

Abstract
Background
Transient receptor potential C3 (TRPC3) has been demonstrated to be involved in the regulation of vascular tone through endothelial cell (EC) hyperpolarization and endothelium‐dependent hyperpolarization–mediated vasodilation. However, the mechanism by which TRPC3 regulates these processes remains unresolved. We tested the hypothesis that endothelial receptor stimulation triggers rapid TRPC3 trafficking to the plasma membrane, where it provides the source of Ca2+ influx for small conductance calcium‐activated K+ (SKCa) channel activation and sustained EC hyperpolarization.
Methods and Results
Pressurized artery studies were performed with isolated mouse posterior cerebral artery. Treatment with a selective TRPC3 blocker (Pyr3) produced significant attenuation of endothelium‐dependent hyperpolarization–mediated vasodilation and endothelial Ca2+ response (EC‐specific Ca2+ biosensor) to intraluminal ATP. Pyr3 treatment also resulted in a reduced ATP‐stimulated global Ca2+ and Ca2+ influx in primary cultures of cerebral endothelial cells. Patch‐clamp studies with freshly isolated cerebral ECs demonstrated 2 components of EC hyperpolarization and K+ current activation in response to ATP. The early phase was dependent on intermediate conductance calcium‐activated K+ channel activation, whereas the later sustained phase relied on SKCa channel activation. The SKCa channel–dependent phase was completely blocked with TRPC3 channel inhibition or in ECs of TRPC3 knockout mice and correlated with increased trafficking of TRPC3 (but not SKCa channel) to the plasma membrane.
Conclusions
We propose that TRPC3 dynamically regulates SKCa channel activation through receptor‐dependent trafficking to the plasma membrane, where it provides the source of Ca2+ influx for sustained SKCa channel activation, EC hyperpolarization, and endothelium‐dependent hyperpolarization–mediated vasodilation.
Keywords: cerebrovascular circulation, endothelium, endothelium‐derived factors, ion channels, vasculature
Introduction
Transient receptor potential (TRP) nonselective cation channels are widely expressed in different tissues and contribute to cytosolic Ca2+ signaling in response to mechanical, thermal, and chemical stimulation.1–2 Several members of the TRP channel family expressed in vascular endothelium play an important role in arterial tone and blood flow regulation.3–6 One important mechanism of such regulation involves small and intermediate conductance calcium‐activated K+ (SKCa and IKCa, respectively) channels, which may be activated by Ca2+ influx through TRP channels at the endothelial plasma membrane (PM).6–9
Endothelial SKCa and IKCa channels play a pivotal role in regulating arterial tone by hyperpolarizing endothelial cells (ECs) in response to increased cytosolic Ca2+. This hyperpolarization is then transferred to surrounding smooth muscle cells via intercellular gap‐junctions, thus providing smooth muscle cell relaxation. This described process of vasodilation is referred to as endothelium‐dependent hyperpolarization (EDH).10–14 Pharmacological activators or blockers of SKCa and IKCa channels can affect resting or active tone in isolated arteries,10–11,13,15 as well as in situ blood flow.13,16–17
TRPC3, a member of the “canonical” subfamily of TRP channels, has been implicated in endothelium‐mediated regulation of arterial tone; however, its regulation and exact role in the EDH‐mediated dilations are not well understood. The mode of channel activation by intracellular lipid messengers such as diacylglycerol suggests a functional link between TRPC3 and the G protein–coupled receptor/phospholipase C (PLC) signaling cascade, activated by hormones and paracrine factors.18–19 Two recent studies have suggested that endothelial TRPC3 contributes to the vasorelaxation response to luminal flow and bradykinin.20–21 In addition, we recently presented evidence for a significant role of TRPC3 in EDH‐mediated dilation through SK/IKCa channel–mediated EC hyperpolarization in mesenteric artery.8 This latter study provided the first evidence linking TRPC3 channel activation and SK/IKCa channel activation in vascular endothelium.
In the present study, we sought to determine the temporal role of TRPC3 in the regulation of SKCa and IKCa channels in vascular endothelium of cerebral artery. From our findings, we now propose a new model in which TRPC3 channels dynamically regulate SKCa channel activation through receptor‐dependent trafficking of TRPC3 to the PM (see Figure 12, later). This regulation of TRPC3 thus contributes to a complex time course of K+ channel activation comprising the overall mechanism of EC hyperpolarization, and thus EDH‐mediated vasodilation.
Figure 12.
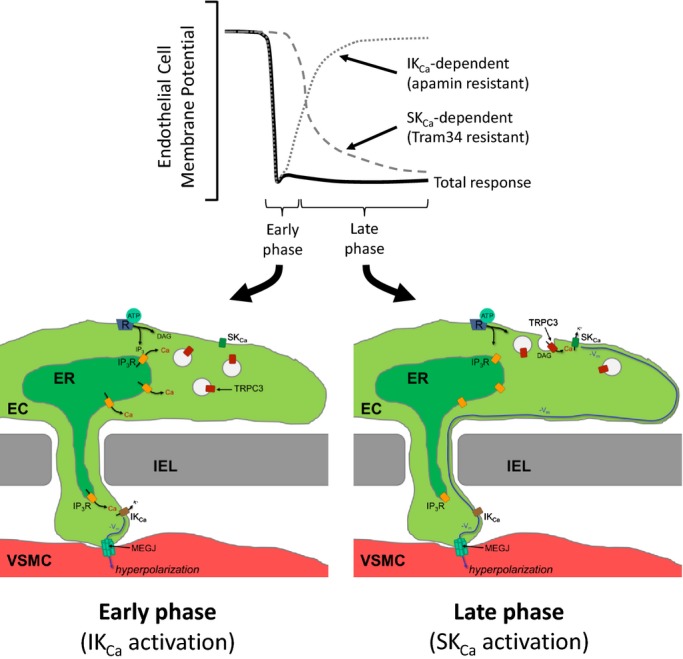
Proposed role of endothelial TRPC3 trafficking in SKCa channel activation to produce sustained EC hyperpolarization and EDH‐mediated vasodilation. Endothelial receptor stimulation leads to EC hyperpolarization (−Vm) through 2 phases. In the early phase, Ca2+ released from IP3 receptors (IP3R) leads to IKCa channel activation and the initial EC hyperpolarization. Ca2+‐dependent trafficking of TRPC3‐containing vesicles to the plasma membrane (PM) is also initiated in this phase. The late phase begins on vesicular fusion with the PM and TRPC3 channel insertion. TRPC3 channel activation (PLC‐derived DAG or constitutively active) promotes sustained Ca2+ influx and SKCa channel activation. This late phase is characterized by the loss of IKCa‐dependent EC hyperpolarization and the gain of SKCa‐dependent EC hyperpolarization. EC hyperpolarization is transferred to the adjacent smooth muscle at regions of heterocellular contact through the internal elastic lamina (IEL) via myoendothelial gap junctions (MEGJ). Smooth muscle hyperpolarization leads to subsequent vasodilation and increased blood flow. EDH indicates endothelium‐dependent hyperpolarization; IKCa channels, intermediate conductance Ca2+‐activated K+ channels; PLC, phospholipase C; DAG, diacylglycerol; VSMC, vascular smooth muscle cell; SKCa channels, small conductance Ca2+‐activated K+ channels.
Methods
Animal Models
All animal experiments were performed in accordance with Baylor College of Medicine Institutional Animal Care and Use Committee guidelines (September 1, 2011, revision). Generation of TRPC3 knockout (KO) mice has been described previously.22 Cx40BAC‐GCaMP2 transgenic mice, expressing calcium biosensor GCaMP2 selectively in ECs,23 were obtained from the Cornell Center for Technology Enterprise and Commercialization. P2Y2 KO mice24 were obtained through Jackson Laboratory (stock 009132). All experiments were performed with male mice.
Pressurized Artery Studies
The posterior cerebral artery (PCA) was dissected from the brain and placed in a perfusion chamber (ChuelTech). The artery was cannulated with 2 micropipettes and secured with ties consisting of individual strands of a polyester thread. Warmed (37°C) and gassed (21% O2–5% CO2, balance N2) Krebs buffer was used for abluminal and luminal perfusion of the artery. Luminal flow (30 to 50 μL/min) was established by setting the luminal inflow and outflow reservoirs at a slight height differential. Arteries were monitored for changes in diameter by using video‐microscopy. L‐NAME (10 μmol/L) and indomethacin (10 μmol/L) were administered to inhibit nitric oxide synthase and cyclooxygenase, respectively. In these conditions, arteries developed spontaneous myogenic tone. In all experiments, ATP (and, in some instances, A23187 at 10 μmol/L) were administered through the lumen of the artery via a miniature remotely operated manifold system to achieve rapid delivery of agents despite very low flow rates. Vasodilation evoked by ATP was calculated as a difference in peak vessel diameter (time averaged over 50 seconds) before and after ATP and normalized to maximal artery diameter (in Ca2+‐free, 3 mmol/L EGTA solution).
EC Ca2+ Measurement in Pressurized Artery
For endothelial Ca2+ measurement within pressurized arteries, the PCA from Cx40BAC‐GCaMP2 mice was prepared as described earlier. Ca2+ measurements were performed in the presence of L‐NAME and indomethacin at 10 μmol/L each. The vessel chamber was placed on the stage of an inverted TE200 fluorescence microscope equipped with a Nikon Super Fluor 10X objective (Nikon Instruments). Photometric measurement of endothelial Ca2+ was performed with a customized UV/photometry system (C&L Instruments), which we have previously used for fura‐2–based measurements.3,25–29 The UV excitation passed through a heat filter, variable aperture, and band pass excitation filter (475/20; Chroma Technology) before being directed into the microscope via a liquid light guide. The emission signal was transmitted through a dichroic mirror and then a second liquid light guide to the photometer equipped with an emission band pass filter (520/40; Chroma Technology). UV lamp output was attenuated approximately 50% to reduce cell damage. Sampling was performed at 20 Hz, and a 5‐second running average transform was applied for final analysis (SigmaPlot).
Ca2+ signal in ECs was detected as an increase in GCaMP2 fluorescence in response to intraluminal application of ATP. The signal was then normalized to the “maximal” Ca2+ response to ionophore A23187 (10 μmol/L). Specificity of the GCaMP2 signal to the endothelium was confirmed by endothelial denudation by air.30 Removal of the endothelium resulted in complete absence of fluorescence change to ATP and A23187. Possible artifact due to artery dilation and constriction was additionally ruled out by using PCA isolated from Tie2‐GFP mice (Ca2+‐insensitive EC‐specific GFP expression) that were run through the same diameter range.
Mouse PCA TRPC3 Immunofluorescence Staining
PCA were removed from both Cx40BAC‐GCaMP2::TRPC3 WT and Cx40BAC‐GCaMP2::TRPC3 KO mice and mounted in a pressurized vessel chamber dedicated for fixation studies (see Figure 10A, later). Additionally, cerebral arteries were removed from WT (C57BL/6) mice and treated as just described (see Figure 10C and 10D, later). Each vessel was pressurized as described earlier and perfused luminally and abluminally with solution containing (in mmol/L): 140 NaCl, 5.6 KCl, 1.6 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.3. In certain instances, the luminal solution additionally contained 100 μmol/L ATP to activate the endothelium. The artery was fixed while pressurized (60 mm Hg) in the chamber with a 10% formalin solution for 10 minutes. At the end of fixation, the artery was cut open along the axis of flow to permit complete access to the endothelial surface for antibody and later imaging. The vessel was then transferred to a bullet tube to perform the permeabilization with 0.2% Triton X for 15 minutes. The vessels were exposed to the blocking solution (10% goat serum, 0.5% BSA, 0.1% Tween‐20 in PBS) for 4 hours at room temperature on a rocker. Tissues were subsequently incubated overnight in rabbit polyclonal anti‐TRPC3 primary antibody (ab75171; Abcam) at 1:200 dilution in block solution. After PBS washes, the arteries were probed for 1 hour with Alexa Fluor 594 goat anti‐rabbit IgG antibody (Invitrogen) at 1:500 dilution. Arteries were then arranged on glass slides in DAPI‐containing mounting media (Vector Labs) for fluorescence microscopy (Olympus IX81 with SlideBook 4.2).
Figure 10.
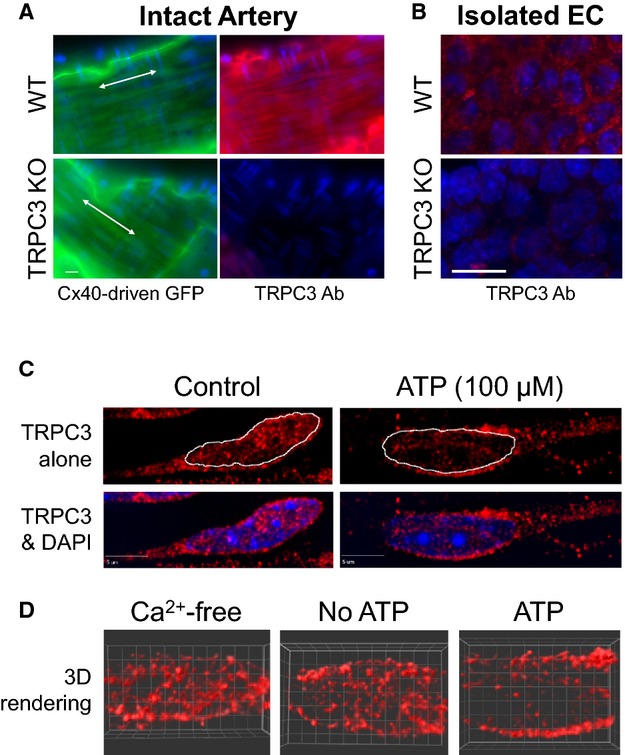
TRPC3 immunofluorescence demonstrating ATP‐stimulated trafficking of TRPC3 channels to plasma membrane in EC of intact cerebral artery. A, Immunofluorescence images of en face preparations of PCA from WT and TRPC3 KO mice acquired with 60× oil objective. WT and KO mice were bred on a Cx40BAC‐GCaMP2 background to provide GFP expression selectively in the endothelium. The arteries were fixed while pressurized and then cut open for direct access to the endothelium. The green fluorescence reflects EC‐specific GFP as well as autofluorescence of the underlying internal elastic lamina (IEL). The GFP signal (green) was used to establish the proper focal plane of the thin endothelial layer for TRPC3 immunofluorescence (red). Nuclei were stained with DAPI (blue). White scale bar=10 μm. B, TRPC3 immunofluorescence images (100× oil objective) of freshly isolated cerebral EC clusters from WT and TRPC3 KO mice (red). Nuclei were stained with DAPI (blue). White scale bar= 10 μm. C, Representative TRPC3 immunofluorescence of cerebral endothelium from pressurized arteries treated with saline (Control) or ATP (100 μmol/L) for 10 minutes. After treatment, pressurized arteries were immediately fixed in the chamber and cut open for TRPC3 immunofluorescence (red). EC were imaged in en face preparations with 100× oil objective. Z stack images were acquired at 0.2‐μm intervals for deconvolution. Images shown represent a maximum projection along the z axis of 5 planes (1 μm thickness). DAPI nuclei (blue) are shown in the bottom panel, whereas the nuclear border is indicated by dotted line in the upper panel. White scale bar = 5 μm. D, Representative 3‐dimensional reconstructions of deconvolved EC TRPC3 immunofluorescence signal (red) from arteries treated with Ca2+‐free buffer, Ca2+‐containing buffer (No ATP), and Ca2+‐containing buffer with 100 μmol/L ATP (ATP). Images represent a 2‐μm‐thick optical section centered about the nucleus. DAPI indicates 4′,6‐Diamidino‐z‐phenylindole dihydrochloride; KO, knockout; PAC, posterior cerebral artery; TRPC3, transient receptor potential C3; WT, wild type.
EC isolation and Recording Conditions
For Ca2+ imaging experiments, ECs were obtained from mouse PCA and middle cerebral artery (MCA) explants grown on BD Matrigel Basement Membrane Matrix (BD Biosciences) as described by Suh et al31 with slight modifications. Briefly, 35‐mm culture dishes were coated with BD Matrigel and diluted 1:1 with DMEM containing glucose and l‐glutamine (Gibco) for 1 to 2 hours at room temperature. Cerebral arteries were carefully excised from brain surface, cleaned of meningeal membranes, washed in Hank's buffer, minced into 2‐mm pieces, and transferred to Matrigel‐coated dishes containing DMEM supplemented with 10% FBS (Gibco), 100 μg/mL endothelial cell growth supplement (BD Biosciences), 10 U/mL heparin (Sigma), 100 U/mL penicillin/streptomycin (Invitrogen), and 2% minimal essential amino acid mixture (Sigma). Vessel pieces attached to the gel surface and were grown for 4 to 12 weeks (37°C and 5% CO2) to allow ECs to proliferate and spread throughout the Matrigel. Purity of the resulting cells was confirmed in preliminary experiments by using Tie2‐GFP transgenic mice. For experiments, ECs were harvested from the gel matrix by using 1‐hour incubation with neutral protease (4 mg/mL; Worthington Biochemical) in PBS, washed twice with DMEM, and plated onto Petri dishes or 12‐mm glass cover‐slips coated with fibronectin (BD Biosciences) 50 μg/mL for 2 hours.
Freshly dispersed ECs used in electrophysiological experiments were obtained from the PCA and MCA through enzymatic dissociation (70 minutes at 37°C) by using neutral protease (4 U/mL) and elastase (1 U/mL) in the following digestion buffer (in mmol/L): 138 NaCl, 5 KCl, 1.5 MgCl2, 0.42 Na2HPO4, 0.44 NaH2PO4, 0.1 CaCl2, 10 HEPES, 4.2 NaHCO3, and 0.3% BSA. The digestion was completed with a 2‐minute incubation with collagenase type 1 (120 U/mL) in the same digestion buffer. All enzymes were obtained from Worthington. After washing, partially digested vessels were triturated using a 200‐μL pipette tip. The resulting EC suspension was placed on ice and typically used at 2 to 6 hours after digestion.
Experiments were performed in an RC‐25 chamber (Warner Instruments). Extracellular bath solution contained (in mmol/L) 140 NaCl, 5.6 KCl, 1.6 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.3. The pipette solution for electrophysiological recordings contained (in mmol/L) 40 KCl, 100 K gluconate, 1 MgCl2, 10 NaCl, 0.1 EGTA, 10 NaCl, 10 HEPES, and 0.1 EGTA, pH 7.2. Pharmacological agents dissolved in bath saline were applied to the cells either via gravity flow or through the large‐bore pipette to the chamber for more rapid solution exchange.
EC Ca2+ Imaging
At the time of experimentation, ECs were loaded with fura‐2AM (TefLabs) for 1 hour at 2.5 μmol/L and imaged by acquiring 340/380‐nm fluorescence ratios (R340/380) every 4 seconds, with an Olympus IX81 fluorescent microscope equipped with a Uplan S‐Apo 20× 0.75 NA lens (Olympus), Lambda LS Xenon Arc lamp, Lambda 10‐2 filter wheel shutter controller (both from Sutter Instruments), and RET‐EXi‐F‐M‐12‐C CCD‐camera (QImaging) and controlled with Slidebook 4.2 Imaging software (Olympus). Background fluorescence was determined from a region without cells and was subtracted before ratio determination. R340/380 time‐lapse data acquired from individual cells were baseline subtracted and expressed as an agonist‐induced ΔR340/380. In Mn2+‐quenching experiments,32 cells were additionally excited at 360 nm (D360/10x‐25 filter; Chroma Technology), the isosbestic point for fura‐2, and the resulting 510‐nm fluorescent images were acquired at 3.6‐second intervals.
Patch‐Clamp Electrophysiology
EC membrane currents or voltage were recorded from EC clusters, typically consisting of 3 to 12 cells, with calculated membrane capacitance ranging from 4 to 30 pF. Recordings were done in either voltage‐ or current‐clamp modes by using the perforated patch configuration of whole‐cell recording with use of a Multiclamp 700B patch‐clamp amplifier, DigiData 1440A computer interface, and Clampex 10.0 acquisition and analysis software (Molecular Devices). The bath was grounded with an Ag‐AgCl pellet via a bridge of 1 mol/L KCl in 2% agarose. Amphotericin B (0.8 mg/mL) was added to the pipette solution immediately before the recording. The series resistance was maintained at or below 30 MΩ and was not compensated. For voltage‐clamp recordings, ECs were held at −70 mV, and whole‐cell currents were evoked by 1.5‐second voltage ramps from −120 to +60 mV every 3 seconds. Potassium currents activated by ATP or NS‐309 were analyzed after subtraction of baseline current, recorded before the agonist application. For membrane capacitance (Cm) measurements, EC Cm was monitored by using the time domain technique33 in which 10‐mV test pulses were applied from a −70‐mV holding potential in whole‐cell mode. Data were acquired by using the membrane test feature in ClampEx.
Statistics
SigmaPlot 11.0 (Systat Software) was used for statistical analyses. All data are expressed as mean±SEM. For pressurized artery studies, n represents the number of animals from which arteries were taken. Calcium imaging recordings from individual cells were averaged for each coverslip (12 to 59 cells per coverslip) representing a minimum of 3 mice for each group. For electrophysiological experiments, individual recordings from EC clusters were combined to calculate an averaged recording. Comparisons between TRPC3 KO and wild‐type (WT) data sets contained samples from at least 3 mice for each group. The average responses were evaluated by using 2‐way repeated‐measures ANOVA with the Holm–Sidak test for pairwise multiple comparison procedures. Whole artery EC Ca2+ measurements were evaluated as the area under the curve and compared by using t test. Vasodilation responses were compared by using 2‐way repeated‐measures ANOVA with the Holm–Sidak test for individual comparisons. A value of P<0.05 was considered significant.
Results
Cerebral Artery Endothelial TRPC3 Contributes to ATP‐Mediated Vasodilation Through Endothelial Ca2+ Regulation
Although TRPC3 has been shown to be involved in endothelium‐mediated vasodilation/vasorelaxation in peripheral arteries,8,20–21 a role for this channel in vasodilation has never been reported for cerebral arteries. Thus, to determine the role of TRPC3 in endothelium‐mediated vasodilation of cerebral artery, we performed experiments in pressurized PCA treated with either vehicle or Pyr3 (1 μmol/L), a blocker of TRPC3. ATP (10 and 100 μmol/L) was delivered through the lumen of the arteries to promote endothelium‐dependent dilation through P2Y receptor activation. Arteries were treated with L‐NAME and indomethacin to isolate the EDH‐mediated component of vasodilation.34 Spontaneous myogenic tone was similar for control and Pyr3‐treated arteries. Application of Pyr3 produced 25±6% tone compared with 21±3% in control arteries (P=0.53); maximum diameters for each group were 190±2 (control) and 188±3 μm (Pyr3‐treated). Figure 1A shows representative PCA diameter measurements in response to cumulative ATP delivery. Note that ATP (100 μmol/L) produced sustained and near‐maximal dilations in control arteries, whereas it produced oscillatory and submaximal dilations in Pyr3‐treated arteries. The dilation responses are summarized in Figure 1B as a percentage of maximal diameter (Ca2+‐free conditions). Note that EDH‐mediated dilation was significantly attenuated in Pyr3‐treated arteries (P=0.024; 2‐way repeated‐measures ANOVA).
Figure 1.
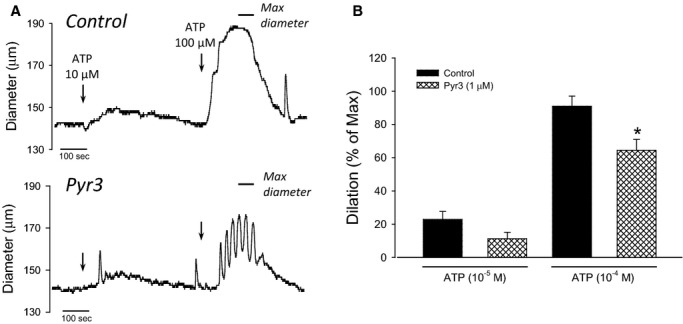
TRPC3 contributes to EDH‐mediated vasodilation in cerebral artery. A, Representative EDH‐mediated dilation responses to ATP (10 to 100 μmol/L) in PCA. L‐NAME and indomethacin were included to isolate the EDH component of vasodilation. Arteries were treated with either vehicle (0.01% DMSO, control) or 1 μmol/L Pyr3 before ATP delivery. Arteries were bathed in Ca2+‐free solution (no Ca2+ and 3 mmol/L EGTA) to obtain maximal relaxation at the end of the experiment (indicated by horizontal bar). B, Average EDH‐mediated dilation to ATP in vehicle or Pyr3 pretreated PCA. Data are presented as a percent of maximal dilation in Ca2+‐free conditions. *P<0.05; 2‐way repeated‐measures ANOVA. EDH indicates endothelium‐dependent hyperpolarization; PCA, posterior cerebral artery; TRPC3, transient receptor potential C3.
Once a role for TRPC3 in EDH‐mediated dilation was demonstrated in the cerebral circulation, we next sought to determine the specific role of this channel in EC Ca2+ regulation and subsequent EC hyperpolarization. We hypothesized that TRPC3 provides the source of Ca2+ influx necessary to sustain EC hyperpolarization after receptor stimulation. To evaluate this possibility, we measured ATP‐mediated changes in endothelial Ca2+ in intact pressurized PCA from mice expressing a fluorescent Ca2+ biosensor selectively in the endothelium (Cx40BAC‐GCaMP2).23,35–36 Because the present study reflects the first reported application of this method in cerebral arteries, we performed a series of validation experiments. In one study, we removed the endothelium with a bolus of air to determine the cell‐specificity of the fluorescent response and rule out possible fluorescence artifact from ATP and A23187. Note that the change in fluorescence to ATP was abolished after endothelial denudation (Figure 2A, bottom trace). Similarly, there was no change in fluorescence to A23187 (not shown). In a separate study, we additionally confirmed that fluorescence change was not due to change in artery diameter. In this separate experiment, we exposed an endothelial GFP‐expressing artery (Tie2‐GFP), in which the fluorescent signal is not Ca2+ dependent, to the full range of artery diameters by changing intraluminal pressure. There was no diameter‐dependent effect on fluorescence in these studies (not shown).
Figure 2.
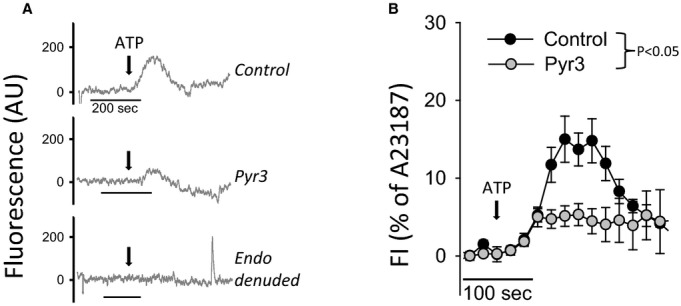
Role of TRPC3 in ATP‐mediated endothelial Ca2+ response in pressurized PCA. A, Representative recordings of endothelial Ca2+ response to ATP in isolated PCA from Cx40BAC‐GCaMP2 mice. Measurements were performed following incubation with vehicle (0.01% DMSO) or Pyr3 (1 μmol/L). ATP was additionally delivered to an artery in which the endothelium was removed by luminal air bubble (endo denuded). Data are shown as a change in GCaMP2 fluorescence after baseline subtraction and correction for photobleaching. B, Summary of endothelial Ca2+ responses to ATP in control (n=4) and Pyr3‐pretreated PCA (n=4), shown as an average increase in GCaMP2 fluorescence normalized to maximal signal in the presence of A23187 ionophore. The arrow indicates the point where ATP reached the artery lumen. The endothelial Ca2+ responses were compared as the AUC of individual recordings from control and Pyr3‐pretreated PCA. *P<0.05 t test comparing group AUC. AUC indicates area under the curve; PCA, posterior cerebral artery; TRPC3, transient receptor potential C3.
Once validated, we used the GCaMP2 biosensor method to determine the EC Ca2+ response to luminal ATP in control and Pyr3‐treated PCA. As shown in Figure 2A, ATP produced an increase in endothelial Ca2+ within intact pressurized arteries. The maximal response was approximately 20% of the full response elicited by luminal A23187 (10 μmol/L). In the presence of Pyr3, however, the Ca2+ response to ATP was significantly attenuated. The summary of ATP responses (normalized to the A23187 response) is shown in Figure 2B and demonstrates significant attenuation after TRPC3 inhibition (P=0.027, t test comparison of area under the curve).
Cerebral EC TRPC3 Contributes to Ca2+ Influx in Response to ATP
In a preliminary set of experiments, we recorded Ca2+ responses to ATP in fura‐2–loaded ECs freshly isolated from PCA and MCA (not shown) and found it problematic for quantitative studies due to considerable cell movement and background artifacts. As a more suitable tool, we used primary cultures of cerebral artery ECs. As shown in Figure 3A, ATP produced a transient (0.5 to 6 minutes) increase in endothelial Ca2+. The Ca2+ responses recorded from individual cells ranged from oscillatory to single prolonged peaks. The response was completely abolished by preincubation with PLC inhibitor U73122 (4 μmol/L), demonstrating critical dependence on a P2Y receptor/PLC–activated cascade rather than direct P2X‐mediated Ca2+ influx. Coapplication of a Ca2+ chelator (4 mmol/L BAPTA) with ATP significantly shortened the Ca2+ response to ATP, confirming a significant role for Ca2+ influx in the ATP‐stimulated response (Figure 3B). Further, the TRPC3 blocker Pyr3 (1 μmol/L) suppressed the Ca2+ response to ATP in duration, similar to the effect of Ca2+ chelation with BAPTA (Figure 3C, top). ECs cultured from TRPC3 KO mice (bottom panel) also demonstrated a more transient Ca2+ response to ATP compared with the described WT response. Application of Pyr3 to the ECs from TRPC3 KO mice did not produce a further reduction in the Ca2+ response, demonstrating the specificity of Pyr3 in the component of the Ca2+ response typically associated with Ca2+ influx.
Figure 3.
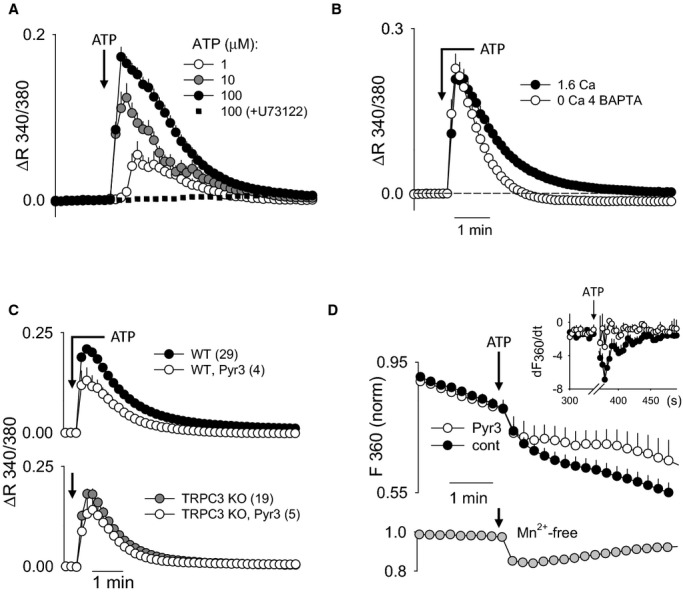
Ca2+ response to ATP in primary cerebral EC cultures. A through C, Ca2+ response as an average change in fura‐2 340/380‐nm ratio per coverslip. A, EC Ca2+ response to 1, 10, and 100 μmol/L ATP (n=6, 13, and 16, respectively) and 100 μmol/L ATP after 20 minutes' pretreatment with PLC inhibitor U73122 at 4 μmol/L (n=2). B, EC Ca2+ response to 100 μmol/L ATP in bath solution Ca2+‐containing buffer (1.6 mmol/L Ca2+) or Ca2+‐free buffer (nominally Ca2+‐free plus 4 mmol/L BAPTA; n=6). For the Ca2+‐free experiments, buffer was switched from Ca2+‐containing to Ca2+‐free at the time of ATP application to avoid depletion of internal Ca2+ stores. The average Ca2+ response to ATP in 1.6 mmol/L Ca2+ saline is shown for comparison (n=10). C, Summary of EC Ca2+ responses to 100 μmol/L ATP with and without a TRPC3 inhibitor (Pyr3, 1 μmol/L) in ECs from WT (upper panel) and TRPC3 KO (lower panel) mice. Number of coverslips is each group is shown in parentheses. D, The effect of Pyr 3 (1 μmol/L) on ATP (100 μmol/L)‐stimulated Mn2+ influx in ECs, as detected by quenching of fura‐2 fluorescence at 360 nm (F360). Summary data are shown as normalized fluorescence changes per cell (n=33 and 3 in control and Pyr3‐treated ECs, respectively). Inset graph shows the summary of fluorescence changes as the first derivative (dF/dt). The arrow indicates the point of ATP application. Note that the rapid initial drop in F360 is due to partial overlap with the prominent F380 signal that decreases on ATP‐stimulated Ca2+ increase. The lower subpanel shows average Mn2+‐independent changes in F360 (n=25), recorded in Mn2+‐free bath saline. KO indicates knockout; PLC, phospholipase C; TRPC3, transient receptor potential C3; WT, wild type.
While these data demonstrated a prominent role of membrane TRPC3 in ATP‐mediated Ca2+ regulation in cerebral ECs, a direct demonstration of this channel in Ca2+ influx was still needed. To specifically evaluate Ca2+ influx, we used the fura‐2/Mn2+–quenching technique.3,32 Addition of 1 mmol/L Mn2+ to the bath solution resulted in some baseline quenching (compare with bottom graph, where Mn2+ was not added) (Figure 3D). This reflection of basal Ca2+ influx did not differ between control and Pyr3‐treated cells. Application of ATP produced a considerable increase in quenching over baseline, indicating activation of Ca2+/Mn2+‐permeable PM channels. The rate of quenching (dF360/dt) is presented in the inset. The rate of quenching was significantly reduced in ECs treated with Pyr3, thus demonstrating the involvement of TRPC3 in ATP‐mediated Ca2+ influx.
SKCa and IKCa Channels Play Distinct Roles in ATP‐Mediated Cerebral EC Hyperpolarization
We examined the roles of SKCa and IKCa channels in ATP‐mediated EC hyperpolarization by patch‐clamp measurements of freshly isolated clusters of ECs from mouse PCA and MCA. EC membrane potential was measured by current‐clamp and whole‐cell K+ currents by voltage clamp in amphotericin B–perforated cells. The isolated clusters of ECs selected for patch‐clamp recording had electrical capacitance in a range of 6 to 30 pF, depending on the number of cells in the cluster. We avoided clusters of greater cell number due to limitations of voltage‐clamping clusters of a larger capacitance. Two types of baseline current in unstimulated EC clusters were revealed. The majority of ECs showed little outward current and a Kir‐like inward current that was sensitive to Ba2+ (100 μmol/L) or a specific Kir2.x blocker ML133 (10 μmol/L)37 at negative voltage (Figure 4A). The reversal potential in resting conditions ranged from −20 to 0 mV, which correlated well with membrane voltage measured from unstimulated cells in the current‐clamp mode. Interestingly, about 20% of cells showed very little inward current and a prominent delayed rectifier‐type outward current at positive voltage (Figure 4B). A similar current was recently described in microvascular ECs from rat brain and attributed to Kv1 family voltage‐dependent K+ channel(s).38 We found that cells of this type did not show any increase in current or change of reversal potential in response to ATP or direct SKCa/IKCa channel activator NS309 (not shown). Note that all remaining studies with ATP are exclusively performed with the more prevalent Kir‐expressing population of ECs.
Figure 4.
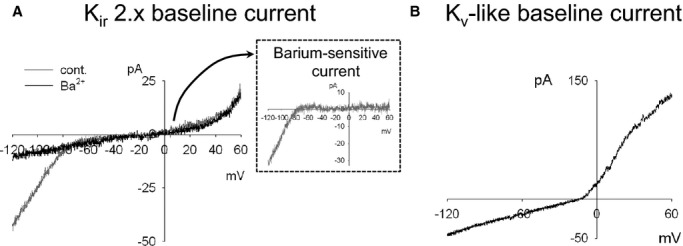
Representative current‐voltage (I‐V) recordings of baseline whole‐cell currents in freshly isolated clusters of cerebral ECs. Whole‐cell I‐V recordings were performed with freshly isolated cerebral ECs. Cell voltage was ramped from −120 to +60 mV (0.18 V/s) from a holding potential of −70 mV. A, Representative recording from an EC displaying a “Kir‐dependent current.” Baseline I‐V response with no treatment (cont) and with a Kir 2.x channel blocker, BaCl2 (100 μmol/L). The Ba2+‐sensitive current is shown in the inset. Similar results were obtained with Kir 2.x channel blocker, ML133 (10 μmol/L). B, Representative recording of an EC displaying a “Kv‐like current.”
Because both P2Y1 and P2Y2 purinergic receptors can mediate endothelium‐mediated vasodilation in cerebral arteries,28,39–40 we tested the receptor specificity of ATP‐mediated SKCa/IKCa current activation by using ECs isolated from WT and P2Y2 KO mice. Note that the ATP‐stimulated K+ current was virtually abolished in ECs from P2Y2 KO mice (Figure 5). To ensure that the functional expression of SKCa/IKCa channels was not altered in P2Y2 KO animals, ECs were treated at the end of each experiment with a direct activator of SKCa/IKCa channels (NS309 10 μmol/L). Application of NS309 produced a robust current increase that was not significantly different between groups, thus demonstrating an equivalent capacity for SK/IKCa‐mediated current activation (data not shown). These data demonstrate that ATP signals primarily through P2Y2 receptors in this preparation.
Figure 5.
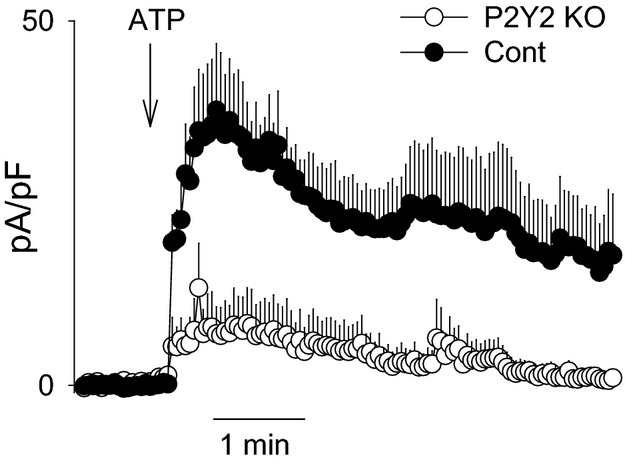
Requirement of P2Y2 receptor in ATP‐mediated current activation in freshly isolated cerebral ECs. ATP (100 μmol/L)‐activated current was recorded during ramps (−120 to +60 mV) in voltage‐clamp mode and is presented as the outward current at +40 mV measured over time. Responses are summarized for ECs isolated from WT (C57Bl/6; n=5) and P2Y2 receptor KO mice (n=7). Current was normalized to cell capacitance and presented as mean current density (pA/pF). All data presented as mean±SEM. Group difference=P<0.01; 2‐way repeated‐measures ANOVA. KO indicates knockout; WT, wild type.
Figure 6A shows a representative hyperpolarization response to ATP, recorded from an EC cluster of 3 cells. About half of recorded clusters, like the one shown, revealed spontaneous negative voltage spikes lasting for a few seconds each. Application of ATP resulted in a profound (to −40 to −60 mV) and long‐lasting (about 10 minutes) hyperpolarization. Figure 6B summarizes the distinct temporal roles of SKCa and IKCa channels in ATP‐mediated EC hyperpolarization. In preliminary experiments, we demonstrated that ATP‐mediated EC hyperpolarization was essentially completely dependent on combined SKCa/IKCa channel activation (not shown). Therefore, we were able to isolate the role of IKCa channel activation by inhibiting SKCa channels (and vice versa). TRAM34 treatment (IKCa blocker) primarily attenuated the peak hyperpolarization response, with little attenuation of the ensuing sustained component. In contrast, UCL1684 treatment (SKCa blocker) had no effect on the peak hyperpolarization response but completely eliminated the sustained component.
Figure 6.
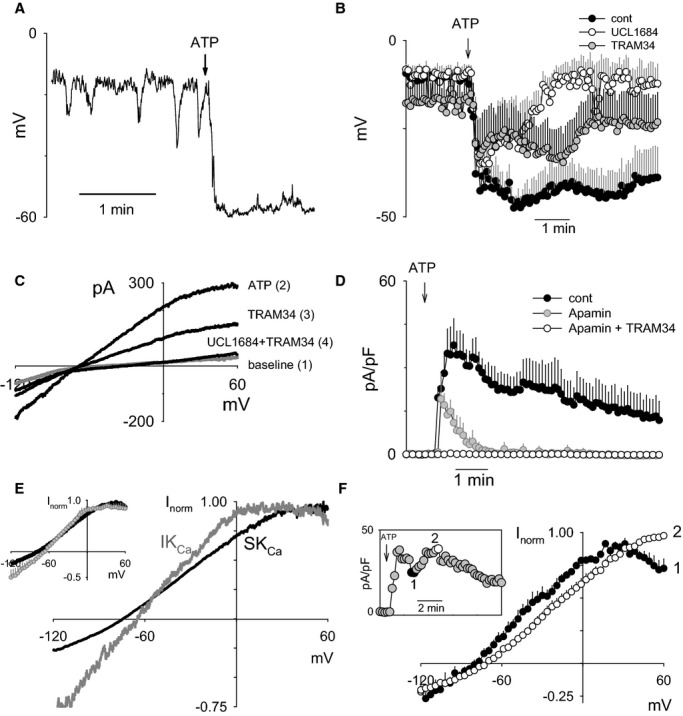
Biphasic contribution of SKCa and IKCa channels in ATP‐mediated hyperpolarization of freshly isolated cerebral ECs. A, Representative current‐clamp measurement of ATP‐induced hyperpolarization in freshly isolated cerebral artery ECs. B, Summary of ATP‐mediated hyperpolarization in the absence of blockers (cont; n=7) and in the presence of SKCa blocker (UCL1684; n=3) and IKCa blocker (TRAM34; n=6). C, Representative I‐V recordings at baseline and then after sequential application of ATP, TRAM34, and combined UCL1684/TRAM34. ATP was present throughout all recordings after the baseline measurement. D, Summary of whole‐cell current measured at +40 mV in response to ATP application. Current was measured from sequential I‐V sweeps following no treatment (control; n=9), SKCa inhibition (apamin, 200 nmol/L; n=4), and combined SKCa/IKCa inhibition (apamin+TRAM34, 1 μmol/L; n=3). E, Representative normalized SKCa (black) and IKCa I‐V response (gray) obtained by determining the UCL1684‐ and TRAM34‐senstive currents. The summary of UCL1684 and TRAM34‐sensitive currents is depicted in the inset (n=4 each). F, Summary of normalized ATP‐stimulated I‐V plots taken from the end of the initial peak response (plot 1) and during the peak of the sustained phase of the response (plot 2). A representative time course of the ATP‐stimulated current measured at +40 mV is shown in the inset (n=4 each). Summary data presented as mean±SEM. IKCa channels indicate intermediate conductance Ca2+‐activated K+ channels; SKCa channels, small conductance Ca2+‐activated K+ channels.
The whole‐cell currents responsible for ATP‐mediated EC hyperpolarization were further evaluated by briefly switching between current‐clamp and voltage‐clamp modes to obtain current‐voltage plots at each condition (Figure 6C). The ATP‐stimulated current was partially attenuated by IKCa blockade (TRAM34 10 μmol/L) and completely abolished by combined SKCa and IKCa blockade (UCL1684 0.2 μmol/L and TRAM34). The temporal contribution of SKCa and IKCa channel activation to ATP‐stimulated whole‐cell current at +40 mV is summarized in Figure 6D. Note that inhibition of SKCa channels with apamin (200 nmol/L) dramatically attenuated the sustained component (similar results obtained with UCL1684). The current‐voltage plots specifically corresponding to IKCa and SKCa channel activation were generated by calculating the TRAM34‐ and UCL1684‐sensitive currents, respectively (Figure 6E). Note the characteristically greater inward rectification at positive voltages for the IKCa current‐voltage relationship compared with that of SKCa.41 The summary of the normalized current‐voltage relationships corresponding to SKCa (black) and IKCa (gray) are depicted in the inset. Figure 6F demonstrates the shift in the whole‐cell current‐voltage relationship of total K+ current without blockers during the course of ATP stimulation. The inset figure is a representative experiment depicting the time points of the response at which current‐voltage relationships were evaluated—at the end of the peak response (denoted as 1) and during the sustained component (denoted as 2). Summary of current‐voltage relationships obtained at the end of the peak response (summary plot 1) strongly resemble IKCa‐dependent relationships, whereas current‐voltage relationships obtained during the sustained component (summary plot 2) strongly resemble SKCa‐dependent relationships. From these aggregate data, it appears that ATP produces EC hyperpolarization through a combination of IKCa and SKCa channel activation, where IKCa channels contribute to the initial hyperpolarization and SKCa channels contribute to the sustained hyperpolarization.
TRPC3 Contributes to SKCa Channel Activation and ATP‐Mediated Cerebral EC Hyperpolarization
Having demonstrated a role of TRPC3 in EC Ca2+ influx and EDH‐mediated vasodilation, we next sought to determine the functional link between TRPC3 activation and EC hyperpolarization. Given that ATP‐mediated EC hyperpolarization is entirely dependent on SKCa/IKCa channel activation, we specifically evaluated the potential of TRPC3 in regulating either of these channels.
The role of TRPC3 in ATP‐mediated EC hyperpolarization was determined by pharmacological inhibition with Pyr3 (1 μmol/L) and in cells isolated from TRPC3 KO mice (Figure 7). Pharmacological inhibition of TRPC3 demonstrated no attenuation of peak hyperpolarization (Figure 7A) or the initial outward current (Figure 7B), whereas the respective sustained components were abolished. Genetic deletion of TRPC3 similarly had no effect on the peak hyperpolarization response (Figure 7C). The sustained component was significantly attenuated, though not as completely as with Pyr3 inhibition. NS309 (SK/IKCa activator) was applied to demonstrate similar potential for hyperpolarization through KCa channel activation in the ECs from TRPC3 KO mice. The effect of Pyr3 was additionally evaluated in the presence of TRAM34 to isolate the role of TRPC3 in SKCa current activation (Figure 7B). Note that combined application of Pyr3 and TRAM34 nearly abolished the ATP‐stimulated current, demonstrating significant TRPC3‐dependence of SKCa current activation. The requirement of TRPC3 in SKCa channel activation is further supported by the virtually identical effect of TRPC3 or SKCa channel inhibition on ATP‐stimulated hyperpolarization and the time course of current activation (compare with Figure 6B and 6D). Together, these results demonstrate a critical role of TRPC3 in the regulation of SKCa channel activation and EC hyperpolarization.
Figure 7.
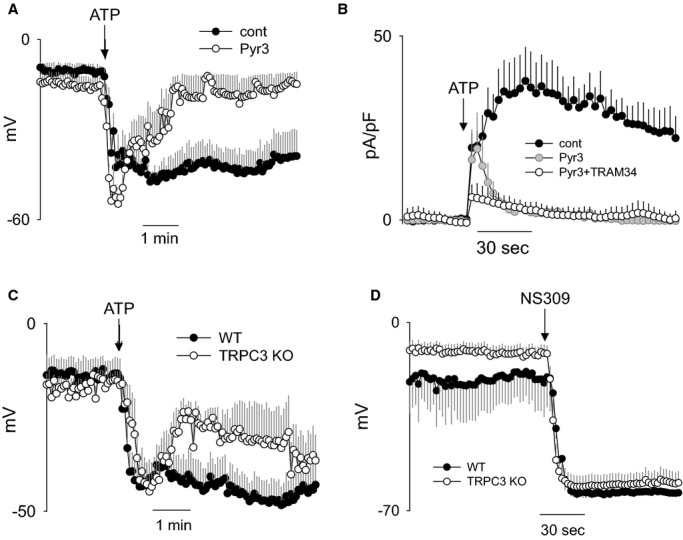
Role of TRPC3 in ATP‐mediated hyperpolarization and SKCa channel activation of freshly isolated cerebral ECs. A, Summary of ATP‐mediated (100 μmol/L) hyperpolarization responses in ECs treated with vehicle (n=7) or TRPC3 blocker (Pyr3, 1 μmol/L; n=5). B, Summary of ATP‐stimulated K+ current at +40 mV obtained from sequential I‐V sweeps. Measurements were performed following no treatment (cont; n=7), TRPC3 blocker (Pyr3; n=3), and TRPC3 blocker plus IKCa blocker (Pyr3+TRAM34; n=4). C, Summary of ATP‐mediated (100 μmol/L) hyperpolarization responses in freshly isolated ECs from WT (n=8) and TRPC3 KO mice (n=5). D, Summary of hyperpolarization response to SK/IKCa channel activator (NS309, 10 μmol/L) in ECs isolated from WT (n=3) and TRPC3 KO mice (n=7). All data presented as mean±SEM. IKCa channels indicate intermediate conductance Ca2+‐activated K+ channels; SKCa channels, small conductance Ca2+‐activated K+ channels; KO, knockout; TRPC3, transient receptor potential C3; WT, wild type.
Evidence of TRPC3 Channel Trafficking in SKCa Channel Activation
In light of the latency between receptor stimulation and SKCa channel activation, we investigated the possibility that channel trafficking was involved in this component of EC hyperpolarization. Figure 8 demonstrates the effect of 2 successive ATP applications on EC current activation. In these experiments, we isolated the SKCa current by maintaining TRAM34 throughout. Figure 8A depicts a representative experiment in which ATP was added twice to the same EC preparation with a 10‐minute washout between exposures. To ensure uniform delivery of ATP, the recording chamber was rapidly flushed with the ATP‐containing solution. With the first application of ATP, SKCa current increased gradually to maximal activation. However, when the ECs were exposed to ATP a second time, SKCa current reached maximum activation almost immediately. SKCa current (normalized to maximum current) after first and second ATP responses is summarized in Figure 8B. These data demonstrate a clear effect of prior receptor stimulation on the rate of SKCa channel activation and suggest a possible role of channel trafficking in response to ATP stimulation.
Figure 8.
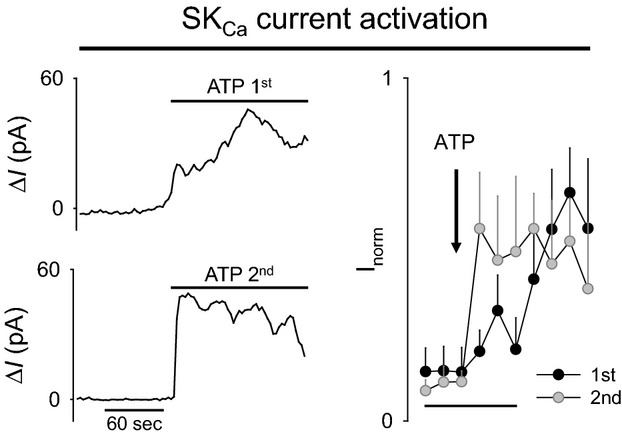
Time course of ATP‐stimulated SKCa current activation in freshly isolated ECs after first and second ATP application (left). Representative ATP‐stimulated SKCa current at +40 mV after first ATP application (top) and after second ATP application (bottom). TRAM34 (1 μmol/L) was present throughout to isolate the SKCa current. ATP was washed out for 10 minutes between first and second exposures (right). Summary of normalized ATP‐stimulated SKCa current for first and second ATP exposures (n=3). Two‐way repeated‐measures ANOVA significant interaction between groups P=0.034. Summary data presented as mean±SEM. SKCa channels indicate small conductance Ca2+‐activated K+ channels.
To explain the differences in rate of SKCa current activation, we first considered the possibility that SKCa channels were trafficked to the PM after ATP stimulation. Precedent for rapid trafficking of SK3 channels has recently been provided in mouse aorta ECs after receptor activation.42 Apamin is an irreversible blocker of SKCa channels and has been used in the demonstration of SKCa channel trafficking to the PM.42 In this method, brief exposure of apamin is used to irreversibly inhibit surface expressed SKCa channels. Return of SKCa current (measured by patch clamp) would thus be reflective of new SKCa channels trafficked to the PM. Figure 9 (left panel) demonstrates a representative experiment in which apamin was applied for 2 minutes and then washed out (n=3). Subsequent ATP application elicited a very transient whole‐cell current, similar to the response obtained in the continued presence of SKCa channel blockers (Figure 6D). When the experiment was performed in the presence of TRAM34 to isolate the SKCa current, ATP application failed to elicit any current response for up to 20 minutes of recording (n=5). Thus, these findings strongly argue against trafficking of the SKCa channel in cerebral ECs over the time course of the delayed SKCa current response.
Figure 9.
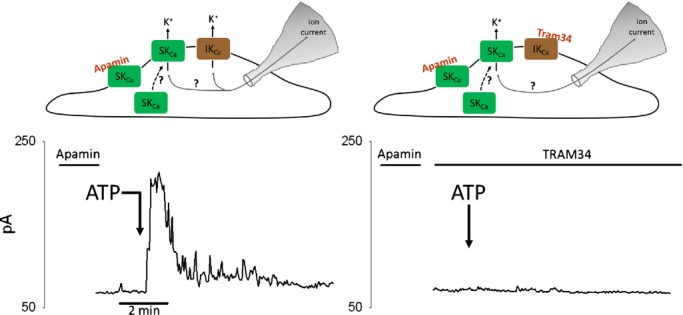
Evidence against SKCa channel trafficking to the plasma membrane following receptor stimulation in cerebral ECs. Representative whole‐cell current recordings from freshly isolated cerebral ECs in response to ATP stimulation (100 μmol/L). Apamin (200 nmol/L) was very briefly applied to freshly isolated ECs (2 minutes) and then washed out before ATP application. (left) Representative recording for ATP response following apamin pretreatment. The resulting current should consist of IKCa current and any potential SKCa current from recruited SKCa channels (see illustration). (right) Representative recording for ATP response following apamin pretreatment plus sustained treatment with TRAM34 (1 μmol/L). The resulting current should consist only of any potential SKCa current from recruited SKCa channels (see illustration). Current was measured at +40 mV. The point of ATP application is indicated by the arrows. Brief exposure to apamin eliminated the sustained current (left) and all SKCa current (right). Traces are representative of 3 to 5 experiments for each group. IKCa channels indicate intermediate conductance Ca2+‐activated K+ channels; SKCa channels, small conductance Ca2+‐activated K+ channels.
Trafficking of TRPC3 channels has also been demonstrated through insertion and removal of intracellular vesicles.43–48 Given that TRPC3 channels appear to be critical to regulating SKCa channel activation, we reasoned that trafficking of TRPC3 could account for delayed SKCa channel activation. Pressurized cerebral arteries and freshly isolated ECs from WT and TRPC3 KO mice were used to demonstrate TRPC3 expression in endothelium and to confirm specificity of the antibody (Figure 10 A and 10B). Next, pressurized arteries were treated with vehicle (control) or ATP (100 μmol/L) for 10 minutes before fixation and processing for TRPC3 immunofluorescence. High magnification (100× oil) deconvolution imaging demonstrated a redistribution of TRPC3 immunofluorescence with ATP stimulation (Figure 10C and 10D). In control arteries, TRPC3 expression was distributed throughout the ECs, with only slight accumulation at the PM. After ATP stimulation, however, TRPC3 accumulated along the PM and was reciprocally decreased throughout the cytosolic compartment. This accumulation was most evident in the PM in the vicinity of the nucleus. Images are representative of artery preparations from 3 mice in each group. The cytosolic‐to‐PM redistribution was even more apparent in 3‐dimensional reconstructions created from planes covering a depth of 2 μm (Figure 10D).
Given that TRPC3 is thought to traffic through vesicular fusion/removal from the PM, we expected that greater TRPC3 trafficking to the PM should be associated with an increase in PM surface area. We therefore measured Cm in cerebral ECs before and after ATP stimulation, as a real‐time measurement of PM surface area. Figure 11 summarizes Cm measurements in response to ATP. Note that application of ATP produced a rapid increase in EC Cm of a similar time course as SKCa current recruitment. When considered with the immunofluorescence data, these findings are consistent with ATP‐stimulated trafficking of TRPC3‐containing vesicles to the PM in the mechanism of SKCa activation.
Figure 11.
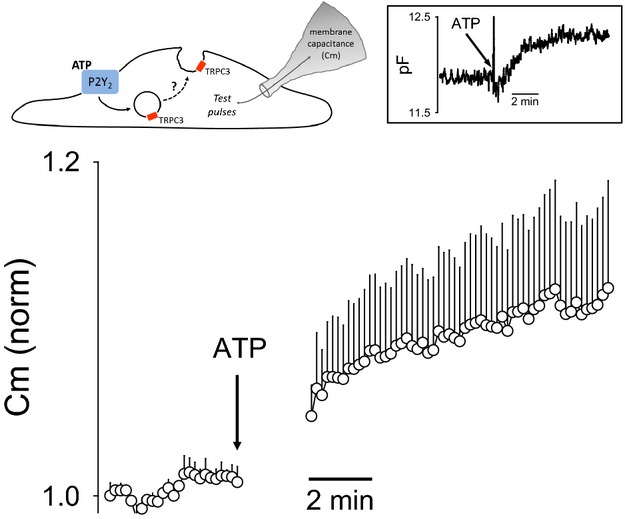
ATP‐stimulated increase in plasma membrane surface area of freshly isolated cerebral ECs measured by whole‐cell capacitance. Real‐time measurement of EC whole‐cell capacitance (Cm) was used to evaluate the effect of ATP stimulation on plasma membrane surface area. The tested model predicts vesicular fusion with the plasma membrane and thus increased membrane surface area/measured Cm following receptor stimulation (see illustration). The data summarizes mean Cm following ATP stimulation (100 μmol/L) in freshly isolated cerebral ECs. The gap in the trace reflects removal of the artifactual data points immediately after bath solution change. Baseline Cm was normalized to unity. Data are presented as mean±SEM (n=3 EC clusters from 3 separate mice). The inset is a representative individual recording of Cm in response to ATP application. TRPC3 indicates transient receptor potential C3.
Discussion
We present the following novel findings regarding the role of TRPC3 in endothelium‐mediated vasodilation in the cerebral circulation: (1) TRPC3 contributes to EDH‐mediated vasodilation of pressurized cerebral artery, (2) TRPC3 contributes to receptor‐mediated Ca2+ regulation in the endothelium intact cerebral artery, (3) receptor‐mediated EC hyperpolarization is initiated by IKCa channel activation and maintained by subsequent SKCa channel activation, and (4) rapid recruitment of TRPC3 to the PM is critical for SKCa channel activation and sustained EC hyperpolarization. We propose a new model in which TRPC3‐containing vesicles are inserted into the PM on receptor stimulation, where the newly inserted TRPC3 then contributes to SKCa channel activation, EC hyperpolarization, and enhanced EDH‐mediated vasodilation (see Figure 12).
TRPC3 Contributes to EDH‐Mediated Vasodilation in Cerebral Artery
In the present study, we specifically tested the role of TRPC3 in EDH‐mediated response to ATP, a potent physiological vasodilator in the cerebral circulation. It has been shown that EDH‐mediated vasodilation plays a major role in controlling blood flow in isolated cerebral arteries and arterioles.40,49 Additionally, recent laser Doppler studies suggest an in vivo role of EDH‐mediated control of cerebral blood flow in mice.13 Our data presented here demonstrate for the first time that the EDH‐dependent mechanism in mouse cerebral artery critically involves TRPC3 for fully functional vasodilation to ATP.
To date, only a few prior studies have linked TRPC3 in the mechanism of endothelium‐dependent vasodilation, though no prior study has demonstrated such a role in the cerebral circulation. In one of the earliest studies linking TRPC3 to endothelial regulation of vasorelaxation, Liu et al20 used antisense oligos delivered in vivo to knock down endothelial expression of TRPC3 in rat mesenteric arteries. They subsequently demonstrated attenuation of flow‐induced vasodilation and bradykinin‐induced vasorelaxation in arteries of antisense‐treated rats. Interestingly, vasorelaxation to histamine, ATP, and cyclopiazonic acid was not affected by TRPC3 knockdown in this preparation. More recently, an isoform‐specific pharmacological blocker of TRPC3 (Pyr3) was developed that has enabled acute pharmacological evaluation of TRPC3 channel function.50 This blocker has since been used to demonstrate the involvement of TRPC3 in bradykinin‐induced relaxation in isolated porcine coronary arteries,21 as well as acetylcholine/carbacholine–induced relaxation in human mammary artery,51 mouse mesenteric artery,8 and mouse aorta.32
TRPC3 Contributes to Sustained Endothelial Ca2+ Influx After Receptor Stimulation
We have demonstrated in the present study that decreased arterial relaxation to ATP after TRPC3 KO or Pyr3 pretreatment correlates with an attenuated endothelial Ca2+ response measured in intact pressurized arteries. Although Ca2+ influx through TRPC3 channels has been previously demonstrated in numerous cell types (see reviews19,52), the specific role of TRPC3 in endothelial Ca2+ regulation is not fully elucidated. Our data show that TRPC3 (evaluated by Pyr3 or TRPC3 KO) primarily affects the duration of the cerebral EC Ca2+ response to ATP. Furthermore, Mn2+‐quenching experiments show relatively long‐lasting Pyr3‐sensitive cation influx after ATP stimulation. Additionally, these data demonstrated no apparent effect of Pyr3 on basal Ca2+ influx, thus suggesting little contribution of constituitively active TRPC3 residing in the PM before receptor stimulation. Such a role of TRPC3 in producing sustained Ca2+ influx after receptor stimulation is consistent with our model in which we propose a requirement for TRPC3 trafficking to provide the source of Ca2+ for SKCa channel activation and EC hyperpolarization.
TRPC3 Is Required for Receptor‐Mediated SKCa Channel Activation
Given our demonstration of TRPC3 in EDH‐mediated vasodilation and EC Ca2+ regulation in this artery, we examined the potential link between TRPC3 activity and SK/IKCa channel activation. Experiments with a TRPC3 blocker (Pyr3) and with ECs from TRPC3 KO mice demonstrated a clear role for TRPC3 in regulating SKCa channel activity. Receptor‐mediated hyperpolarization was virtually identically affected by TRPC3 blockade/ablation as with SKCa channel inhibition—namely, there was no effect on the peak hyperpolarization response and significant attenuation of the sustained component (compare Figures 6B and 7A). The effect on TRPC3 blockade/ablation similarly mimicked the effect of SKCa inhibition on K+ current activation (compare Figures 6D and 7B). These studies suggested a clear requirement of functional TRPC3 channel for SKCa channel activation. We have recently reported a role for TRPC3 in SKCa and IKCa channel activation in rat mesenteric artery,8 indicating that a role for TRPC3 in regulating Ca2+‐activated K channels is not unique to the cerebral circulation. However, unlike our findings in mesenteric artery, we found no evidence for significant TRPC3‐dependent regulation of IKCa channels in the cerebral circulation.
Receptor‐Mediated Activation of ECs Leads to Biphasic Activation of IKCa and SKCa Channels
EDH‐mediated vasodilation in peripheral circulations is often found to require activation of endothelial SKCa and IKCa channels.53 For instance, combined inhibition of SKCa and IKCa channels is required to block EDH‐mediated vasodilation, whereas inhibition of either channel alone is insufficient to block the response.54 In the cerebral circulation, however, inhibition of IKCa channels alone has been shown to substantially blunt or eliminate EDH‐mediated vasodilation.25,55–56 While SKCa channels do not appear to be critical for initiating EDH‐mediated vasodilation, the full role of SKCa channels in this mechanism is yet to be resolved. Our present study corroborates the critical role of IKCa channels in providing the initial EC hyperpolarization that is required for EDH‐mediated vasodilation; however, it also establishes an important role for SKCa channels in sustaining the hyperpolarization. Our claim of 2 temporal components of EC hyperpolarization is supported by our measurements of hyperpolarization and K+ current activation in the presence of specific blockers to isolate SKCa and IKCa effects. In these studies, we found that IKCa channel activation and IKCa channel–mediated hyperpolarization occurred immediately after receptor stimulation but began rapidly decaying within the first minute (see Figure 6B and 6D). SKCa channel activation occurred more gradually but was sustained for several minutes (see Figure 8). The 2 distinct currents were also evident in the current‐voltage plots recorded at early and later times after receptor stimulation (see Figure 6F). In this experiment, current‐voltage plots appeared to be initially dominated by IKCa‐like currents before shifting to predominantly SKCa‐like currents during the sustained phase. Together, these findings demonstrate a time‐dependent transition from IKCa to SKCa channel dependence. We propose that the initial EC hyperpolarization requires IKCa channel activation, while sustaining that hyperpolarization requires SKCa channel activation through prolonged Ca2+ influx via TRPC3 channels.
Role of Vesicular Trafficking in TRPC3‐Dependent Activation of SKCa Channels
The delayed onset of the SKCa current could have multiple explanations; however, in the context of our studies, 2 possible scenarios seemed initially plausible. In the first scenario, SKCa channels could be trafficked to the PM in response to receptor stimulation, where they would then be activated by TRPC3 channels already present in the membrane. Precedent for SKCa channel trafficking has recently been established for SK3 channels in mouse aortic ECs.42 In the second scenario, TRPC3 channels could be trafficked to the PM where they would then contribute to SKCa channel activation. There is considerable recent evidence for TRPC3 channel trafficking in a variety of neuronal cell and EC preparations,43–44,43–48,57 as well as one study suggesting TRPC3 trafficking in cultured porcine coronary ECs.21 We discuss the evidence for each of these scenarios further next.
SKCa channel trafficking
Lin et al reported trafficking of SKCa channels (presumed SK3) in mouse aorta ECs.42 In that study, they used brief apamin exposure to irreversibly inhibit surface SKCa channels and then monitored for recovery of SKCa current on washout. Within minutes of washout, they found rapid recovery of SKCa current that was abolished by disruption of Ca2+‐dependent vesicular trafficking. Our data in cerebral ECs, however, were not consistent with similar SKCa trafficking. In particular, after brief application of apamin, the sustained component (after ATP challenge) remained absent during the apamin washout period. In addition, cells treated with TRAM34 (to isolate the SKCa current) failed to produce any current in response to ATP during the apamin washout period for up to 20 minutes (see Figure 9). From our studies in which apamin or UCL1684 was not included, we found that the SKCa current otherwise appeared in less than 1 minute and peaked within 5 minutes. In short, trafficking of SKCa does not appear to account for the gradually increasing SKCa current after initial receptor stimulation or the immediately responsive SKCa current after prior ATP stimulation.
TRPC3 channel trafficking
Recent evidence indicates that TRPC3 channels can be actively trafficked to and from the PM in a variety of cell types43–44,43–48,57 through distinct constitutive and receptor‐mediated trafficking pathways.47 TRPC3 has been localized to vesicles and associates with several key proteins involved in SNARE‐mediated vesicular trafficking.48,57–58 The SNARE proteins and Ca2+‐sensitive SNARE associating proteins provide the necessary components for docking of vesicles at the subplasmalemmal surface and ultimate vesicular fusion with the PM (exocytosis).59–60 In the present study, we demonstrated consolidation of TRPC3 at the EC PM of pressurized arteries after ATP stimulation, consistent with increased forward trafficking of TRPC3‐containing vesicles to the PM (Figure 10). We also demonstrated rapid increase in cell capacitance (correlates with PM surface area) following ATP stimulation of freshly isolated ECs, consistent with the fusion of exocytotic vesicles with the PM (see Figure 11). The time course of this increase in PM surface area correlated very well with the corresponding increase in SKCa‐mediated current and EC hyperpolarization. When the evidence is considered as a whole, it appears that receptor‐mediated trafficking of TRPC3 channels occurs and contributes to SKCa channel regulation in cerebral endothelium.
Limitations
In the present studies, we used a combination of global TRPC3 KO mice and Pyr3 to evaluate the role of endothelial TRPC3 in the regulation of EC calcium, EC SK/IKCa channels, and ultimately vasodilation of cerebral artery. Use of global TRPC3 KO mice is appropriate for studies involving isolated ECs; however, these mice are not ideal for pressurized artery experiments due to the additional loss of TRPC3 expression in vascular smooth muscle, perivascular nerves, etc. For the pressurized artery experiments, we instead used Pyr3 at a concentration that we found to produce TRPC3‐specific inhibition of endothelial Ca2+ responses (Figure 3C) and that we previously found to target endothelial function over smooth muscle function in mesenteric arteries. Nevertheless, we cannot fully exclude nonendothelial effects of Pyr3 in the attenuation of ATP‐mediated vasodilation. For a more specific determination of the role of endothelial TRPC3 channels in intact arteries and in vivo blood flow, we are currently developing EC‐specific TRPC3 KO mice.
With regard to the proposed mechanism of TRPC3 regulation of SKCa channels, we have provided multiple pieces of evidence for the functional interaction of these channels. However, we have not directly demonstrated a close physical association between these 2 ion channels. Our proposed trafficking model would be strengthened by future demonstration of physical association and disassociation of these channels in a P2Y2 receptor–dependent fashion. We have also assumed that TRPC3 channels are shuttled to and from the PM through forward and reverse vesicular trafficking as in other cell types. Although defining the exact mechanism of TRPC3 trafficking is not critical to our overall model of TRPC3‐dependent regulation of SKCa channel activity, further study into receptor‐mediated TRPC3 trafficking in cerebrovascular endothelium could demonstrate additional targets for SKCa channel regulation and the ultimate regulation of cerebrovascular tone.
Summary
We demonstrate that TRPC3 channels are functionally expressed in cerebral ECs and play a critical role in regulating Ca2+ influx and ATP‐induced hyperpolarization. TRPC3 channel activation is essential for the SKCa channel–dependent EC hyperpolarization that develops subsequent to the initial IKCa channel–mediated hyperpolarization. Furthermore, TRPC3‐mediated regulation of SKCa channels appears to involve trafficking of TRPC3 channels to the PM, where they then provide the source of Ca2+ influx for SKCa channel activation. Ultimately, TRPC3‐dependent regulation of SKCa channel activation appears to be an important component of sustained EDH‐mediated vasodilation in cerebral arteries.
Sources of Funding
This study was supported by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute grant R01 HL088435 to Dr Marrelli and by the Intramural Research Program of the NIH (project Z01‐ES101684 to Dr Birnbaumer).
Disclosures
None.
References
- 1.Pan Z, Yang H, Reinach PS. Transient receptor potential (TRP) gene superfamily encoding cation channels. Hum Genomics. 2011; 5:108-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu LJ, Sweet TB, Clapham DE. International union of basic and clinical pharmacology. LXXVI. current progress in the mammalian TRP ion channel family. Pharmacol Rev. 2010; 62:381-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marrelli SP, O'neil RG, Brown RC, Bryan RM., Jr PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. Am J Physiol Heart Circ Physiol. 2007; 292:H1390-H1397. [DOI] [PubMed] [Google Scholar]
- 4.Brayden JE, Earley S, Nelson MT, Reading S. Transient receptor potential (TRP) channels, vascular tone and autoregulation of cerebral blood flow. Clin Exp Pharmacol Physiol. 2008; 35:1116-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang DX, Gutterman DD. Transient receptor potential channel activation and endothelium‐dependent dilation in the systemic circulation. J Cardiovasc Pharmacol. 2011; 57:133-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Earley S, Gonzales AL, Crnich R. Endothelium‐dependent cerebral artery dilation mediated by TRPA1 and Ca2+‐activated K+ channels. Circ Res. 2009; 104:987-994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Earley S. Endothelium‐dependent cerebral artery dilation mediated by transient receptor potential and ca(2+)‐activated K(+) channels. J Cardiovasc Pharmacol. 2011; 57:148-153. [DOI] [PubMed] [Google Scholar]
- 8.Senadheera S, Kim Y, Grayson TH, Toemoe S, Kochukov MY, Abramowitz J, Housley GD, Bertrand RL, Chadha PS, Bertrand PP, Murphy TV, Tare M, Birnbaumer L, Marrelli SP, Sandow SL. Transient receptor potential canonical type 3 channels facilitate endothelium‐derived hyperpolarization‐mediated resistance artery vasodilator activity. Cardiovasc Res. 2012; 95:439-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill‐Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012; 336:597-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dora KA, Gallagher NT, McNeish A, Garland CJ. Modulation of endothelial cell KCa3.1 channels during endothelium‐derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res. 2008; 102:1247-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chadha PS, Liu L, Rikard‐Bell M, Senadheera S, Howitt L, Bertrand RL, Grayson TH, Murphy TV, Sandow SL. Endothelium‐dependent vasodilation in human mesenteric artery is primarily mediated by myoendothelial gap junctions intermediate conductance calcium‐activated K+ channel and nitric oxide. J Pharmacol Exp Ther. 2011; 336:701-708. [DOI] [PubMed] [Google Scholar]
- 12.Behringer EJ, Segal SS. Tuning electrical conduction along endothelial tubes of resistance arteries through ca(2+)‐activated K(+) channels. Circ Res. 2012; 110:1311-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hannah RM, Dunn KM, Bonev AD, Nelson MT. Endothelial SK(ca) and IK(ca) channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J Cereb Blood Flow Metab. 2011; 31:1175-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasenau AL, Nielsen G, Morisseau C, Hammock BD, Wulff H, Kohler R. Improvement of endothelium‐dependent vasodilations by SKA‐31 and SKA‐20, activators of small‐ and intermediate‐conductance Ca2+ ‐activated K+ ‐channels. Acta Physiol (Oxf). 2011; 203:117-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dalsgaard T, Kroigaard C, Bek T, Simonsen U. Role of calcium‐activated potassium channels with small conductance in bradykinin‐induced vasodilation of porcine retinal arterioles. Invest Ophthalmol Vis Sci. 2009; 50:3819-3825. [DOI] [PubMed] [Google Scholar]
- 16.Kurian MM, Berwick ZC, Tune JD. Contribution of IKCa channels to the control of coronary blood flow. Exp Biol Med (Maywood). 2011; 236:621-627. [DOI] [PubMed] [Google Scholar]
- 17.Mishra RC, Belke D, Wulff H, Braun AP. SKA‐31, a novel activator of SK(ca) and IK(ca) channels, increases coronary flow in male and female rat hearts. Cardiovasc Res. 2013; 97:339-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albert AP. Gating mechanisms of canonical transient receptor potential channel proteins: role of phosphoinositols and diacylglycerol. Adv Exp Med Biol. 2011; 704:391-411. [DOI] [PubMed] [Google Scholar]
- 19.Smedlund K, Bah M, Vazquez G. On the role of endothelial TRPC3 channels in endothelial dysfunction and cardiovascular disease. Cardiovasc Hematol Agents Med Chem. 2012; 10:265-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu CL, Huang Y, Ngai CY, Leung YK, Yao XQ. TRPC3 is involved in flow‐ and bradykinin‐induced vasodilation in rat small mesenteric arteries. Acta Pharmacol Sin. 2006; 27:981-990. [DOI] [PubMed] [Google Scholar]
- 21.Huang JH, He GW, Xue HM, Yao XQ, Liu XC, Underwood MJ, Yang Q. TRPC3 channel contributes to nitric oxide release: significance during normoxia and hypoxia‐reoxygenation. Cardiovasc Res. 2011; 91:472-482. [DOI] [PubMed] [Google Scholar]
- 22.Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron. 2008; 59:392-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tallini YN, Brekke JF, Shui B, Doran R, Hwang SM, Nakai J, Salama G, Segal SS, Kotlikoff MI. Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC GCaMP2 transgenic mice. Circ Res. 2007; 101:1300-1309. [DOI] [PubMed] [Google Scholar]
- 24.Homolya L, Watt WC, Lazarowski ER, Koller BH, Boucher RC. Nucleotide‐regulated calcium signaling in lung fibroblasts and epithelial cells from normal and P2Y(2) receptor (‐/‐) mice. J Biol Chem. 1999; 274:26454-26460. [DOI] [PubMed] [Google Scholar]
- 25.Marrelli SP, Eckmann MS, Hunte MS. Role of endothelial intermediate conductance KCa channels in cerebral EDHF‐mediated dilations. Am J Physiol Heart Circ Physiol. 2003; 285:H1590-H1599. [DOI] [PubMed] [Google Scholar]
- 26.Golding EM, Ferens DM, Marrelli SP. Altered calcium dynamics do not account for attenuation of endothelium‐derived hyperpolarizing factor‐mediated dilations in the female middle cerebral artery. Stroke. 2002; 33:2972-2977. [DOI] [PubMed] [Google Scholar]
- 27.Marrelli SP. Altered endothelial Ca2+ regulation after ischemia/reperfusion produces potentiated endothelium‐derived hyperpolarizing factor‐mediated dilations. Stroke. 2002; 33:2285-2291. [DOI] [PubMed] [Google Scholar]
- 28.Marrelli SP. Mechanisms of endothelial P2Y(1)‐ and P2Y(2)‐mediated vasodilatation involve differential [Ca2+]i responses. Am J Physiol Heart Circ Physiol. 2001; 281:H1759-H1766. [DOI] [PubMed] [Google Scholar]
- 29.Marrelli SP. Selective measurement of endothelial or smooth muscle [ca(2+)](i) in pressurized/perfused cerebral arteries with fura‐2. J Neurosci Methods. 2000; 97:145-155. [DOI] [PubMed] [Google Scholar]
- 30.Marrelli SP, Khorovets A, Johnson TD, Childres WF, Bryan RM., Jr P2 purinoceptor‐mediated dilations in the rat middle cerebral artery after ischemia‐reperfusion. Am J Physiol. 1999; 276:H33-H41. [DOI] [PubMed] [Google Scholar]
- 31.Suh SH, Vennekens R, Manolopoulos VG, Freichel M, Schweig U, Prenen J, Flockerzi V, Droogmans G, Nilius B. Characterisation of explanted endothelial cells from mouse aorta: electrophysiology and Ca2+ signalling. Pflugers Arch. 1999; 438:612-620. [DOI] [PubMed] [Google Scholar]
- 32.Kochukov MY, Balasubramanian A, Noel RC, Marrelli SP. Role of TRPC1 and TRPC3 channels in contraction and relaxation of mouse thoracic aorta. J Vasc Res. 2013; 50:11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindau M, Neher E. Patch‐clamp techniques for time‐resolved capacitance measurements in single cells. Pflugers Arch. 1988; 411:137-146. [DOI] [PubMed] [Google Scholar]
- 34.Namiranian K, Lloyd EE, Crossland RF, Marrelli SP, Taffet GE, Reddy AK, Hartley CJ, Bryan RM., Jr Cerebrovascular responses in mice deficient in the potassium channel, TREK‐1. Am J Physiol Regul Integr Comp Physiol. 2010; 299:R461-R469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tallini YN, Ohkura M, Choi BR, Ji G, Imoto K, Doran R, Lee J, Plan P, Wilson J, Xin HB, Sanbe A, Gulick J, Mathai J, Robbins J, Salama G, Nakai J, Kotlikoff MI. Imaging cellular signals in the heart in vivo: cardiac expression of the high‐signal Ca2+ indicator GCaMP2. Proc Natl Acad Sci USA. 2006; 103:4753-4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI, Nelson MT. Functional architecture of inositol 1,4,5‐trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci USA. 2008; 105:9627-9632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang HR, Wu M, Yu H, Long S, Stevens A, Engers DW, Sackin H, Daniels JS, Dawson ES, Hopkins CR, Lindsley CW, Li M, McManus OB. Selective inhibition of the K(ir)2 family of inward rectifier potassium channels by a small molecule probe: the discovery, SAR, and pharmacological characterization of ML133. ACS Chem Biol. 2011; 6:845-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Millar ID, Wang S, Brown PD, Barrand MA, Hladky SB. Kv1 and Kir2 potassium channels are expressed in rat brain endothelial cells. Pflugers Arch. 2008; 456:379-391. [DOI] [PubMed] [Google Scholar]
- 39.Horiuchi T, Dietrich HH, Hongo K, Dacey RG., Jr Comparison of P2 receptor subtypes producing dilation in rat intracerebral arterioles. Stroke. 2003; 34:1473-1478. [DOI] [PubMed] [Google Scholar]
- 40.You J, Johnson TD, Marrelli SP, Bryan RM., Jr Functional heterogeneity of endothelial P2 purinoceptors in the cerebrovascular tree of the rat. Am J Physiol. 1999; 277:H893-H900. [DOI] [PubMed] [Google Scholar]
- 41.Ledoux J, Bonev AD, Nelson MT. Ca2+‐activated K+ channels in murine endothelial cells: block by intracellular calcium and magnesium. J Gen Physiol. 2008; 131:125-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin MT, Adelman JP, Maylie J. Modulation of endothelial SK3 channel activity by ca(2)+dependent caveolar trafficking. Am J Physiol Cell Physiol. 2012; 303:C318-C327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fauconnier J, Lanner JT, Sultan A, Zhang SJ, Katz A, Bruton JD, Westerblad H. Insulin potentiates TRPC3‐mediated cation currents in normal but not in insulin‐resistant mouse cardiomyocytes. Cardiovasc Res. 2007; 73:376-385. [DOI] [PubMed] [Google Scholar]
- 44.Goel M, Sinkins WG, Zuo CD, Hopfer U, Schilling WP. Vasopressin‐induced membrane trafficking of TRPC3 and AQP2 channels in cells of the rat renal collecting duct. Am J Physiol Renal Physiol. 2007; 293:F1476-F1488. [DOI] [PubMed] [Google Scholar]
- 45.Goel M, Zuo CD, Schilling WP. Role of cAMP/PKA signaling cascade in vasopressin‐induced trafficking of TRPC3 channels in principal cells of the collecting duct. Am J Physiol Renal Physiol. 2010; 298:F988-F996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim EY, Alvarez‐Baron CP, Dryer SE. Canonical transient receptor potential channel (TRPC)3 and TRPC6 associate with large‐conductance Ca2+‐activated K+ (BKCa) channels: role in BKCa trafficking to the surface of cultured podocytes. Mol Pharmacol. 2009; 75:466-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smyth JT, Lemonnier L, Vazquez G, Bird GS, Putney JW., Jr Dissociation of regulated trafficking of TRPC3 channels to the plasma membrane from their activation by phospholipase C. J Biol Chem. 2006; 281:11712-11720. [DOI] [PubMed] [Google Scholar]
- 48.van Rossum DB, Oberdick D, Rbaibi Y, Bhardwaj G, Barrow RK, Nikolaidis N, Snyder SH, Kiselyov K, Patterson RL. TRP_2, a lipid/trafficking domain that mediates diacylglycerol‐induced vesicle fusion. J Biol Chem. 2008; 283:34384-34392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cipolla MJ, Smith J, Kohlmeyer MM, Godfrey JA. SKCa and IKCa channels, myogenic tone, and vasodilator responses in middle cerebral arteries and parenchymal arterioles: effect of ischemia and reperfusion. Stroke. 2009; 40:1451-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, Hamachi I, Mori Y. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci USA. 2009; 106:5400-5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao G, Bai XY, Xuan C, Liu XC, Jing WB, Novakovic A, Yang Q, He GW. Role of TRPC3 channel in human internal mammary artery. Arch Med Res. 2012; 43:431-437. [DOI] [PubMed] [Google Scholar]
- 52.Birnbaumer L. The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular ca(2+) concentrations. Annu Rev Pharmacol Toxicol. 2009; 49:395-426. [DOI] [PubMed] [Google Scholar]
- 53.de Wit C, Griffith TM. Connexins and gap junctions in the EDHF phenomenon and conducted vasomotor responses. Pflugers Arch. 2010; 459:897-914. [DOI] [PubMed] [Google Scholar]
- 54.Zygmunt PM, Hogestatt ED. Role of potassium channels in endothelium‐dependent relaxation resistant to nitroarginine in the rat hepatic artery. Br J Pharmacol. 1996; 117:1600-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong H, Jiang Y, Cole WC, Triggle CR. Comparison of the pharmacological properties of EDHF‐mediated vasorelaxation in guinea‐pig cerebral and mesenteric resistance vessels. Br J Pharmacol. 2000; 130:1983-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McNeish AJ, Sandow SL, Neylon CB, Chen MX, Dora KA, Garland CJ. Evidence for involvement of both IKCa and SKCa channels in hyperpolarizing responses of the rat middle cerebral artery. Stroke. 2006; 37:1277-1282. [DOI] [PubMed] [Google Scholar]
- 57.Singh BB, Lockwich TP, Bandyopadhyay BC, Liu X, Bollimuntha S, Brazer SC, Combs C, Das S, Leenders AG, Sheng ZH, Knepper MA, Ambudkar SV, Ambudkar IS. VAMP2‐dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist‐stimulated Ca2+ influx. Mol Cell. 2004; 15:635-646. [DOI] [PubMed] [Google Scholar]
- 58.Lockwich T, Pant J, Makusky A, Jankowska‐Stephens E, Kowalak JA, Markey SP, Ambudkar IS. Analysis of TRPC3‐interacting proteins by tandem mass spectrometry. J Proteome Res. 2008; 7:979-989. [DOI] [PubMed] [Google Scholar]
- 59.Di Giovanni J, Iborra C, Maulet Y, Leveque C, El Far O, Seagar M. Calcium‐dependent regulation of SNARE‐mediated membrane fusion by calmodulin. J Biol Chem. 2010; 285:23665-23675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012; 490:201-207. [DOI] [PMC free article] [PubMed] [Google Scholar]