

Abstract
Background
Chronic hypersecretion of the pancreatic hormone amylin is common in humans with obesity or prediabetic insulin resistance and induces amylin aggregation and proteotoxicity in the pancreas. We recently showed that hyperamylinemia also affects the cardiovascular system. Here, we investigated whether amylin aggregates interact directly with cardiac myocytes and whether controlling hyperamylinemia protects the heart.
Methods and Results
By Western blot, we found abundant amylin aggregates in lysates of cardiac myocytes from obese patients, but not in controls. Aggregated amylin was elevated in failing hearts, suggesting a role in myocyte injury. Using rats overexpressing human amylin in the pancreas (HIP rats) and control myocytes incubated with human amylin, we show that amylin aggregation at the sarcolemma induces oxidative stress and Ca2+ dysregulation. In time, HIP rats developed cardiac hypertrophy and left‐ventricular dilation. We then tested whether metabolites with antiaggregation properties, such as eicosanoid acids, limit myocardial amylin deposition. Rats were treated with an inhibitor of soluble epoxide hydrolase, the enzyme that degrades endogenous eicosanoids. Treatment doubled the blood concentration of eicosanoids, which drastically reduced incorporation of aggregated amylin in cardiac myocytes and blood cells, without affecting pancreatic amylin secretion. Animals in the treated group showed reduced cardiac hypertrophy and left‐ventricular dilation. The cardioprotective mechanisms included the mitigation of amylin‐induced cardiac oxidative stress and Ca2+ dysregulation.
Conclusions
The results suggest blood amylin as a novel therapeutic target in diabetic heart disease and elevating blood levels of antiaggregation metabolites as a pharmacological strategy to reduce amylin aggregation and amylin‐mediated cardiotoxicity.
Keywords: amyloid, calcium, circulation, diabetes mellitus, heart diseases
Introduction
The mechanisms by which obesity and prediabetic insulin resistance predispose to cardiac dysfunction are complex and not completely understood.1–9 We have recently8 identified toxic amylin deposits in heart samples from individuals with heart failure (HF) and obesity or type‐2 diabetes (T2D), but not in controls. However, determining the underlying cardiotoxic mechanism remains a key knowledge gap, especially since such deposits may represent a previously unknown potential therapeutic target. Here, we focus on (1) characterizing a possible link between cardiac amylin pathology and HF in humans with obesity; and (2) testing clinically relevant therapeutic strategies in an animal model of the pathological process.
Amylin, also known as islet amyloid polypeptide, is an amyloidogenic hormone cosecreted with insulin by pancreatic β‐cells.10 Amylin plays a paracrine role in restraining insulin secretion and is also known to bind to brain neuron receptors, inducing an anorexic effect.10 People with obesity or prediabetic insulin resistance show increased secretion of amylin.10–13 With increased secretion (hyperamylinemia), human amylin forms small aggregates (or oligomers) in the secretory vesicles of β‐cells14 and amyloid fibrils extracellularly, in the islet.10,14–18 The oligomerized amylin is cytotoxic, disrupts cellular membranes, and may also act as nonselective ion channels.14–18 Clumping together amylin oligomers of various sizes may form amyloid fibrils and plaques, which are believed to be relatively inert.14–18 Oligomerization of amylin induces oxidative19–20 and inflammatory21 stress in the pancreas, thus contributing to β‐cell apoptosis and development of T2D. The vast majority of patients with T2D show amylin deposition in pancreatic islets,10 demonstrating the exposure to hyperamylinemia. Recent studies8–9,22 indicate that hyperamylinemia may also contribute to the complications of diabetes by promoting toxic accumulation of amylin aggregates outside the pancreas, in kidneys,9 heart,8 and brain.22 In the heart, amylin deposition is specific to obesity or T2D.8 Neither nonfailing nor failing hearts from lean, nondiabetic patients showed amylin accumulation.8 Using a rat model of T2D expressing human amylin in the pancreas (the HIP rat), we found that cardiac accumulation of oligomerized amylin begins in prediabetes and is associated with diastolic dysfunction and cardiac hypertrophy.8 We have also shown that the transition to full‐blown T2D exacerbates amylin‐induced cardiac effects in HIP rats.23 In contrast, age‐ and blood glucose–matched rats expressing only wild‐type (nonamyloidogenic) rat amylin, which develop T2D associated with other pathological defects in the pancreatic β‐cells,24 display cardiac dysfunction after the onset of T2D.8,24 These studies8,24 suggest that myocardial accumulation of oligomerized amylin may accelerate diabetic heart dysfunction in humans.
Epoxyeicosatrienoic acids (EETs) are signaling molecules produced from arachidonic acid by cytochrome P450 epoxygenases25 and have several isomers with various functions.24–29 EETs are primarily degraded by soluble epoxide hydrolase (sEH), which transforms them to less lipophilic and more readily conjugated 1,2‐diols (dihydroxyeicosatrienoic acids).28 Pharmacological inhibition or genetic deletion of sEH elevates EETs, which was shown to have various therapeutic effects in animal models of metabolic disorders and cardiovascular disease.24,28–31 For example, we showed recently24 that pharmacologically increased levels of EETs improve glucose homeostasis in diabetic rats.24 Because some EET isomers have antiaggregation effects and reduce proteinaceous deposition on blood vessels,26–27 we hypothesized that increasing the blood levels of EETs may also limit the infiltration of toxic amylin aggregates in the heart.
The aims of the present study were twofold. First, we tested whether amylin aggregates attach to/incorporate into human cardiac myocytes and how incorporated amylin levels correlate with heart function (i.e., failing/nonfailing) and diabetes status in humans. Secondly, we used the HIP rat model to test the hypothesis that EETs limit cardiovascular accumulation of aggregated amylin, thus preserving myocardial structure and function. We anticipate that the results are of relevance to heart failure in diabetes.
Methods
Human Samples
Failing human hearts were obtained at the time of orthotopic heart transplantation at the Hospital of University of Pennsylvania. Nonfailing hearts from obese and lean individuals were from organ donation. Tissue specimens were obtained in accordance with Institutional Review Board approval, and informed consent was obtained prospectively in all cases. The study includes both transmural left ventricular myocardial samples and isolates of enriched cardiac myocytes obtained from 20 hearts divided into 4 pathologically distinct groups. O‐HF forms the group of obese patients (body mass index ≥30) who developed overt T2D within 1 year post‐transplantation (N=5), when all patients are treated with systemic corticosteroids as part of the standard immunosuppressive regimen. Hence, we speculated that these patients were in the prediabetic state at the time of heart transplant. This group included individuals with both ischemic and nonischemic cardiomyopathy. We also assessed the level of oligomerized amylin in myocyte lysates from nonfailing hearts of obese humans (O‐NF; N=6). Nonfailing hearts from healthy, nonobese (body mass index <30) individuals (L‐NF; N=4) and failing hearts from lean patients (L‐HF; N=5) without T2D served as negative controls. All patients in the L‐HF group had nonischemic dilated cardiomyopathy. All cardiac myocyte isolates used in the present study were also used in the cohorts reported in our previous publication.8
Experimental Animals and Treatment
In contrast to human amylin, rodent amylin does not form amyloid and is not cytotoxic.10 Therefore, to address mechanistically the impact of a “human” hyperamylinemia on myocardial structure and function, we used a diabetic rat model transgenic for human amylin (the HIP rat; Charles River Laboratory). HIP rats express human amylin in the pancreatic β‐cells on the insulin II promoter. By ≈5 months of age, they show impaired fasting glucose and have a ≈3‐fold increase of amylin secretion,32 similar to humans with hyperamylinemia. This causes amylin aggregation and deposition in pancreas32 and peripheral organs, including the heart, as we showed recently.8 Pancreatic deposition of amylin leads to gradual decline of β‐cell mass and T2D by 10 to 12 months of age.32 Age‐matched wild‐type (WT) littermates were used as nondiabetic controls. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and was approved by the Institutional Animal Care and Use Committees at University of Kentucky and University of California, Davis. A total of n=17 prediabetic male HIP rats (7 months of age) and n=15 age‐matched WT rats were used in this study.
To increase plasma eicosanoids levels, the animals were treated with APAU (1‐(1‐acetypiperidin‐4‐yl)‐3‐adamantanylurea, UC1153, AR9281), a sEH inhibitor synthesized by the method of Jones et al33 Animals in the APAU treatment group (HIP‐T group; N=11) received APAU in their drinking water for 13 weeks, starting from the onset of the prediabetic state (ie, when the blood glucose level was in the 150±30 mg/dL, or 8.3±1.5 mmol/L, range for 2 consecutive readings). APAU was formulated in polypropylene glycol at a concentration of 10 mg/mL with 30 minutes sonication and then added to drinking water at a final concentration of 1%. The animals had unrestricted access to water. Prediabetic HIP rats in the untreated group (HIP‐UT; n=6) received the same amount of polyethylene glycol, a neutral biocompatible polymer. We calculated the minimum sample size with a 2‐tail t test considering (1) the standardized mean difference in cardiac amylin level derived from Figure 4B in reference 8 (1.5); (2) α=0.05; and (3) 1−β=0.8. The calculation was performed using GPower 3.1.9.2 software (University of Dusseldorf) and yielded 9 animals per treatment group. Thus, our study was adequately powered.
Figure 4.
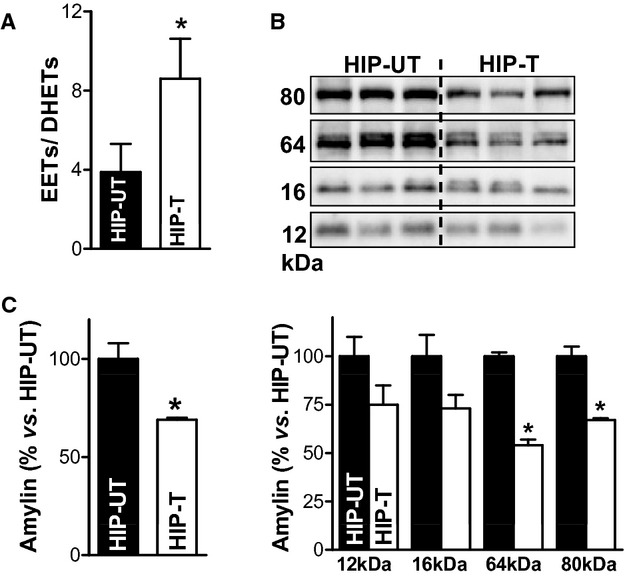
EETs limit accumulation of oligomeric amylin at cardiac myocyte sarcolemma. A, The ratio of total EETs to DHETs in plasma from APAU‐treated and untreated HIP rats at the end of the 13 weeks study period. B, Top panel shows representative (of 4 experiments) Western blot with an antiamylin antibody on ventricular myocyte lysates from treated (HIP‐T) vs (HIP‐UT) untreated HIP rats. Bottom panel displays the intensity analysis of the molecular weight bands corresponding to amylin trimers (≈12 kDa), tetramers (≈16 kDa), 16‐mers (≈64 kDa), and 20‐mers (≈80 kDa) in the top panel. C, Total oligomerized amylin in myocyte lysates from rats in HIP‐T and HIP‐UT groups obtained by integrating all molecular weight bands in (B). *P<0.05. APAU indicates (1‐(1‐acetypiperidin‐4‐yl)‐3‐adamantanylurea; DHETs, dihydroxyeicosatrienoic acids; EETs, epoxyeicosatrienoic acids; HIP, human amylin in the pancreas.
Nonfasting blood glucose levels were measured biweekly (morning time) with a glucometer (One Touch Blood Glucose Meter; LifeScan, Inc). Plasma levels of insulin and amylin (total) in fasted rats were measured by enzyme‐linked immunosorbent assay. At the end of the 13‐week treatment period, blood levels of APAU and EET were measured by liquid chromatography/tandem mass spectrometry as before.24
In Vivo Echocardiography
In vivo echocardiography was performed at the beginning and at the end of the 13‐week treatment period using M‐mode imaging as previously described.8
Immunochemistry
Western blot analysis and immunohistochemistry were used to test amylin accumulation in cardiac specimens. Western blot analysis was performed on tissue (heart and pancreas) homogenates and lysates from cardiac myocytes and blood cells. Tissue was homogenized in buffer containing 10 mmol/L Tris‐HCl, pH 7.4, 150 mmol/L NaCl, 0.1% sodium dodecyl sulfate, 1% Triton X‐100, 1% sodium deoxycholate, 5 mmol/L EDTA, 1 mmol/L NaF, 1 mmol/L sodium orthovanadate, and protease and phosphatase inhibitor cocktail (Calbiochem, San Diego, CA). Isolated cardiac myocytes were lysed in lysis buffer containing 1% NP‐40, 150 mmol/L NaCl, 10 mmol/L Tris‐HCl, 2 mmol/L EGTA, 50 mmol/L NaF, and protease and phosphatase inhibitor cocktail (Calbiochem). Blood samples were centrifuged at 2000 g to separate the plasma from the blood cells, which were then lysed with the buffer described above. For heart samples, equal loading was verified by reprobing with anti‐GAPDH (6C5) antibody (monoclonal, ab8245; Abcam Inc, Cambridge, MA). The primary antibody was a polyclonal antiamylin antibody (T‐4157; Bachem‐Peninsula Laboratories, San Carlos, CA) that recognizes both human and rat amylin (the latter with higher avidity8). The results were verified using a second antihuman amylin antibody (polyclonal, raised in rabbit, LS‐C20521; Lifespan Biosciences, Seattle, WA). To verify specific staining of protein bands, samples were loaded onto a gel in duplicate and, after blotting and blocking, the membrane was cut and one half was incubated with the antiamylin antibody while the other half was incubated without the primary. Both halves were then incubated with the secondary antibody (biotinylated goat antirabbit IgG antibody BA‐1000; Vector Laboratories, Burlingame, CA) and developed and imaged together. No staining was apparent in the absence of the primary antibody (not shown; for a detailed analysis, see reference 8). The specificity of the antiamylin staining in immunohistochemistry studies was demonstrated by incubating human pancreas sections only with the secondary antibody (not shown; for a detailed analysis, see reference 8).
Signal intensity analysis was performed in Image J. For each gel, we averaged the signal intensity of the corresponding bands for the control samples. Then, we normalized the signal intensity in all lanes to this average. This procedure was repeated on at least 4 gels. For each sample, the normalized signal intensity was averaged.
Quantitative Real‐Time Polymerase Chain Reaction
Quantitative real‐time polymerase chain reaction was used to assess the expression level of human and rat amylin in the pancreas and heart as previously described.22
Adult Cardiac Myocyte Isolation and Measurement of Cell Size
Rats were anesthetized by intraperitoneal injection of pentobarbital (≈1 mg/g). When deep anesthesia was reached, hearts were excised quickly, placed on a Langendorff perfusion apparatus, and perfused with buffer containing 1 mg/mL collagenase. When the heart became flaccid, the left ventricular tissue was cut into small pieces, dispersed, and filtered. Human cardiac myocytes were also isolated by collagenase perfusion, as described previously.34 The standard Tyrode's solution used in experiments with live myocytes contained (in mmol/L): 140 NaCl, 4 KCl, 1 MgCl2, 10 glucose, 5 HEPES, and 1 CaCl2 (pH=7.4). All experiments were done at room temperature (23 to 25°C). To measure myocyte size, the midsection was imaged with a confocal microscope and we measured the cell length and width using Image J. Myocyte area was calculated both as length×width and by tracing the external membrane of the cell and measuring the area inside with ImageJ, which produced essentially similar results.
Incubation of Isolated Myocytes With Recombinant Human Amylin
The amylin oligomerization reaction was started in saline solution at 37°C using 50 μmol/L recombinant human amylin, which is the concentration at which this peptide forms oligomers under physiological conditions in the pancreas.15–16 Myocytes were then incubated with the preformed amylin oligomers for 2 hours at room temperature.
Electron Microscopy Imaging
Transmission electron microscopy imaging was done as previously described.24 Briefly, myocytes were frozen rapidly and embedded in Eponate‐12 resin. Blocks were cut into 70‐nm‐thick sections. Serial sections were collected on slot grids covered with a 50‐nm Formvar support film and visualized on the transmission electron microscope (JEM‐1230; JEOL) operated at 120 keV. Images of each section (5 images per block) were recorded at ≈2 μm defocus on a 2048×2048‐pixel charge‐coupled device camera (TVIPS, Gauting, Germany) and at nominal magnification of either ×1000 or ×2500, yielding an effective pixel size of 11 or 4.4 nm, respectively.
Intracellular Ca2+ Measurements
Intracellular Ca2+ measurements were performed with Fluo‐4. Data are expressed as F/F0, where F0 is the fluorescence signal in resting myocytes, to make the signal independent of the amount of dye loaded into myocytes.
ROS Production Measurements
Myocytes were loaded with the fluorescent indicator 5‐(and‐6)‐chloromethyl‐20,70‐dichlorodihydrofluorescein diacetate acetyl ester (CM‐H2DCFA; Invitrogen), 10 μmol/L for 15 minutes, and imaged with a confocal microscope (excitation=488 nm, emission >505 nm). Myocytes were electrically stimulated at 1 Hz in the presence of isoproterenol (1 μmol/L), and reactive oxygen species (ROS) production was calculated as the slope of the increase in CM‐H2DCFA fluorescence.
Statistical Analysis
Data are expressed as mean±SEM. For data that passed the D'Agostino and Pearson normality test, statistical discriminations were performed using (1) 2‐tailed unpaired Student t test when comparing 2 groups; (2) 1‐way analysis of variance with the Bonferroni's post‐hoc test when comparing multiple groups; and (3) 2‐way analysis of variance when comparing multiple groups for multiple conditions. When the sample size was too small to perform the normality test, data were analyzed with nonparametric tests (Mann–Whitney when comparing 2 columns, Kruskal–Wallis followed by Dunn's post‐test when comparing multiple columns). In Figures 3E, 7, 8, and 9B, data from multiple myocytes from the same animal were averaged and the resulting values per animal were averaged for each group. Statistical analysis was done in GraphPad Prism version 5.0 for Windows (GraphPad Software, La Jolla, CA). P<0.05 was considered significant.
Figure 3.
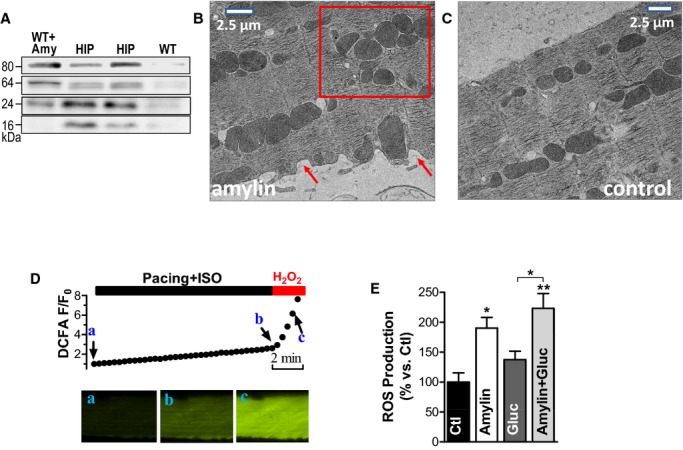
The interaction of oligomeric amylin with sarcolemma causes oxidative stress in cardiac myocytes. A, Western blot with an antiamylin antibody on lysates from control myocytes incubated with preformed amylin aggregates (WT+Amy), HIP rats myocytes (HIP), and control WT cells (WT). Oligomers were preformed by incubating 50 μmol/L exogenous human amylin for 2 hours at room temperature. B and C, Representative transmission EM images of WT rat cardiac myocytes incubated with 50 μmol/L oligomerized human amylin for 2 hours (B) and control myocytes (C). Arrows point to alterations in sarcolemmal structure. The square guides the eye to the mitochondrial disarrangement. D, Representative example of ROS production measurement with the fluorescent indicator CM‐H2DCFA. Myocytes loaded with CM‐H2DCFA were electrically stimulated at 1 Hz in the presence of isoproterenol (ISO, 1 μmol/L), which increased CM‐H2DCFA fluorescence (from point a to point b), indicating ROS (H2O2) production. At the end, pacing was stopped and H2O2 (1 mmol/L) was washed in to verify the specificity of the signal (increased fluorescence from b to c). ROS production was measured as the slope of fluorescence increase between the points a and b. E, ROS production was compared in control myocytes (Ctl; 11 myocytes from 3 rats) and myocytes incubated with preformed amylin oligomers (amylin; 8 myocytes, 3 rats), 400 mg/dL glucose (gluc; 7 myocytes, 3 rats), and a combination of glucose (400 mg/dL) and preformed amylin oligomers (amylin+gluc; 7 myocytes, 3 rats). *P<0.05, **P<0.01. CM‐H2DCFA indicates 5‐(and‐6)‐chloromethyl‐20,70‐dichlorodihydrofluorescein diacetate acetyl ester; EM, electron microscopy; HIP, human amylin in the pancreas; ROS, reactive oxygen species; WT, wild‐type.
Figure 7.
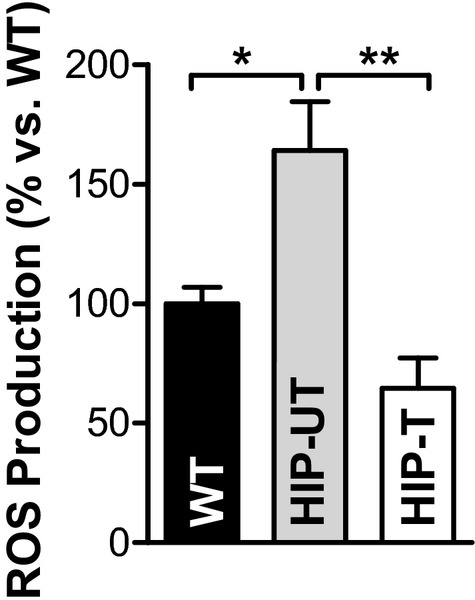
APAU treatment reduces amylin oligomer‐induced oxidative stress. ROS production measured with CM‐H2DCFA in myocytes from WT (8 myocytes from 4 rats), untreated HIP (HIP‐UT; 13 cells, 4 rats), and treated HIP (HIP‐T; 9 cells, 3 rats) rats. *P<0.05, **P<0.01. APAU indicates (1‐(1‐acetypiperidin‐4‐yl)‐3‐adamantanylure; CM‐H2DCFA,5‐(and‐6)‐chloromethyl‐20,70‐dichlorodihydrofluo‐rescein diacetate acetyl ester; ROS, reactive oxygen species; WT, wild‐type.
Figure 8.
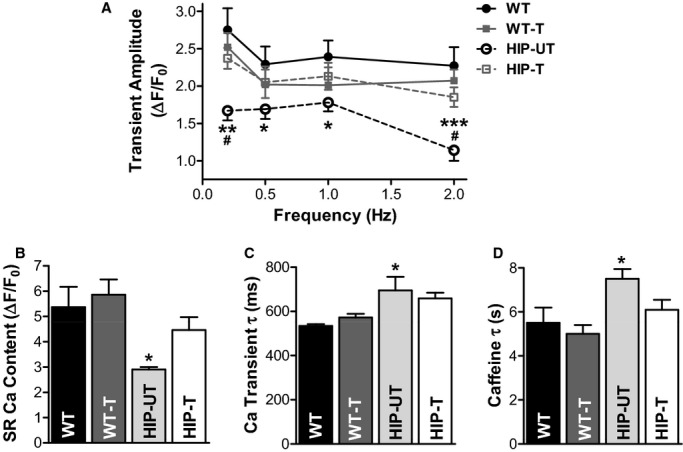
Treatment reduces cardiac Ca2+ cycling alterations in HIP rats. A, Mean Ca2+ transient amplitude, (B) SR Ca2+ content, (C) decay time of Ca2+ transient induced by electrical stimulation at 0.5 Hz, and (D) decay time of caffeine‐induced Ca2+ transient in cardiac myocytes from WT (15 cells, 4 rats), APAU‐treated WT (17 myocytes, 3 rats), untreated HIP (16 cells, 4 rats), and treated HIP rats (25 cells, 4 rats). *P<0.05, **P<0.01, ***P<0.001 vs untreated WT. #P<0.05 vs treated HIP. APAU indicates (1‐(1‐acetypiperidin‐4‐yl)‐3‐adamantanylurea; HIP, human amylin in the pancreas; SR, sarcoplasmic reticulum; WT, wild‐type.
Figure 9.
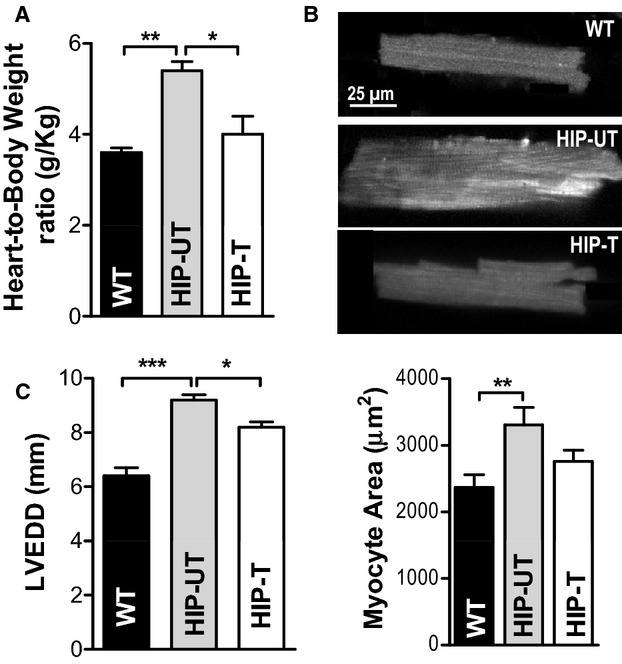
Treatment reduces cardiac hypertrophy and left‐ventricular dilation in HIP rats. A, Heart‐weight‐to‐body‐weight ratio in WT, untreated HIP, and treated HIP rats. B, Top—representative examples of myocytes from WT, HIP‐UT, and HIP‐T groups; Bottom—average surface area (length×width) for WT, untreated HIP, and treated HIP rat myocytes. C, Left‐ventricular end‐diastolic diameter in WT, untreated HIP, and treated HIP rats at the end of the 13‐week study period. *P<0.05, **P<0.01, ***P<0.001. HIP indicates human amylin in the pancreas; LVEDD, left‐ventricular end‐diastolic diameter; WT, wild‐type.
Results
Oligomerized Amylin Incorporates Within Myocytes in Prediabetic Humans and HIP Rats
To test whether aggregated amylin incorporates directly within cardiac myocytes and how the level of incorporated amylin correlates with heart function (failing/nonfailing) and diabetes status in humans, we analyzed left ventricular myocyte lysates from 4 pathologically distinct groups of patients. The O‐HF group included obese patients (body mass index ≥30) with heart failure who developed overt T2D during the first year post‐transplantation. Patients in this group were likely in the prediabetic state at the time of heart transplant. The O‐NF group consisted of obese individuals without heart failure. Nonfailing hearts from lean individuals (L‐NF) and failing hearts from nonobese (body mass index <30) nondiabetic patients (L‐HF) served as negative controls. Western blot analysis showed that the level of oligomerized amylin (ie, trimers, tetramers, and octamers) was significantly higher in myocyte lysates from the O‐HF group compared to those from L‐NF and L‐HF control groups (Figure 1A and 1B). Aggregated amylin was even elevated in the O‐NF group (Figure 1A and 1B), indicating an early process of amylin aggregation. The Western blot experiments were performed under several conditions (with and without dithiothreitol and sodium dodecyl sulfate) to examine soluble and insoluble fractions of aggregated amylin in myocytes (Figure 1C). The results show that nonreducing and nondenaturing conditions preserve large amylin aggregates (>150 kDa) that may “intrinsically” form in cells and tissues. Adding dithiothreitol and sodium dodecyl sulfate in the sample buffer and gel breaks amylin aggregates, resulting in lower‐molecular‐weight bands as in Figure 1A and 1B. The lower‐molecular‐weight amylin aggregates observed in Figure 1A and 1B remain insoluble to dithiothreitol and sodium dodecyl sulfate and are likely formed by intermolecular interactions.
Figure 1.
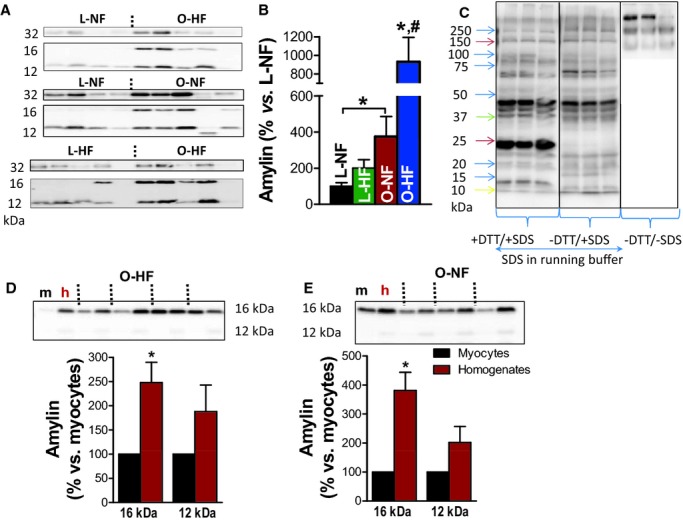
Oligomerized amylin incorporates within cardiac myocytes in prediabetic humans. A and B, Western blot analysis with an antiamylin antibody on left ventricular myocyte lysates from human hearts demonstrates the presence of amylin trimers (12 kDa), tetramers (16 kDa), and octamers (32 kDa) attached to the sarcolemma or inside the myocytes (representative of 4 experiments). Nonfailing hearts (L‐NF) and failing hearts (L‐HF) from nonobese patients served as negative controls. C, Western blot experiments with antiamylin antibody on HIP myocyte lysates performed with and without DTT and SDS in the sample and running buffers. D and E, Western blot analysis with an antiamylin antibody showing the presence of amylin aggregates (trimers and tetramers) in both left ventricle protein homogenates (h) and myocyte lysates (m) from the same patient from O‐HF (C) and O‐NF (D) groups. *P<0.05 vs L‐NF. #P<0.05 vs O‐NF. DTT indicates dithiothreitol; HIP, human amylin in the pancreas; O‐HF, obese patients with heart failure; O‐NF, obese patients with nonfailing hearts; SDS, sodium dodecyl sulfate.
Figure 1D and 1E shows that amylin trimers and tetramers are present in both left ventricular protein homogenates and myocyte lysates from the same patient. This result suggests that amylin aggregates are also prominent in nonmyocytes and/or the extracellular matrix. In fact, the level of aggregated amylin is higher in protein homogenates, suggesting that aggregated amylin is even more prevalent outside of the myocytes. Indeed, immunohistochemistry data show amylin‐positive plaques in heart parenchyma from diabetic patients (Figure 2A; blue arrows) but not in healthy hearts from nondiabetic individuals (Figure 2C). Serial staining with amylin and Congo Red suggested that amylin may form amyloid‐like structures in the diabetic human heart (Figure 2B). The presence of amylin outside of cardiac myocytes may not be static and could promote cardiac myocyte uptake. For example, amylin is known to bind to the calcitonin gene‐related peptide receptor,10 which could mediate the amylin uptake in cardiac myocytes.
Figure 2.
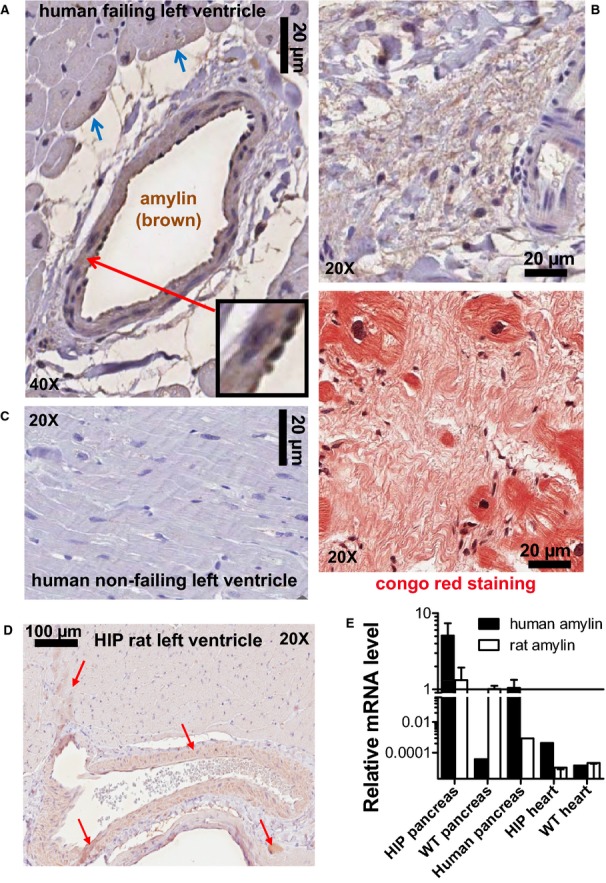
Amylin incorporates in blood vessels and parenchyma in failing hearts from diabetic humans and HIP rat hearts. A, Immunohistochemistry analysis of failing hearts from T2D patients with an antiamylin antibody shows amylin deposits (brown) incorporated in the blood vessel wall, perivascular space, and on the sarcolemma of neighboring cardiac myocytes (blue arrows). The inset shows amylin deposition in the basement membranes and co‐localization with neutrophils. B, Amylin (brown) is present in atherosclerotic lesions. Staining of the next section with Congo red (lower panel) indicates that amylin deposition may form amyloid‐like structures (pink) within myocardial interstices. C, Cardiac parenchyma from nonfailing human hearts shows no amylin deposits. D, Immunohistochemistry with an antiamylin antibody shows that amylin (brown; red arrows) incorporates into blood vessel walls and heart parenchyma in HIP rat. E, Quantitative real‐time PCR shows no presence of human amylin mRNA in the HIP rat heart. Note the logarithmic scale and scale break. N=8 HIP rat heart samples and N=4 human pancreas and rat samples were analyzed in this study. WT rat heart samples (N=8) served as negative controls. HIP indicates human amylin in the pancreas; PCR, polymerase chain reaction; T2D, type 2 diabetes; WT, wild‐type.
Amylin also accumulates in coronary arteries and cardiac parenchyma in the HIP rat (Figure 2D). Transcript analysis by quantitative real‐time polymerase chain reaction showed no presence of human amylin mRNA in HIP rat hearts (Figure 2E; note logarithmic scale and scale break). Thus, the amylin incorporated in HIP rat hearts comes from the circulation, originating in the pancreas, as in humans. Hence, the HIP rat is a clinically relevant animal model to examine potential therapeutic strategies to limit cardiovascular accumulation of aggregated amylin.
Amylin Deposition Causes Oxidative Stress in Cardiac Myocytes
The interaction of aggregated amylin with sarcolemma may affect myocyte function through various mechanisms. We have previously shown that aggregated amylin incorporates in the sarcolemma, inducing sarcolemmal Ca2+ leak and activation of Ca2+‐mediated hypertrophy signaling pathways.8 Amylin aggregation is also known as a potent generator of oxidative stress in both cell culture and animal models.10,19–20 Therefore, we measured the production of ROS in WT rat cardiac myocytes incubated with preformed amylin aggregates for 2 hours. The level and characteristic molecular weights of amylin aggregates found in lysates from myocytes incubated under these conditions are comparable to those found in HIP rat myocyte lysates (Figure 3A). Thus, short‐term incubation of myocytes with exogenous human amylin aggregates mimics the in vivo attachment of amylin aggregates to myocytes in HIP rat hearts. Moreover, the interaction of oligomerized human amylin with cardiac myocytes alters cardiac myocyte structure. Electron microscopy images of isolated WT cardiac myocytes incubated with exogenous human amylin aggregates show significant alterations of cardiac myocyte structure, including sarcolemmal damage (Figure 3B, arrows) and mitochondrial disarrangement (Figure 3B, square). In contrast, control WT cardiac myocytes present intact sarcolemmal and internal structures (Figure 3C).
Myocyte ROS production was assessed with CM‐H2DCFA. Dye‐loaded cells were electrically stimulated in the presence of isoproterenol (ISO) and the slope of the fluorescence increase was taken as a measure of ROS production (Figure 3D). At the end, pacing was stopped and H2O2 (1 mmol/L) was washed in to verify the specificity of the signal. Incubation with amylin greatly increased myocyte ROS production (by 87±21%; Figure 3E). By comparison, incubation of cardiac myocytes with 400 mg/dL (22.2 mmol/L) glucose, for the same duration, increased ROS production by only 40±12% (Figure 3E). Thus, in addition to altering Ca2+ cycling,8 the interaction of amylin aggregation with the sarcolemma induces sarcolemmal damage and oxidative stress.
Eicosanoids Limit Cardiovascular Accumulation of Aggregated Amylin
Because EETs reduce proteinaceous deposition on blood vessels and have antiaggregation properties,26–27 we hypothesized that increasing the EETs level in the blood may limit the infiltration of aggregated amylin in the heart. To elevate blood levels of EETs, HIP rats were treated with the sEH inhibitor APAU33 for 13 weeks (HIP‐T group). The study was started when rats were in the prediabetic state (nonfasting blood glucose in the 150 to 200 mg/dL range [http://www.nlm.nih.gov/medlineplus/ency/article/000313.htm]). Age‐matched HIP rats in the untreated group received the same amount of polyethylene glycol, a neutral biocompatible polymer (HIP‐UT group). sEH inhibition increased plasma levels of EETs in treated HIP rats (Figure 4A and Table 1), with the EETs/dihydroxyeicosatrienoic acids ratio being almost double in treated compared to untreated HIP rats at the end point of the study (Figure 4A). Thus, APAU efficiently increases EETs level, consistent with our previous results in a different diabetic model.24 Western blot analysis of ventricular myocyte lysates from treated and untreated HIP rats shows that the treatment significantly reduced the incorporation of aggregated amylin into cardiac myocytes (Figure 4B and 4C). Thus, elevating the blood level of pro‐fibrinolytic EETs lowers cardiac accumulation of aggregated amylin.
Table 1.
Plasma Levels of EETs and Inactive DHETs in Untreated and APAU‐Treated HIP Rats
| Treated | Untreated | |
|---|---|---|
| Epoxyeicosatrienoic acids, nmol/L | ||
| 14,15‐EET | 18.3±5.2 | 7.6±2.8 |
| 11,12‐EET | 18.2±6.8 | 8.3±3.6 |
| 8,9‐EET | 6.7±1.8 | 4.2±1.9 |
| 5,6‐EET | 31.6±9.5 | 13.7±6.7 |
| Dihydroxyeicosatrienoic acids, nmol/L | ||
| 14,15‐DHET | 1.8±0.1 | 2.3±1.0 |
| 11,12‐DHET | 2.2±0.3 | 3.1±1.6 |
| 8,9‐DHET | 1.1±0.2 | 1.9±0.4 |
| 5,6‐DHET | 3.2±0.5 | 3.4±0.6 |
APAU indicates (1‐(1‐acetypiperidin‐4‐yl)‐3‐adamantanylurea; DHETs, dihydroxyeicosatrienoic acids; EETs, epoxyeicosatrienoic acids.
Reduced cardiac accumulation of aggregated amylin in HIP‐T rats is not due to a lower level of amylin secreted in the blood. Plasma amylin (Figure 5A), along with insulin (Figure 5B) and glucose (Figure 5C), were compared in animals from WT, HIP‐T, and HIP‐UT groups at various time points. Animals in HIP‐T and HIP‐UT groups had similar plasma levels of amylin (and insulin), suggesting that amylin secretion from pancreatic β‐cells is not altered by inhibiting sEH. Intriguingly, the levels of aggregated amylin in the blood pellet (ie, amylin aggregates attached to blood cells) are significantly lower in animals from the HIP‐T group (Figure 6A). Thus, EETs may either dissociate the amylin aggregates and/or inhibit their attachment to membranes. To test this hypothesis, we incubated WT cardiac myocytes with preformed amylin aggregates in the presence or absence of 5 μmol/L 14,15‐EET. Figure 6B shows that co‐incubation with 14,15‐EET considerably reduced the deposition of amylin in the sarcolemma. Adding 14,15‐EET after incubating the myocytes with amylin aggregates for 2 hours disentangled some small amylin aggregates from the sarcolemma (the Amy/EET column in Figure 6C). Intriguingly, preconditioning the myocytes by exposure to 14,15‐EET prior to incubation with aggregated amylin potentiated the amylin attachment to the sarcolemma (the EET/Amy column in Figure 6C). To test whether EETs are also able to dissociate amylin aggregates, we investigated the effect of 14,15‐EET in solution (Figure 6D). Co‐incubation of aggregated amylin with 5 μmol/L 14,15‐EET reduced the levels of amylin dimers and trimers in solution. Similar effects were produced by plasmin (Figure 6D), another protein known for its profibrinolytic properties. These results suggest that EETs limit cardiovascular accumulation of aggregated amylin by 2 likely mechanisms: (1) binding to amylin aggregates, which blocks the attachment to cell membranes; and (2) disentangling circulating amylin aggregates, consistent with the antiaggregation properties of EETs.26–27 Still, further investigation is needed to elucidate the mechanisms by which EETs reduce amylin accumulation.
Figure 5.
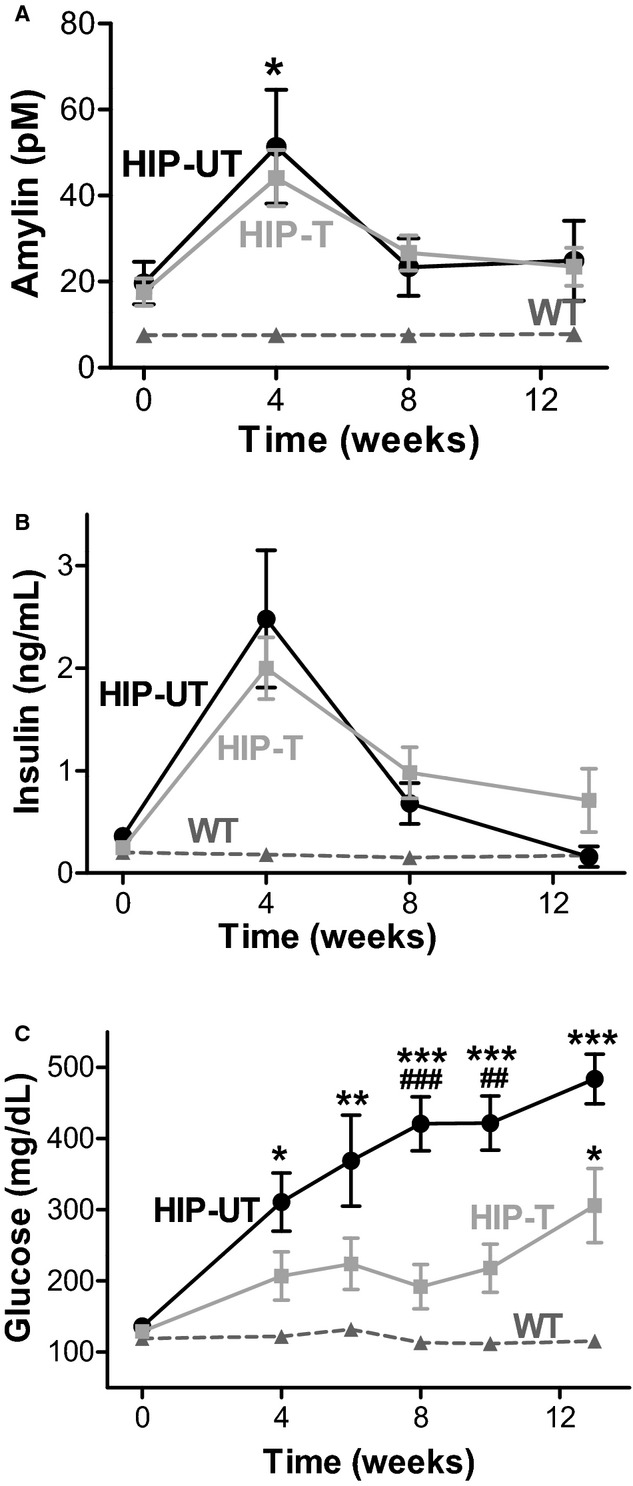
Plasma amylin (A), insulin (B), and glucose (C) levels in WT, HIP‐T, and HIP‐UT animal groups at various time points during the study period. The initial increase of plasma amylin and insulin is due to the onset of prediabetic insulin resistance in HIP rats, which triggers compensatory secretion of the 2 hormones (ie, hyperinsulinemia and hyperamylinemia). N=10 HIP rats for treated group, N=7 untreated HIP rats, and N=10 nondiabetic WT rats. ***P<0.001 vs WT; ###P<0.001 vs HIP‐treated. HIP indicates human amylin in the pancreas; WT, wild‐type.
Figure 6.
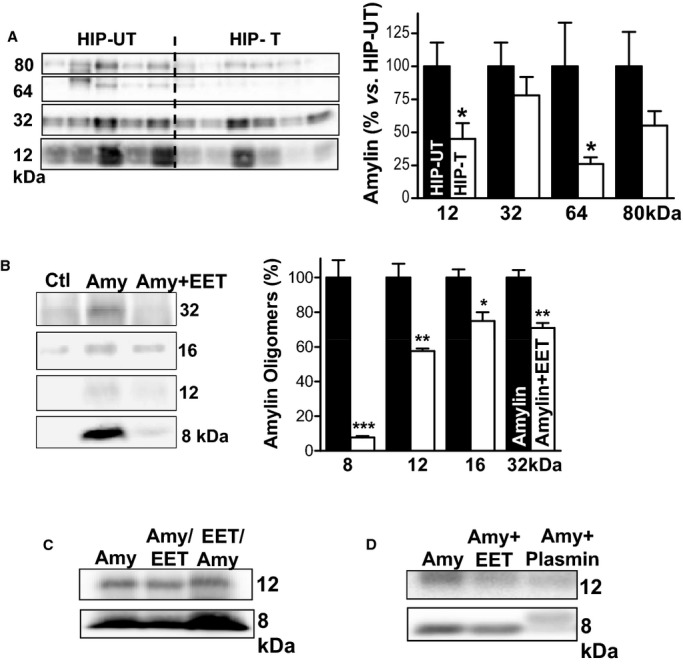
EETs reduce the level of amylin oligomers circulating in the blood. A, Representative (of 4 experiments) Western blot with an antiamylin antibody on whole blood cell lysates from HIP rats in the treated (T) and untreated (UT) groups. Bottom panel shows the band intensity analysis for these experiments. B, Representative (of 3 experiments) Western blot with an antiamylin antibody on ventricular myocyte lysates from control rats. Before lysis, myocytes were incubated for 2 hours in the absence (Ctl) or in the presence of 50 μmol/L recombinant human amylin (Amy). Myocytes in the Amy+EET column were incubated with 50 μmol/L human amylin and 5 μmol/L 14,15‐EET. Co‐incubation with EETs reduced the attachment of amylin oligomers to cardiac myocytes. Bottom panel shows the band intensity analysis. C, The effect of 5 μmol/L 14,15‐EET on amylin attachment to the sarcolemma when added before (EET/Amy) or after (Amy/EET) incubation of myocytes with oligomerized amylin. D, Representative Western blot showing that 14,15‐EETs and plasmin (both 5 μmol/L) limit oligomerization of recombinant human amylin in saline solution (50 μmol/L amylin; 2 to 3 hours incubation time). *P<0.05; **P<0.01; ***P<0.001. EETs indicates epoxyeicosatrienoic acids; HIP, human amylin in the pancreas.
Rodent amylin does not form amyloid.10 However, rodent amylin may interact with human amylin in the transgenic rat forming differently packed, mixed rodent amylin–human amylin oligomers. A different packing of mixed amylin oligomers may explain the variability in molecular weights of amylin aggregates identified in HIP rats (Figure 4B) versus amylin oligomers formed in an ex vivo formulation (Figure 3A) versus oligomerized amylin identified in cardiac tissue from humans (Figure 1A).
Limiting Myocardial Accumulation of Aggregated Amylin Reduces the Oxidative Stress and Preserves Myocyte Ca2+ Cycling
The mechanism underlying the beneficial effects of reducing myocardial accumulation of toxic aggregated amylin is likely multimodal. Because aggregated amylin is a source of cardiac oxidative stress and Ca2+ dysregulation, we investigated the treatment outcome on these 2 pathological processes.
ROS production was significantly increased in myocytes from untreated HIP rats versus WT (by 70±16%, Figure 7), in agreement with data in Figure 3E showing oxidative stress in myocytes acutely exposed to amylin oligomers. Lowering cardiac accumulation of aggregated amylin by APAU treatment greatly reduced ROS production in HIP rat myocytes (Figure 7). Alterations in myocyte [Ca2+]i regulation play an important role in the pathophysiology of diabetic heart disease.35–38 We have previously shown that dysregulation of myocyte Ca2+ cycling is one of the early, direct effects induced by accumulation of aggregated amylin in the heart.8,35 We now compared Ca2+ transients and sarcoplasmic reticulum Ca2+ load in cardiac myocytes from HIP‐UT and HIP‐T rat groups and age‐matched WT rats at the end of the 13‐week treatment period. Ca2+ transients were significantly smaller in myocytes from HIP‐UT rats compared to WT rats for all stimulation frequencies (0.2, 0.5, 1, and 2 Hz; Figure 8A), indicating impaired myocyte contractility in HIP‐UT rats. Deficient myocyte contractility in HIP‐UT rats is also suggested by the lower sarcoplasmic reticulum Ca2+ content, which was measured as the amplitude of the Ca2+ transient induced by 10 mmol/L caffeine (Figure 8B). Limiting the incorporation of aggregated amylin in cardiac myocytes restored both Ca2+ transient amplitude and sarcoplasmic reticulum Ca2+ load (Figure 8A and 8B). sEH inhibition had no significant effect in WT rats (WT‐T group; Figure 8A and 8B). The decline of Ca2+ transients induced by electrical stimulation and caffeine is mediated mainly by the sarcoplasmic reticulum Ca2+‐ATPase and the sarcolemmal Na+/Ca2+ exchanger, respectively. In both cases, Ca2+ transient decay was significantly slower in myocytes from HIP‐UT rats versus WT rats (Figure 8C and 8D), in agreement with our prior data8 showing reduced sarcoplasmic reticulum Ca2+‐ATPase and Na+/Ca2+ exchanger expression in diabetic HIP rats. Both effects were partially mitigated by reducing cardiac accumulation of aggregated amylin (Figure 8C and 8D).
Thus, the cardioprotective mechanisms of reducing accumulation of toxic amylin aggregates in the heart include the mitigation of amylin‐induced oxidative stress and myocyte Ca2+ alterations.
sEH Inhibition Reduces Cardiac Hypertrophy and Left‐Ventricular Dilation
Using the HIP rat model, we have previously shown that cardiac accumulation of aggregated amylin leads to cardiac hypertrophy and left‐ventricular dilation even in the prediabetic state.8,35 At the end of the treatment period, heart‐weight‐to‐body‐weight (HW/BW) ratio (Figure 9A) and myocyte surface area (Figure 9B) were markedly increased in HIP‐UT rats compared to WT littermates, consistent with the development of cardiac hypertrophy. The larger myocyte area was mainly due to an increase in cell length (125±5 μm in HIP versus 101±6 μm in WT rats), while cell width was not significantly affected (26.3±1.8 μm in HIP versus 22.8±1.5 μm in WT rats), consistent with eccentric hypertrophy. In addition, M‐mode echocardiography showed significantly increased left ventricular end‐diastolic diameter in HIP‐UT rats compared to WT rats (Figure 9C). Full echocardiography data are included in Table 2. Limiting cardiovascular accumulation of aggregated amylin significantly reduced both cardiac hypertrophy (Figure 9A and 9B) and left ventricular remodeling (Figure 9C) in HIP‐T rats. Thus, controlling hyperamylinemia by reducing cardiovascular accumulation of aggregated amylin with EETs preserves myocardial structure and function in HIP rats.
Table 2.
Echocardiographic Parameters Before Starting the Treatment (Baseline) and at the End of the 13‐Week Treatment Period in Treated (n=8) and Untreated (n=6) HIP Rats
| Baseline | Treated | Untreated | |
|---|---|---|---|
| LVEDD, mm | 8.0±0.4 | 8.2±0.2 | 9.0±0.2* |
| LVESD, mm | 4.4±0.3 | 4.4±0.2 | 5.1±0.3 |
| Fractional shortening, % | 45.9±2.4 | 46.6±1.7 | 43.3±2.6 |
| Ejection fraction, % | 81.8±2.2 | 82.4±1.5 | 78.8±2.7 |
The mean echo parameters at baseline were similar between the treated and untreated groups, so those data were pulled together. LVEDD indicates left‐ventricular end‐diastolic diameter; LVESD, left‐ventricular end‐systolic diameter. Fractional shortening=([LVEDD−LVESD]/LVEDD)×100%.
P<0.05.
Discussion
Present results indicate that amylin aggregation affects directly cardiac myocytes in humans and that this pathological process starts in prediabetes (Figure 1A and 1B). Moreover, significantly higher levels of amylin incorporated in cardiac myocytes from O‐HF individuals suggest a link of hyperamylinemia and amylin aggregation with the development of heart disease in humans. These data are consistent with the pathological role of amylin aggregation within (1) pancreas,10,14,19–21 kidneys,9 and brain22 in humans; and (2) pancreas32 and heart8 in HIP rats. Using the HIP rat model, we now demonstrated that limiting cardiovascular accumulation of aggregated amylin by increasing eicosanoid activity protects the heart (Figures 7 through 9). This study provides proof of the concept that reducing the level of aggregated amylin in the cardiovascular system ameliorates amylin's deleterious effects on heart function.
Cytotoxicity of “human” hyperamylinemia is associated with the high propensity of human amylin to form amyloids,10 which is common to all amyloid‐assembling proteins.39 Previous data40 demonstrated that cardiac myocyte‐restricted expression and accumulation of the amyloidogenic peptide polyglutamine induces heart failure in mice. Presenilin, another amyloidogenic peptide found in the heart, is also associated with cardiac pathologies in humans.41 Similarly, adding exogenous human amylin in cell culture promotes fibril growth at the cell membrane,18 altering cell viability and function, an effect that is also suggested by present results (Figure 3B). Not surprisingly, even small human amylin aggregates (trimers, tetramers) can induce cytotoxicity, which may be triggered by their great ability to quickly and massively insert into cell membranes and leading to the formation of nonselective pores.8 For example, we have previously8 shown that incubation of intact cardiac myocytes with human amylin for 1 to 2 hours leads to infiltration of small amylin oligomers in the sarcolemma, which considerably increased sarcolemmal Ca2+ leak and altering vital Ca2+‐mediated signaling. In contrast, rodent amylin is not amyloidogenic due to 6 proline substitutions in the amylin amino acid sequence.42 Hypersecretion of rodent amylin in mice does not induce amyloid deposition, β‐cell loss, or T2D.43 Using the HIP rat model, we determined mechanistically the impact of a “human” hyperamylinemia on the heart and demonstrated that controlling the “human” hyperamylinemia protects the heart. Hence, our study may potentially reveal a novel and important therapeutic strategy in reducing/preventing cardiac complications of T2D in humans.
The present results indicate that pro‐fibrinolytic EETs can control hyperamylinemia by mitigating cardiovascular accumulation of aggregated amylin. The level of aggregated amylin incorporated within cardiac myocytes is considerably lower in HIP‐T rats compared to HIP‐UT rats (Figure 4). Moreover, aggregated amylin was also reduced in blood cell lysates from HIP‐T rats compared to HIP‐UT rats (Figure 6). Because animals in treated and untreated groups have similar plasma levels of amylin (Figure 5A) and insulin (Figure 5B), we infer that (1) treatment does not alter pancreatic β‐cell secretory function; and (2) EETs interact directly with circulating amylin aggregates and increase their solubility. In support to this proposed mechanism, we found that EETs block both the aggregation of recombinant human amylin in saline solution (Figure 6D) and the acute incorporation of preformed amylin aggregates in myocytes (Figure 6B). These results are consistent with the profibrinolytic and antiaggregation properties of EETs.26–27
The mechanism underlying the beneficial effects of EETs on the cardiovascular system is likely multimodal. EETs mediate endothelium‐dependent vasodilation,31 which lowers the afterload and may contribute to reduced hypertrophy in the treated rats, and are anti‐inflammatory.31 Elevated EETs also attenuate hyperglycemia,24,29–30 which we observed here as well (Figure 5C). Indeed, only 40% of HIP‐T rats developed overt hyperglycemia (ie, nonfasting blood glucose >200 mg/dL) at the end of the 13 weeks of treatment. In contrast, all rats in the HIP‐UT group progressed rapidly to overt hyperglycemia. It is also possible that EETs reduced the vascular deposition of amylin oligomers. Nevertheless, our data indicate that reducing the incorporation of aggregated amylin in cardiac myocytes is a key component of the multimodal mechanism31 of cardioprotection mediated by EETs in T2D (Figure 10), by limiting amylin‐induced cardiac oxidative stress (Figure 7) and myocyte Ca2+ dysregulation (Figure 8). At the whole‐heart level, HIP rats show abnormalities in left ventricular structure (Figure 9). Such abnormalities typically precede the onset of heart failure in individuals with cardiac infiltrative diseases44 and are also important pathological changes leading to the development of diabetic heart disease.2–8 Elevated EETs blood levels preserve myocardial structure and function in HIP rats. Our results are particularly interesting in the context of recent data45 showing cardioprotection in T2D by dietary supplementation with docosahexaenoic acid (DHA), a naturally derived omega‐3 fatty acid and counterpart of EETs. EETs and DHA may have similar mechanisms of cardioprotection that involve reducing pathological cardiac hypertrophy.
Figure 10.
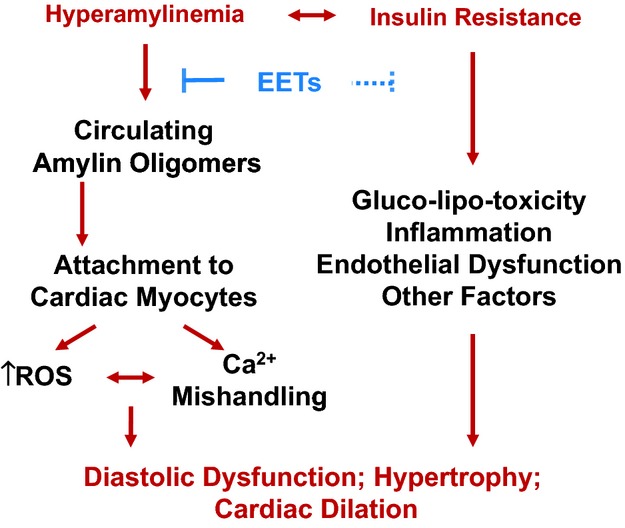
Proposed mechanism: hyperamylinemia, a condition secondary to insulin resistance, promotes amylin oligomer attachment/incorporation to cardiac myocytes, leading to heart dysfunction. The mechanism underlying the beneficial effects of EETs on heart function is likely multimodal and includes an efficient diminution of infiltration of oligomerized amylin in cardiac myocytes. EETs indicates epoxyeicosatrienoic acids; ROS, reactive oxygen species.
In summary, our study provides evidence in support of the hypothesis that hyperamylinemia is directly implicated in diabetic heart injury in humans and presents proof of concept data for reducing the level of aggregated amylin in the cardiovascular system and its deleterious effects on cardiac function in an animal model. Our study also points to circulating aggregated amylin as a novel therapeutic target in diabetic cardiovascular disease and elevating EETs as an innovative therapeutic strategy to limit circulation of amylin aggregates and thus, the risk of cardiovascular disease in patients with obesity and insulin resistance.
Sources of Funding
Work was supported by NIH (1R01‐HL118474‐01A1 to F. Despa; R01‐HL109501 to S. Despa; R01‐HL85844 and R01‐HL85727 to Chiamvimonvat; 5R01‐HL061483 to Taegtmeyer; R01‐HL089847 and R01‐HL105993 to Margulies; R01‐ES002710, P42‐ES004699 and U24‐DK097154 to Hammock), AHA (13GRNT16470034 to F. Despa), ADA (1‐13‐IN‐70 to F. Despa), and NSF (CBET 1133339 to F. Despa). Hammock is a George and Judy Marcus Senior Fellow of the American Asthma Foundation. Harris is supported by NIH T32 Training Grant in Basic and Translational Cardiovascular Science (T32‐HL86350).
Disclosures
Hammock is associated with EicOsis in developing soluble epoxide hydrolase inhibitors for human therapy.
References
- 1.Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol. 2006; 68:123-158. [DOI] [PubMed] [Google Scholar]
- 2.Young LH, Wackers FJ, Chyun DA, Davey JA, Barrett EJ, Taillefer R, Heller GV, Iskandrian AE, Wittlin SD, Filipchuk N, Ratner RE, Inzucchi SE. Cardiac outcomes after screening for asymptomatic coronary artery disease in patients with type 2 diabetes: the DIAD study: a randomized controlled trial. JAMA. 2009; 301:1547-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopaschuk GD. Metabolic abnormalities in the diabetic heart. Heart Fail Rev. 2002; 7:149-159. [DOI] [PubMed] [Google Scholar]
- 4.Peterson LR. Obesity and insulin resistance: effects on cardiac structure, function, and substrate metabolism. Curr Hypertens Rep. 2006; 8:451-456. [DOI] [PubMed] [Google Scholar]
- 5.Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Endocr Metab Disord. 2010; 11:31-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taegtmeyer H, McNulty P, Young ME. Adaptation and maladaptation of the heart in diabetes: part I: general concepts. Circulation. 2002; 105:1727-1733. [DOI] [PubMed] [Google Scholar]
- 7.Guha A, Harmancey R, Taegtmeyer H. Nonischemic heart failure in diabetes mellitus. Curr Opin Cardiol. 2008; 23:241-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Despa S, Margulies KB, Chen L, Knowlton AA, Havel PJ, Taegtmeyer H, Bers DM, Despa F. Hyperamylinemia contributes to cardiac dysfunction in obesity and diabetes: a study in humans and rats. Circ Res. 2012; 110:598-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gong W, Liu ZH, Zeng CH, Peng A, Chen HP, Zhou H, Li LS. Amylin deposition in the kidney of patients with diabetic nephropathy. Kidney Int. 2007; 72:213-218. [DOI] [PubMed] [Google Scholar]
- 10.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011; 91:795-826. [DOI] [PubMed] [Google Scholar]
- 11.Johnson KH, O'Brien TD, Jordan K, Westermark P. Impaired glucose tolerance is associated with increased islet amyloid polypeptide (IAPP) immunoreactivity in pancreatic beta cells. Am J Pathol. 1989; 135:245-250. [PMC free article] [PubMed] [Google Scholar]
- 12.Enoki S, Mitsukawa T, Takemura J, Nakazato M, Aburaya J, Toshimori H, Matsukara S. Plasma islet amyloid polypeptide levels in obesity, impaired glucose tolerance and non‐insulin‐dependent diabetes mellitus. Diabetes Res Clin Pract. 1992; 15:97-102. [DOI] [PubMed] [Google Scholar]
- 13.Westermark GT, Steiner DF, Gebre‐Medhin S, Engstrom U, Westermark P. Pro islet amyloid polypeptide (ProIAPP) immunoreactivity in the islets of Langerhans. Ups J Med Sci. 2000; 105:97-106. [DOI] [PubMed] [Google Scholar]
- 14.Gurlo T, Ryazantsev S, Huang CJ, Yeh MW, Reber HA, Hines OJ, O'Brien TD, Glabe CG, Butler PC. Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol. 2010; 176:861-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lukinius A, Wilander E, Westermark GT, Engstrom U, Westermark P. Co‐localization of islet amyloid polypeptide and insulin in the β‐cell secretory granules of the human pancreatic islets. Diabetologia. 1989; 32:240-244. [DOI] [PubMed] [Google Scholar]
- 16.Wiltzius JJ, Sievers SA, Sawaya MR, Eisenberg D. Atomic structures of IAPP (amylin) fusions suggest a mechanism for fibrillation and the role of insulin in the process. Protein Sci. 2009; 18:1521-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luca S, Yau WM, Leapman R, Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid‐state NMR. Biochemistry. 2007; 46:13505-13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engel MF, Khemtemourian L, Kleijer CC, Meeldijk HJ, Jacobs J, Verkleij AJ, de Kruijff B, Killian JA, Hoppener JW. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc Natl Acad Sci USA. 2008; 105:6033-6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janciauskiene S, Ahren B. Fibrillar islet amyloid polypeptide differentially affects oxidative mechanisms and lipoprotein uptake in correlation with cytotoxicity in two insulin‐producing cell lines. Biochem Biophys Res Commun. 2000; 267:619-625. [DOI] [PubMed] [Google Scholar]
- 20.Zraika S, Hull RL, Udayasankar J, Aston‐Mourney K, Subramanian SL, Kisilevsky R, Szarek WA, Kahn SE. Oxidative stress is induced by islet amyloid formation and time‐dependently mediates amyloid‐induced beta cell apoptosis. Diabetologia. 2009; 52:626-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westwell‐Roper C, Dai DL, Soukhatcheva G, Potter KJ, van Rooijen N, Ehses JA, Verchere CB. IL‐1 blockade attenuates islet amyloid polypeptide‐induced proinflammatory cytokine release and pancreatic islet graft dysfunction. J Immunol. 2011; 187:2755-2765. [DOI] [PubMed] [Google Scholar]
- 22.Jackson K, Barisone GA, Diaz E, Jin LW, DeCarli C, Despa F. Amylin deposition in the brain: a second amyloid in Alzheimer's disease? Ann Neurol. 2013; 74:517-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycemia activates CaMKII and arrhythmias by O linked glycosylation. Nature. 2013; 502:372-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guglielmino K, Jackson K, Harris TR, Vu V, Dong H, Dutrow G, Evans JE, Graham J, Cummings BP, Havel PJ, Chiamvimonvat N, Despa S, Hammock BD, Despa F. Pharmacological inhibition of soluble epoxide hydrolase provides cardioprotection in hyperglycemic rats. Am J Physiol Heart Circ Physiol. 2012; 303:H853-H862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oliw EH. Oxygenation of polyunsaturated fatty acids by cytochrome P450 monooxygenases. Prog Lipid Res. 1994; 33:329-354. [DOI] [PubMed] [Google Scholar]
- 26.Fitzpatrick FA, Ennis MD, Baze ME, Wynalda MA, McGee JE, Liggett WF. Inhibition of cyclooxygenase activity and platelet aggregation by epoxyeicosatrienoic acids: influence of stereochemistry. J Biol Chem. 1986; 261:15334-15338. [PubMed] [Google Scholar]
- 27.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004; 43:55-90. [DOI] [PubMed] [Google Scholar]
- 28.Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res. 2005; 44:1-51. [DOI] [PubMed] [Google Scholar]
- 29.Luo P, Chang HH, Zhou Y, Zhang S, Hwang SH, Morisseau C, Wang CY, Inscho EW, Hammock BD, Wang MH. Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J Pharmacol Exp Ther. 2010; 334:430-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luria A, Bettaieb A, Xi Y, Shieh GJ, Liu HC, Inoue H, Tsai HJ, Imig JD, Haj FG, Hammock BD. Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc Natl Acad Sci USA. 2011; 108:9038-9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012; 92:101-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matveyenko AV, Butler PC. Beta‐cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type 2 diabetes. Diabetes. 2006; 55:2106-2114. [DOI] [PubMed] [Google Scholar]
- 33.Jones PD, Tsai HJ, Do ZN, Morisseau C, Hammock BD. Synthesis and SAR of conformationally restricted inhibitors of soluble epoxide hydrolase. Bioorg Med Chem Lett. 2006; 16:5212-5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dipla K, Mattiello JA, Jeevanandam V, Houser SR, Margulies KB. Myocyte recovery after mechanical circulatory support in humans with end‐stage heart failure. Circulation. 1998; 97:2316-2322. [DOI] [PubMed] [Google Scholar]
- 35.Despa S, Bers DM, Despa F. Accumulation of islet amyloid polypeptide (IAPP) oligomers in the heart in type 2 diabetes alters Ca cycling in myocytes. Circulation. 2010; 122:A18345 [Google Scholar]
- 36.Lebeche D, Davidoff AJ, Hajjar RJ. Interplay between impaired calcium regulation and insulin signaling abnormalities in diabetic cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008; 5:715-724. [DOI] [PubMed] [Google Scholar]
- 37.Netticadan T, Temsah RM, Kent A, Elimban V, Dhalla NS. Depressed levels of Ca2+‐cycling proteins may underlie sarcoplasmic reticulum dysfunction in the diabetic heart. Diabetes. 2001; 50:2133-2138. [DOI] [PubMed] [Google Scholar]
- 38.Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, Richard S, Gomez AM. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. 2006; 55:608-615. [DOI] [PubMed] [Google Scholar]
- 39.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006; 75:333-366. [DOI] [PubMed] [Google Scholar]
- 40.Pattison JS, Sanbe A, Maloyan A, Osinska H, Klevitsky R, Robbins J. Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation. 2008; 117:2743-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gianni D, Li A, Tesco G, McKay KM, Moore J, Raygor K, Rota M, Gwathmey JK, Dec GW, Aretz T, Leri A, Semigran MJ, Anversa P, Macgillivray TE, Tanzi RE, Del Monte F. Protein aggregates and novel presenilin gene variants in idiopathic dilated cardiomyopathy. Circulation. 2010; 121:1216-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci USA. 1990; 87:5036-5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang CJ, Haataja L, Gurlo T, Butler AE, Wu X, Soeller WC, Butler PC. Induction of endoplasmic reticulum stress‐induced beta‐cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. Am J Physiol Endocrinol Metab. 2007; 293:1656-1662. [DOI] [PubMed] [Google Scholar]
- 44.Seward JB, Casaclang‐Verzosa G. Infiltrative cardiovascular diseases: cardiomyopathies that look alike. J Am Coll Cardiol. 2010; 55:1769-1779. [DOI] [PubMed] [Google Scholar]
- 45.Khairallah RJ, O'Shea KM, Brown BH, Khanna N, Des Rosiers C, Stanley WC. Treatment with docosahexaenoic acid, but not eicosapentaenoic acid, delays Ca2+‐induced mitochondria permeability transition in normal and hypertrophied myocardium. J Pharmacol Exp Ther. 2010; 335:155-162. [DOI] [PMC free article] [PubMed] [Google Scholar]