase; SGLT1, sodium‐dependent glucose co‐transporter 1; WT, wildtype.
SGLT1 Knockdown Attenuates Glycogen Storage, Cardiac Hypertrophy, and LV Dysfunction in PRKAG2 Cardiomyopathy
We have previously reported that pharmacological inhibition of SGLT1 by the competitive inhibitor phlorizin decreased cardiac glycogen content and inhibited cardiac glucose uptake in TGT400N mice.13 However, phlorizin is not completely selective for SGLT1, and has systemic effects in noncardiac tissue. In order to ascertain the role of SGLT1 in PRKAG2 cardiomyopathy, we generated TGT400N/TGSGLT1‐DOWN mice. Relative to TGT400N mice, TGT400N/TGSGLT1‐DOWN mice exhibited significant reductions in heart‐to‐body‐weight ratio, and in Nppa and Nppb transcript levels (Figure 4A through 4C). The significant 23% reduction in cardiac glycogen content in TGT400N/TGSGLT1‐DOWN mice relative to TGT400N mice was similar to our previous finding using phlorizin (Figure 4D). Histological examination with hematoxylin and eosin confirmed a decrease in glycogen‐laden myocytes in TGT400N/TGSGLT1‐DOWN mice relative to TGT400N mice (Figure 4E).
In addition, TGT400N/TGSGLT1‐DOWN mice at 7 weeks of age had increased fractional shortening relative to all other groups of mice (Table 3). At 12 weeks of age, TGT400N/TGSGLT1‐DOWN mice had a trend towards improved LV fractional shortening (32±3 versus 13±4, P=0.09) and significantly less LV end diastolic dilation compared to TGT400N. There was no significant difference in LV wall thickness between TGT400N/TGSGLT1‐DOWNand TGT400N mice at 7 or 12 weeks of age.
Membrane‐Associated GLUT1 and GLUT4 Levels are Similar in TGT400N/TGSGLT1‐DOWN and TGT400NMice
Since SGLT1 knockdown may be associated with compensatory changes in GLUT1 and GLUT4, we assessed their expression in TGT400N/TGSGLT1‐DOWN and TGT400N hearts. There was no significant qualitative difference in cardiac membrane‐associated GLUT1 and GLUT4 between TGT400N/TGSGLT1‐DOWN and TGT400N mice (Figure 5).
Conditional Cardiac Overexpression of SGLT1 Leads to Cardiac Glycogen Accumulation, Hypertrophy, Fibrosis, and Contractile Dysfunction
The conditional system showed robust control of cardiac SGLT1 expression by doxycycline. TGSGLT1‐ON mice exhibited an increase in SGLT1 transcript and membrane protein levels at 10 and 20 weeks of age relative to WT and TGSGLT1‐OFF mice (Figure 7). In addition, TGSGLT1‐ON/OFF mice had a reduction in SGLT1 transcript and membrane protein expression at 20 weeks of age to levels comparable to baseline. Furthermore, TGSGLT1‐ON hearts at 10 weeks of age exhibited no differences in AMPK activity relative to WT mice, as reflected by expression of phospho‐Thr172 AMPKα (Figure 6).
Figure 7.
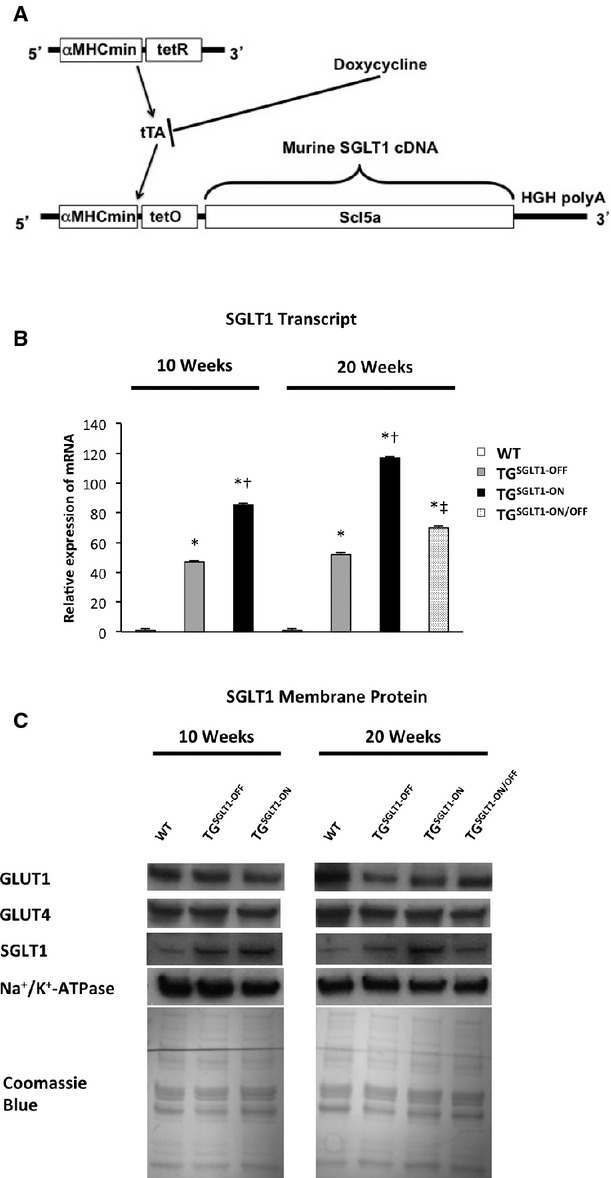
Conditional cardiomyocyte‐specific transgenic overexpression of SGLT1. A, The wildtype sequence for Slc5a1 (SGLT1) cDNA was placed under the control of a cardiomyocyte‐specific myosin heavy‐chain promoter (αMHCmin), which was dependent on the doxycycline‐controlled transcriptional activator (tTA) system for expression. B, Relative total cardiac SGLT1 mRNA expression, and (C) membrane SGLT1, GLUT1, and GLUT4 protein expression in 10‐ and 20‐week‐old WT mice, mice with SGLT1 suppression by administration of doxycycline in chow (TGSGLT1‐OFF), and mice with SGLT1 overexpression (TGSGLT1‐ON), and in 20‐week‐old mice with SGLT1 overexpression from 4 to 10 weeks of age followed by SGLT1 suppression from 10 to 20 weeks of age (TGSGLT‐ON/OFF). QPCR at 10 weeks: WT, N=5; TGSGLT1‐OFF, N=4; TGSGLT1‐ON, N=4. QPCR at 20 weeks: WT, N=5; TGSGLT1‐OFF, N=4; TGSGLT1‐ON, N=3; TGSGLT1‐ON/OFF, N=4. Immunoblots protein from multiple mice was pooled into 1 lane; WT, N=8; TGSGLT1‐OFF, N=5; TGSGLT1‐ON, N=5; TGSGLT1‐ON/OFF, N=5. *P<0.05 vs WT; †P<0.05 vs TGSGLT1‐OFF; ‡P<0.05 vs TGSGLT1‐ON. Data are expressed as mean±SE. QPCR indicates quantitative polymerase chain reaction; SGLT1, sodium‐dependent glucose co‐transporter 1; WT, wildtype.
At 10 weeks of age, TGSGLT1‐ON mice showed significant increases in heart‐to‐body‐weight ratio relative to WT mice (Figure 8A) that were associated with significant increases in the expression of the hypertrophy biomarkers Nppa and Nppb (Figure 8B and 8C). Furthermore, they exhibited a 16.5‐fold increase in cardiac glycogen content (Figure 8D). On echocardiography at 10 weeks, TGSGLT1‐ON mice relative to WT mice had an increase in LV end‐diastolic dimension (P<0.005), and a decrease in fractional shortening (P<0.005) indicating contractile dysfunction (Table 4).
Figure 8.

TGSGLT1‐ON mice exhibited increases relative to WT mice in heart‐to‐body‐weight ratio (A), glycogen content (B), and markers of cardiac hypertrophy (C and D) at 10 and 20 weeks of age. There was a significant reduction in heart‐to‐body‐weight ratio, glycogen content, and markers of hypertrophy in TGSGLT1‐ON/OFF relative to TGSGLT1‐ON mice at age 20 weeks. Heart‐to‐body‐weight ratio: WT, N=4; TGSGLT1‐OFF, N=4;TGSGLT1‐ON, N=3; TGSGLT1‐ON/OFF, N=4. Glycogen content and QPCR at 10 weeks: WT, N=5; TGSGLT1‐OFF, N=4; TGSGLT1‐ON, N=4. Glycogen content and QPCR at 20 weeks: WT, N=5; TGSGLT1‐OFF, N=4; TGSGLT1‐ON, N=3; TGSGLT1‐ON/OFF, N=4. *P<0.05 vs WT; †P<0.005 vs WT; ‡P<0.05 vs TGSGLT1‐OFF; §P<0.005 vs TGSGLT1‐OFF; ∥P<0.05 vs TGSGLT1‐ON; #P<0.005 vs TGSGLT1‐ON; **P<0.01 TGSGLT1‐ON (10 weeks) vs TGSGLT1‐ON/OFF. Data are expressed as mean±SE. QPCR indicates quantitative polymerase chain reaction; SGLT1, sodium‐dependent glucose co‐transporter 1; WT, wildtype.
Table 4.
Echocardiography of WT, TGSGLT1‐OFF, and TGSGLT1‐ON Mice at 10 and 20 Weeks of Age and TGSGLT1‐ON/OFF Mice at 20 Weeks of Age
| WT | TGSGLT1‐OFF | TGSGLT1‐ON | TGSGLT1‐ON/OFF | |
|---|---|---|---|---|
| 10 weeks | ||||
| N | 7 | 5 | 8 | |
| LVAWT, mm | 0.91±0.05 | 0.80±0.03 | 0.81±0.03 | |
| LVEDD, mm | 2.84±0.12 | 2.79±0.19 | 4.03±0.15†§ | |
| FS, % | 63±2 | 63±4 | 35±4*‡ | |
| HR, bpm | 617±57 | 549±26 | 416±5*§ | |
| 20 weeks | ||||
| N | 5 | 4 | 3 | 4 |
| LVAWT, mm | 0.92±0.06 | 0.93±0.07 | 0.98±0.07 | 0.93±0.08 |
| LVEDD, mm | 3.04±0.20 | 3.01±0.26 | 4.73±0.42†‡ | 3.16±0.10# |
| FS, % | 56±5 | 59±5 | 37±6 | 58±7** |
| HR, bpm | 558±22 | 578±26 | 420±9†§ | 510±29∥** |
FS indicates fractional shortening; HR, heart rate; LVAWT, left ventricular anterior wall thickness; LVEDD, left ventricular end‐diastolic diameter; WT, wildtype.
*P<0.01 relative to same age WT; †P<0.001 relative to same age WT; ‡P<0.05 relative to same‐age TGSGLT1‐OFF; §P<0.01 relative to same‐age TGSGLT1‐OFF; ∥P<0.05 relative to 20‐week‐old TGSGLT1‐ON; #P<0.005 relative to 20‐week‐old TGSGLT1‐ON; **P<0.05 relative to 10‐week‐old TGSGLT1‐ON.
At 20 weeks of age, TGSGLT1‐ON mice continued to show significant increases in heart‐to‐body‐weight ratio (Figure 8A), and markers of cardiac hypertrophy (Figure 8B and 8C), along with a significant 9.1‐fold increase in cardiac glycogen content (Figure 8D) relative to WT mice. On echocardiography at 20 weeks of age, TGSGLT1‐ON continued to show LV dilation compared to WT mice.
On histopathological examination, TGSGLT1‐ON relative to WT and TGSGLT1‐OFF mice exhibited increased myocyte size, increased nuclear size, and apparent cytoplasmic inclusions that were evident on hematoxylin and eosin staining at 20 weeks of age, as well as increased interstitial fibrosis on Masson trichrome staining at 10 and 20 weeks of age (Figure 9). Histomorphometric analysis confirmed an increase in myocyte size in TGSGLT1‐ON mice relative to WT and TGSGLT1‐OFF mice at 10 and 20 weeks of age. In addition, procollagen‐1 transcript expression was significantly increased at 10 and 20 weeks of age.
Figure 9.
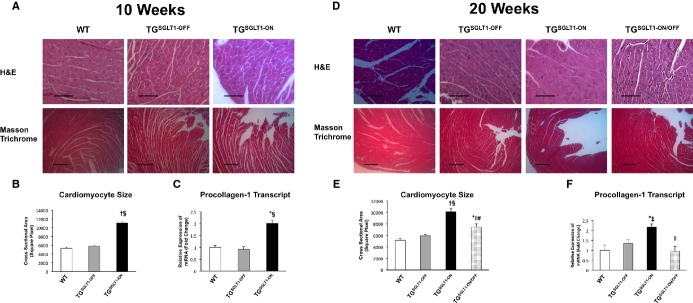
Histopathological changes at 10 and 20 weeks of age. A and D, Representative transverse sections of left ventricular tissue stained with hematoxylin and eosin (H&E) (top row) and Masson trichrome (bottom row) obtained from WT, TGSGLT1‐OFF, TGSGLT1‐ON, and TGSGLT1‐ON/OFF mice. Fibrosis, sarcoplasmic inclusions, and increased nuclear size were evident in TGSGLT1‐ON mice compared to WT, TGSGLT1‐OFF, and TGSGLT1‐ON/OFF mice. B and E, Histomorphometric assessment showed an increase in myocyte size in TGSGLT1‐ON mice compared to WT and TGSGLT1‐OFF mice. In addition, TGSGLT1‐ON/OFF mice showed reduced myocyte size compared to both 10‐ and 20‐week‐old TGSGLT1‐ON mice. N=25/group. C and F, QPCR showed an increase in collagen expression in TGSGLT1‐ON mice compared to WT and TGSGLT1‐OFF mice. TGSGLT1‐ON/OFF mice showed a reduction in collagen expression compared to 20‐week‐old TGSGLT1‐ON mice. QPCR at 10 weeks: WT, N=5; TGSGLT1‐OFF, N=4; TGSGLT1‐ON, N=4. QPCR at 20 weeks: WT, N=5; TGSGLT1‐OFF, N=4; TGSGLT1‐ON, N=3; TGSGLT1‐ON/OFF, N=4. *P<0.05 vs WT; †P<0.001 vs WT; ‡P<0.05 vs TGSGLT1‐OFF; §P<0.001 vs TGSGLT1‐OFF; ∥P<0.01 vs 20‐week‐old TGSGLT1‐ON; #P<0.001 vs10‐week‐old TGSGLT1‐ON. Data are expressed as mean±SE. H&E bar=3 μm; trichrome bar=1 μm. QPCR indicates quantitative polymerase chain reaction; SGLT1, sodium‐dependent glucose co‐transporter1; WT, wildtype.
Effects of Cardiac SGLT1 Overexpression Are Reversible
To determine whether the effects of SGLT1 overexpression in the heart can be reversed, the SGLT1 transgene was suppressed from 10 to 20 weeks of age by the administration of doxycycline in chow (TGSGLT1‐ON/OFF). TGSGLT1‐ON/OFFmice exhibited a reduction in glycogen content compared to 10‐week‐old and 20‐week‐old TGSGLT1‐ON mice (Figure 8). In addition, heart‐to‐body‐weight ratios and markers of cardiac hypertrophy were decreased in TGSGLT1‐ON/OFFcompared to TGSGLT1‐ON at 20 weeks of age. Echocardiography showed improvement of LV end diastolic dimension in TGSGLT1‐ON/OFF mice relative to TGSGLT1‐ON mice at 20 weeks of age. In addition, there was improvement of LV fractional shortening in TGSGLT1‐ON/OFF relative to TGSGLT1‐ON mice at 10 weeks of age (Table 4). On histomorphometric analysis, TGSGLT1‐ON/OFF mice exhibited a reduction in myocyte size relative to TGSGLT1‐ON mice at both 10 and 20 weeks of age (Figure 9). In addition, TGSGLT1‐ON/OFF mice showed a qualitative decrease in interstitial fibrosis on Masson's trichrome stain and a significant decrease in procollagen‐1 transcript level compared to 20‐week‐old TGSGLT1‐ON mice.
Discussion
Transgenic mouse models of the Thr400Asn (TGT400N)4 and the Asn488Ile (TGN488I)18 mutations in PRKAG2 recapitulate the human glycogen storage cardiomyopathy. These mice exhibit an initial inappropriate activation of AMPK. In vivo cardiac glucose uptake is elevated early in TGT400N mice, and in TGN488I hearts the increased glucose entry is directed toward glycogen synthesis.8 The mechanism of increased cardiac glucose uptake had remained uncertain until our recent discovery that SGLT1 is upregulated in TGT400N cardiomyocytes.13
Our data establish a role for SGLT1 in the pathophysiology of PRKAG2 cardiomyopathy. Cardiomyocyte‐specific knockdown of SGLT1 by transgenic RNA interference led to a reduction in cardiac mass, cardiac glycogen content, markers of hypertrophy, histopathological abnormalities, and structural and functional abnormalities on echocardiogram. Although cardiac membrane GLUT1 protein expression was mildly decreased in TGSGLT1‐DOWN relative to WT mice, no difference was observed in TGT400N/TGSGLT1‐DOWN relative to TGT400N mice. Therefore, the attenuation in phenotype in TGT400N/TGSGLT1‐DOWN mice was not attributable to changes in GLUT1 expression. The reduction in glycogen content with SGLT1 knockdown was modest but similar to the reduction we previously observed with chronic administration of phlorizin, a pharmacological inhibitor of SGLT1. However, despite modest changes in glycogen content and no significant change in wall thickness, we observed significant histological and functional improvements in TGT400N/TGSGLT1‐DOWN relative to TGT400N mice. PRKAG2 cardiomyopathy is associated with increased fractional shortening early in the course of the disease, which is followed by worsening LV function and dilation.4 The TGT400N/TGSGLT1‐DOWN mice exhibited a delay in the progression of LV dysfunction. These mice had increased fractional shortening at 7 weeks of age and a trend towards improved fractional shortening compared to TGT400N mice at 12 weeks of age.
Although our data provide strong evidence for a role of SGTL1 in PRKAG2 cardiomyopathy, SGLT1 knockdown only partially attenuates the phenotype, and TGT400N/TGSGLT1‐DOWN mice still exhibit LV hypertrophy and eventual dysfunction. It is likely that some of the excess glucose entry is mediated by other glucose transporters, such as the facilitative glucose transporters GLUT1 and GLUT4. Moreover, the reduction in SGLT1 expression is incomplete, in the range of 80%. A cardiac‐specific SGLT1 knockout model may exhibit greater attenuation of the phenotype. However, the knockdown model may more closely mirror clinical practice, in the setting of drug therapy for PRKAG2 cardiomyopathy. It is also likely that the cardiac hypertrophy in PRKAG2 cardiomyopathy is not completely attributable to glycogen deposition. We and others previously showed that much of the hypertrophy may be related to activation of hypertrophic signaling pathways such as NF‐κβ and Akt.9–10 SGLT1 knockdown probably does not inhibit all of these multiple downstream signaling pathways. SGLT1 upregulation likely represents 1 of several maladaptive processes responsible for PRKAG2 cardiomyopathy.
To investigate the effects of increased cardiac SGLT1 expression, we constructed a transgenic mouse model conditionally overexpressing SGLT1 in cardiomyocytes. TGSGLT1‐ON mice exhibited an increase in hypertrophic signaling and eventual LV dysfunction, similar to TGT400N mice, but had only a modest increase in glycogen content and no increase in wall thickness. These results complement the findings in TGT400N/TGSGLT1‐DOWN mice, suggesting that SGLT1 affects cardiac function and hypertrophic signaling with a modest effect on glycogen storage. Thus, the phenotype of PRKAG2 cardiomyopathy is likely in part attributable to upregulation of SGLT1, but is also dependent on other mechanisms.
Our data also suggest that the deleterious effects of SGLT1 overexpression in the heart can be reversed if the overexpression is suppressed. TGSGLT1‐ON/OFF mice had decreased glycogen content and cardiomyocyte size, and improved LV function relative to TGSGLT1‐ON mice at 10 weeks of age. This observation has potential clinical relevance, since the cardiac remodeling activated by SGLT1 overexpression has potential for reversal.
The mechanism of increased SGLT1 expression in PRKAG2 cardiomyopathy remains to be elucidated. Inappropriate activation of AMPK is likely to be the initiating event. We have previously reported13 that pharmacological activation of AMPK in WT hearts results in an increase of SGLT1 transcript levels (and unpublished data). Furthermore, we have found that other acquired heart disease associated with AMPK activation, such as ischemia, is associated with increased SGLT1 expression.12
Classically, it has been thought that cardiac glucose uptake is mediated primarily by GLUT1 and GLUT4. Although SGLT1 protein has been known to be expressed in the small intestine and renal cells,20 we have only recently determined that it is highly expressed in the heart.12 The role that SGLT1 plays in cardiac physiology and pathophysiology is still largely unknown. Recently, it has been reported that inhibition of SGLT1 in isolated human cardiomyocytes has a negative inotropic effect.21–22 In addition, Balteau et al have shown that NOX2 activation in primary cultures of adult rat cardiomyocytes results from glucose transport through SGLT1.23 Our current report is the first to describe a role of SGLT1 in cardiac disease.
In this study, we demonstrate that knockdown of SGLT1 in the murine heart by the transgenic overexpression of cardiomyocyte‐specific short hairpin RNA to SGLT1 is feasible. To our knowledge, this report is the first description of the successful use of this strategy, and also the first description of transgenic knockdown of cardiac SGLT1 showing benefit in an animal model of human cardiac disease.
While this study focused on the role of SGLT1 in PRKAG2 cardiomyopathy, other forms of human cardiac disease may potentially involve SGLT1.12 With the ongoing development of new drug inhibitors for the sodium glucose co‐transporter family, primarily for diabetes, there may be a potential role for these drugs in the treatment of cardiac disease.
Sources of Funding
This work was supported by grants R21 HL109812 and U01 HL108642 from the NIH (FA).
Disclosures
None.
References
- 1.Kemp BE, Mitchelhill KI, Stapleton D, Michell BJ, Chen ZP, Witters LA. Dealing with energy demand: the AMP‐activated protein kinase. Trends Biochem Sci. 1999; 24:22-25. [DOI] [PubMed] [Google Scholar]
- 2.Hardie DG. Minireview: the AMP‐activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003; 144:5179-5183. [DOI] [PubMed] [Google Scholar]
- 3.Arad M, Benson DW, Perez‐Atayde AR, McKenna WJ, Sparks EA, Kanter RJ, McGarry K, Seidman JG, Seidman CE. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest. 2002; 109:357-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banerjee SK, Ramani R, Saba S, Rager J, Tian R, Mathier MA, Ahmad F. A PRKAG2 mutation causes biphasic changes in myocardial AMPK activity and does not protect against ischemia. Biochem Biophys Res Commun. 2007; 360:381-387. [DOI] [PubMed] [Google Scholar]
- 5.Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez‐Atayde AR, Sawyer DB, Walter M, Li GH, Burgon PG, Maguire CT, Stapleton D, Schmitt JP, Guo XX, Pizard A, Kuperchmidt S, Roden DM, Berul CI, Seidman CE, Seidman JG. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff‐Parkinson‐White syndrome in glycogen storage cardiomyopathy. Circulation. 2003; 107:2850-2856. [DOI] [PubMed] [Google Scholar]
- 6.Sidhu JS, Rajawat YS, Rami TG, Gollob MH, Wang Z, Yuan R, Marian AJ, DeMayo FJ, Weilbacher D, Taffet GE, Davies JK, Carling D, Khoury DS, Roberts R. Transgenic mouse model of ventricular preexcitation and atrioventricular reentrant tachycardia induced by an AMP‐activated protein kinase loss‐of‐function mutation responsible for Wolff‐Parkinson‐White syndrome. Circulation. 2005; 111:21-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies JK, Wells DJ, Liu K, Whitrow HR, Daniel TD, Grignani R, Lygate CA, Schneider JE, Noel G, Watkins H, Carling D. Characterization of the role of gamma2 R531G mutation in AMP‐activated protein kinase in cardiac hypertrophy and Wolff‐Parkinson‐White syndrome. Am J Physiol Heart Circ Physiol. 2006; 290:H1942-H1951. [DOI] [PubMed] [Google Scholar]
- 8.Luptak I, Shen M, He H, Hirshman MF, Musi N, Goodyear LJ, Yan J, Wakimoto H, Morita H, Arad M, Seidman CE, Seidman JG, Ingwall JS, Balschi JA, Tian R. Aberrant activation of AMP‐activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J Clin Invest. 2007; 117:1432-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerjee SK, McGaffin KR, Huang XN, Ahmad F. Activation of cardiac hypertrophic signaling pathways in a transgenic mouse with the human PRKAG2 Thr400Asn mutation. Biochim Biophys Acta. 2010; 1802:284-291. [DOI] [PubMed] [Google Scholar]
- 10.Kim M, Hunter RW, Garcia‐Menendez L, Gong G, Yang YY, Kolwicz SC, Jr, Xu J, Sakamoto K, Wang W, Tian R. Mutation in the gamma2‐subunit of AMP‐activated protein kinase stimulates cardiomyocyte proliferation and hypertrophy independent of glycogen storage. Cir Res. 2014; 114:966-975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwenk RW, Luiken JJ, Bonen A, Glatz JF. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovasc Res. 2008; 79:249-258. [DOI] [PubMed] [Google Scholar]
- 12.Banerjee SK, McGaffin KR, Pastor‐Soler NM, Ahmad F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc Res. 2009; 84:111-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banerjee SK, Wang DW, Alzamora R, Huang XN, Pastor‐Soler NM, Hallows KR, McGaffin KR, Ahmad F. SGLT1, a novel cardiac glucose transporter, mediates increased glucose uptake in PRKAG2 cardiomyopathy. J Mol Cell Cardiol. 2010; 49:683-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr HL‐1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998; 95:2979-2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gulick J, Subramaniam A, Neumann J, Robbins J. Isolation and characterization of the mouse cardiac myosin heavy chain genes. J Biol Chem. 1991; 266:9180-9185. [PubMed] [Google Scholar]
- 16.Ahmad F, Banerjee SK, Lage ML, Huang XN, Smith SH, Saba S, Rager J, Conner DA, Janczewski AM, Tobita K, Tinney JP, Moskowitz IP, Perez‐Atayde AR, Keller BB, Mathier MA, Shroff SG, Seidman CE, Seidman JG. The role of cardiac troponin T quantity and function in cardiac development and dilated cardiomyopathy. PLoS ONE. 2008; 3:e2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac‐specific transgenesis with an attenuated myosin heavy chain promoter. Cir Res. 2003; 92:609-616. [DOI] [PubMed] [Google Scholar]
- 18.Ahmad F, Arad M, Musi N, He H, Wolf C, Branco D, Perez‐Atayde AR, Stapleton D, Bali D, Xing Y, Tian R, Goodyear LJ, Berul CI, Ingwall JS, Seidman CE, Seidman JG. Increased alpha2 subunit‐associated AMPK activity and PRKAG2 cardiomyopathy. Circulation. 2005; 112:3140-3148. [DOI] [PubMed] [Google Scholar]
- 19.Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, Minnemann T, Boers ME, Hadro E, Oberste‐Berghaus C, Quist W, Lowell BB, Ingwall JS, Kahn BB. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest. 1999; 104:1703-1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wright EM. Renal Na(+)‐glucose cotransporters. Am J Physiol Renal Physiol. 2001; 280:F10-F18. [DOI] [PubMed] [Google Scholar]
- 21.von Lewinski D, Gasser R, Rainer PP, Huber MS, Wilhelm B, Roessl U, Haas T, Wasler A, Grimm M, Bisping E, Pieske B. Functional effects of glucose transporters in human ventricular myocardium. Eur J Heart Fail. 2010; 12:106-113. [DOI] [PubMed] [Google Scholar]
- 22.von Lewinski D, Rainer PP, Gasser R, Huber MS, Khafaga M, Wilhelm B, Haas T, Machler H, Rossl U, Pieske B. Glucose‐transporter‐mediated positive inotropic effects in human myocardium of diabetic and nondiabetic patients. Metabolism. 2010; 59:1020-1028. [DOI] [PubMed] [Google Scholar]
- 23.Balteau M, Tajeddine N, de Meester C, Ginion A, Des Rosiers C, Brady NR, Sommereyns C, Horman S, Vanoverschelde JL, Gailly P, Hue L, Bertrand L, Beauloye C. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc Res. 2011; 92:237-246. [DOI] [PubMed] [Google Scholar]