

Abstract
Background
Angiopoietin‐like‐2 (angptl2) is produced by several cell types including endothelial cells, adipocytes and macrophages, and contributes to the inflammatory process in cardiovascular diseases. We hypothesized that angptl2 impairs endothelial function, and that lowering angptl2 levels protects the endothelium against high‐fat diet (HFD)‐induced fat accumulation and hypercholesterolemia.
Methods and Results
Acute recombinant angptl2 reduced (P<0.05) acetylcholine‐mediated vasodilation of isolated wild‐type (WT) mouse femoral artery, an effect reversed (P<0.05) by the antioxidant N‐acetylcysteine. Accordingly, in angptl2 knockdown (KD) mice, ACh‐mediated endothelium‐dependent vasodilation was greater (P<0.05) than in WT mice. In arteries from KD mice, prostacyclin contributed to the overall dilation unlike in WT mice. After a 3‐month HFD, overall vasodilation was not altered, but dissecting out the endothelial intrinsic pathways revealed that NO production was reduced in arteries isolated from HFD‐fed WT mice (P<0.05), while NO release was maintained in KD mice. Similarly, endothelium‐derived hyperpolarizing factor (EDHF) was preserved in mesenteric arteries from HFD‐fed KD mice but not in those from WT mice. Finally, the HFD increased (P<0.05) total cholesterol–to–high‐density lipoprotein ratios, low‐density lipoprotein–to–high‐density lipoprotein ratios, and leptin levels in WT mice only, while glycemia remained similar in the 2 strains. KD mice displayed less triglyceride accumulation in the liver (P<0.05 versus WT), and adipocyte diameters in mesenteric and epididymal white adipose tissues were smaller (P<0.05) in KD than in WT fed an HFD, while inflammatory gene expression increased (P<0.05) in the fat of WT mice only.
Conclusions
Lack of angptl2 expression limits the metabolic stress induced by an HFD and maintains endothelial function in mice.
Keywords: adipokines, endothelium‐derived relaxing factors, inflammation, isolated arteries
Introduction
Dietary imbalance is well known to cause obesity favoring with time, the development of insulin resistance, dyslipidemia, diabetes and ultimately atherosclerosis.1 Altered levels of cholesterol, especially high low‐density lipoprotein (LDL)–to–high‐density lipoprotein (HDL) ratios, as well as heightened insulin levels in diabetes, promote inflammation and endothelial dysfunction, which are at the root of atherogenesis.2 Recently, a member of the angiopoietin‐like (angptl) family, angiopoietin‐like‐2 (angptl2), has been identified as 1 of the key inflammatory mediators that regulate obesity‐related insulin sensitivity,3 dyslipidemia, and atherogenesis.4–5 Angptl2 seems to play a major pro‐inflammatory role in a variety of pathologies, including atherosclerosis,4–5 diabetes,6 abdominal aortic aneurysm,7 neointimal hyperplasia,8 rheumatoid arthritis,9 dermatomyositis,10 and even tumor progression.11 Increased angptl2 expression has been reported in endothelial cells from chronic atherosclerotic smokers,12 while its circulating level correlates with adiposity,3 C‐reactive protein levels,3 and tumor necrosis factor (TNF)α levels.8 Little is known, however, about the role of angptl2 in lipid profiling and endothelial function. It was suggested that angptl2 replenishment could restore insulin sensitivity and improve lipid levels with decreased serum triglycerides (TGs) and free fatty acids (FFAs) in genetically diabetic (db/db) mice13; in contrast, another study in overweight subjects revealed lower circulating angptl2 levels with lifestyle intervention in association with changes in TG metabolism.14 Our recent study, which used infusion of recombinant angptl2 in preatherosclerotic young mice, further increased cholesterol and LDL in the plasma,4 suggesting deleterious effects of angptl2 on lipid profiling. In contrast, no changes in lipid profiles of angptl2 knockout mice fed a high‐fat diet (HFD) compared with wild‐type (WT) mice were reported.3,5 Taken together, there are discrepancies in recent literature regarding the role of angptl2 in lipid handling. In terms of endothelial function, a recent study demonstrated that endothelium‐specific overexpression of angptl2 in mice led to vasodilatory dysfunction and vascular inflammation.5 In the same study, it was also shown that in angptl2‐deficient mice, HFD‐induced endothelial dysfunction was ameliorated in the aorta, which was associated with greater expression levels of phospho–endothelial nitric oxide synthase (eNOS) compared with WT mice.5 With the knowledge that endothelial cells express and produce angptl24–5,12 and that the endothelium is a dynamic and complex organ, with diverse phenotypes depending on vascular beds, it is important to better characterize the role of angptl2 in regulating endothelial function.
Healthy vascular function involves the balance of endothelium‐derived relaxing factors (EDRFs) and contracting factors, while imbalance of these contributes to endothelial dysfunction. There are 3 major EDRFs—nitric oxide (NO),15 prostacyclin (PGI2),16 and endothelium‐derived hyperpolarizing factor (EDHF).17 In conductance arteries, NO is the main EDRF, while in resistance arteries, the main contributor to dilation is EDHF.18 Reduced endothelium‐dependent vasodilation due to impairment of 1 of these EDRFs contributes to endothelial dysfunction,19–20 which can ultimately lead to cardiovascular disease. We and others reported, however, that the expression of the EDHF pathway compensated for the decreased NO‐ and PGI2‐dependent vasodilatory contribution in femoral arteries from dyslipidemic mice21 and carotid arteries from rabbits22 but then deteriorated with age. In eNOS−/− mice, the loss of NO during acetylcholine (ACh)‐ and flow‐mediated vasodilation is compensated by the expression of EDHF,23–24 while the contribution of PGI2 is increased.25 Additional compensatory pathways such as increased contribution of dilatory H2O2 have also been reported in coronary arteries from patients with coronary artery disease, which may eventually contribute to the endothelial dysfunction associated with metabolic stress.18 The vascular endothelium is therefore plastic and adapts to the metabolic environment up to a decompensation phase revealing its irreversible damage and dysfunction. Based on the pro‐inflammatory4–5 and pro‐oxidative26 properties of angptl2, we hypothesized that angptl2 modulates endothelial function and that lowering angptl2 levels protects the endothelium against HFD‐induced fat accumulation and hypercholesterolemia.
To test our hypothesis, we examined EDRF contribution and lipid handling using our newly generated angptl2 knockdown (KD) mice, fed either a regular diet (RD) or an HFD, an established method to induce adiposity, metabolic stress, and endothelial dysfunction.27 Our results suggest that lowering angptl2 is beneficial for the vascular endothelium by maintaining its respective EDRF contribution in conductance and resistance arteries, in addition to a more favorable lipid profile in KD mice fed an HFD. To the best of our knowledge, this is the first report of the impact of angptl2 in the contribution of the various EDRFs and their resistance against a stress induced by an HFD.
Materials and Methods
Animals
All animal experiments were performed in accordance with the “Guide for the Care and Use of Experimental Animals of the Canadian Council on Animal Care” and the “Guide for the Care and Use of Laboratory Animals” of the US National Institutes of Health (NIH Publication No. 85‐23, revised 1996) and was approved by the Montreal Heart Institute Ethics Committee (ET 2010‐62‐1). Generation of the angptl2 KD mouse model was achieved through a microinjection of a construct generated via retroviral gene trap vectors (developed at Texas A&M Institute for Genomic Medicine) performed in C57Bl/6J mice (Figure 1A) purchased from The Jackson Laboratory. A β‐geo cassette was inserted between bp 5305 and 5390 of the angptl2 gene. KD mice were subsequently bred at the Institute for Research in Immunology and Cancer (Montreal, Quebec, Canada). All mice used in this study were genotyped by PCR analysis of genomic DNA isolated from ear clips to select both KD and WT animals (see Table 1 for primer sequences). Negligible levels of angptl2 mRNA and protein levels were confirmed in various tissues (Figure 1B and 1C). In characterizing fasting plasma profile and endothelial function in mice at 3 to 4 months of age, angptl2+/+ littermates were used as WT and no significant differences were observed between angptl2+/+ littermates and C57Bl/6J WT mice. Subsequently, WT mice purchased from The Jackson Laboratory were used for the diet study. Male mice were used for all experiments. Mice were fed ad libitum either a regular diet (RD, 2018; Harlan Teklad Laboratories) or a high‐fat diet (HFD, TD.88137; Harlan Teklad Laboratories), starting at 3 months until 6 months of age. Mice were kept under standard conditions (24°C; 12:12‐hour light/dark cycle), and during the 3 months of diet treatment, blood pressure and heart rate were recorded weekly by using tail‐cuff plethysmography (Kent Scientific Corporation), after training to limit stress, as previously described.28 Mice were fasted 16 hours before sacrifice for experiments by using terminal anesthesia (44 mg/kg ketamine and 2.2 mg/kg xylazine). Plasma and tissues (liver, adipose tissues, heart, soleus muscle) were collected and kept at −80°C, while the femoral artery and mesenteric arterial bed were harvested and placed in ice‐cold physiological saline solution (pH 7.4, in mmol/L: NaCl 119, KCl 4.7, KH2PO4 1.18, MgSO4 1.17, NaHCO3 24.9, CaCl2 1.6, EDTA 0.023, and glucose 10) for endothelial function studies.21,29 Liver and adipose tissue samples were immediately fixed in formaldehyde and paraffin embedded for subsequent hematoxylin‐eosin (H&E) staining. A segment of the femoral artery was immediately embedded in OCT for subsequent dihydroethidium (DHE, D7008; Sigma Aldrich) staining.
Figure 1.
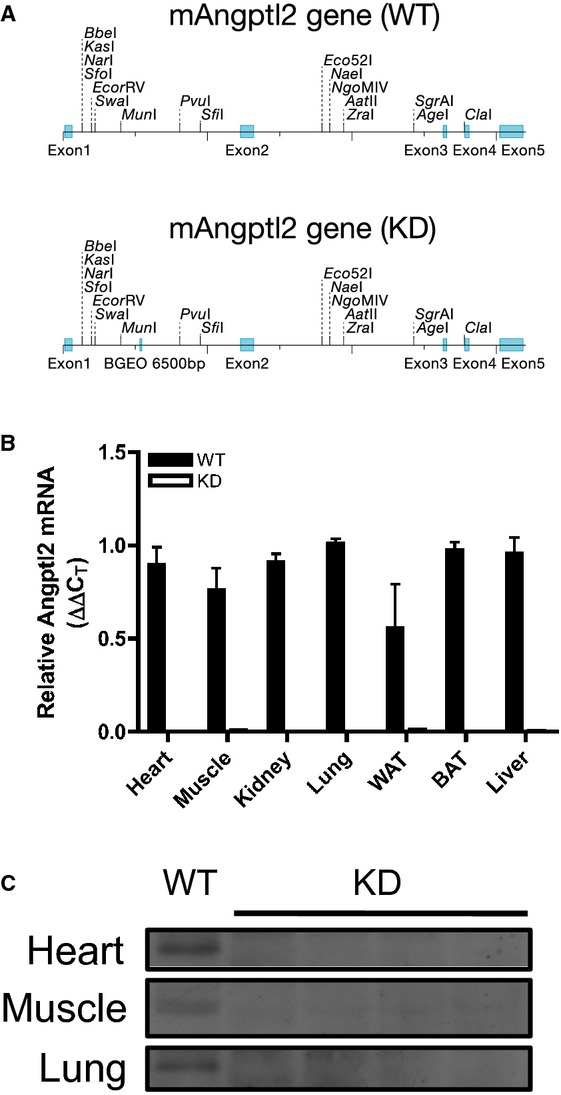
A, Schematic representation showing insertion of a promoterless trapping β‐geo cassette of 6500 bp in size into the mouse angptl2 locus, downstream of exon 1; top and bottom representation show angptl2 wild‐type (WT) and knock‐down (KD) scheme, respectively. B, Verification of angptl2 knock‐down in various mouse tissues by qPCR analysis; white adipose tissue (WAT), brown adipose tissue (BAT); n=3 to 4. C, Verification of angptl2 knock‐down in various mouse tissues by Western blot. qPCR indicates quantitative polymerase chain reaction.
Table 1.
Primer Sequences Used in Quantitative RT‐PCR
| Gene | Forward Sequence (5′ to 3′) | Reverse Sequence (5′ to 3′) |
|---|---|---|
| Angplt2 | GATCCAGAGTGACCAGAATC | TCTCAGGCTTCACCAGGTAG |
| Angptl2 V76 | CTTGCAAAATGGCGTTACTTAAGC | CCAATAAACCCTCTTGCAGTTGC |
| TNFα | TGATCCGCGACGTGGAACTGG | CGACGTGGGCTACAGGCTTGTCA |
| IL‐6 | CCATAGCTACCTGGAGTACATGA | GTCCTTAGCCACTCCTTCTGTGA |
| TGFβ | ATTCCTGGCGTTACCTTGG | CCTGTATTCCGTCTCCTTGG |
| LPL | GGCTCTGCCTGAGTTGTAGAA | TCACTCGGATCCTCTCGATGA |
| Adiponectin | GTCAGTGGATCTGACGACACCAA | ATGCCTGCCATCCAACCTG |
| Leptin | CAGGATCAATGACATTTCACACAC | CTGGTCCATCTTGGACAAACTC |
| Cyclophilin A | CCGATGACGAGCCCTTGG | GCCGCCAGTGCCATTATG |
| HSL | GGCACAGACCTCTAAATCCC | CCGCTCTCCAGTTGAACC |
| SREBP1c | GAACAGACACTGGCCGAGATG | GAGGCCAGAGAAGCAGAAGAGAAG |
| SREBP2 | GTTCTGGAGACCATGGAG | AAACAAATCAGGGAACTCTC |
| Angptl3 | AGCACCAAGAACTACTCCCC | ATAAACGGCAGAGCAGTCGG |
| Angptl4 | TCCGTGGGGACCTTAACTGT | GTAGCGGCCCTTCCATGTTT |
| Citrate synthase | GCCAGTGCTTCTTCCACGAAT | CATGCCACCGTACATCATGTC |
| Cyp7a1 | AACGATACACTCTCCACCTTTG | CTGCTTTCATTGCTTCAGGG |
| PPARα | CTATTCGGCTGAAGCTGGTGTA | CAGGTCGTGTTCACAGGTAAGA |
| HMG‐CoA reductase | AGTACATTCTGGGTATTGCTGG | ACTCGCTCTAGAAAGGTCAATC |
| CD36 | GGCCAAGCTATTGCGACATGA | CAGATCCGAACACAGCGTAGA |
| FXR | TGGAGAACTCAAAATGACTCAGG | CTTTTGTAGCACATCAAGCAGG |
| Sirtuin‐1 | ATCCAGCTCAGGTGGAGGAAT | TTGACCGATGGACTCCTCACT |
| PPARγ | CCTGAAGCTCCAAGAATACC | GGTTCTTCATGAGGCCTGTT |
FXR indicates farnesoid X receptor; HMG‐CoA, 3‐hydroxy‐3‐methylglutaryl coenzyme‐A; HSL, hormone‐sensitive lipase; LPL, lipoprotein lipase; PPAR, peroxisome proliferator‐activated receptor; SREBP, sterol regulatory element binding protein; TGF, transforming growth factor; TNF, tumor necrosis factor; IL, interleukin.
Plasma Parameters
Plasma lipid profile (total cholesterol, LDL cholesterol, HDL cholesterol), glucose, and TG levels were measured at the Biochemistry Laboratory of the Montreal Heart Institute (Montreal, Quebec, Canada). Adiponectin (MRP300; R&D Systems), leptin (90030; Crystal Chem), FFAs (K612‐100; BioVision), fasting insulin (80‐INSMSU‐E01; Alpco Diagnostics), and angptl2 (sE91919Mu; Uscn Life Science Inc) were quantified by using ELISA kits according to the manufacturers' protocol.
Western Blots
Proteins isolated from mouse tissues (heart, soleus muscle, and lung) in lysis buffer (50 mmol/L Tris‐HCl, pH 7.45, 5 mmol/L EDTA, 10 mmol/L EGTA, 1% v/v Triton) were subjected to SDS‐PAGE followed by Western blotting to detect angptl2 (1:200, AF2084; R&D Systems).
Endothelial Function of the Femoral Artery According to Pressurized Arteriography
Segments of 2 to 3 mm of the left or right gracilis artery were dissected in ice‐cold physiological saline solution; surrounding fat and tissues were removed, after which the segment was cannulated at both ends (average internal diameter=283±3 μm; 165 segments) and pressurized at 80 mm Hg under no‐flow conditions in a pressurized arteriograph (Living Systems Instrumentation) as described previously.21 The artery segment was aerated with 12% O2/5% CO2/83% N2 and equilibrated at 37°C for 45 minutes before the addition of phenylephrine (PE, 1 to 3 μmol/L) to obtain preconstriction of 30% to 50% of maximal diameter and single cumulative concentration‐response curves to ACh (1 nmol/L to 30 μmol/L). The acute effects (1 hour) of angptl2‐Glutathione S‐transferase (GST) (50 nmol/L) on ACh‐induced dilation, combined or not with the antioxidant N‐acetylcysteine (NAC; 10 μmol/L), were assessed and compared with exposure to an equivalent aliquot of the last dialysis bath used for purification of the recombinant protein (Tris‐buffered Saline EDTA [TBSE]; 50 mmol/L Tris‐base, 150 mmol/L NaCl, 1 mmol/L EDTA). Recombinant angptl2‐GST protein was produced as detailed previously.4 For other studies of endothelial function, Nω‐nitro‐l‐arginine (LNNA, 100 μmol/L), indomethacin (Indo, 10 μmol/L), or the combination of both drugs was placed in the bath throughout equilibration and experiment, to inhibit NOS or cyclooxygenase or to reveal EDHF, respectively. To study vascular smooth muscle cell function, the endothelium was removed by passing an air bubble through the lumen of the artery and confirmed by loss of response to ACh. After incubation, PE (0.1 nmol/L to 30 μmol/L) was used to preconstrict the artery, followed by single cumulative concentration‐response curves to sodium nitroprusside (0.1 nmol/L to 30 μmol/L).
Fluorescence Studies
Isolated femoral artery segments of 2 mm from 6‐month‐old WT or angptl2 KD mice fed with either an RD or HFD were incubated in oxygenated physiological saline solution at 37°C along with 10 μmol/L 4,5‐diaminoflorescein diacetate (DAF‐2, a fluorescent dye for NO detection30) with or without LNNA (100 μmol/L) for 30 minutes, after which the artery segment was washed 3 times with physiological saline solution, preconstricted with 3 μmol/L PE, and dilated with 1 μmol/L ACh. During the experiment, changes in fluorescence intensities reflecting NO production during vasodilation were measured, as previously described.30
Oxidative Stress Quantification in the Femoral Artery
Frozen femoral artery segments in OCT were cut into 7‐μm‐thick sections, and sections were double stained with 5 μmol/L DHE and 2 μmol/L To‐Pro‐3 (T3605; Molecular Probes), as previously described.31 DHE fluorescence was visualized by using confocal microscopy (Zeiss LSM 510; Carl Zeiss; objective ×20) in which DHE was excited with the HeNe laser at 543 nm and resulting emitted light was collected between 565 and 615 nm, while acquisition settings were kept constant for all samples. ImageJ (National Institutes of Health) software was used to analyze DHE fluorescence intensities based on this equation: I=Σ I/A, where I is the DHE‐fluorescence intensity, Σ I is the summation of To‐Pro‐3 nuclei stain intensities, and A is the total area of the nuclei.
Endothelial Function of Mesenteric Arteries According to Wire Myography
Mesenteric arteries (third‐order branches) were dissected in ice‐cold physiological saline solution. Segments of 2 mm in length were mounted on 20‐μm tungsten wires in microvessel myographs (IMF, University of Vermont, Burlington) as previously described.29 Mesenteric segments were first equilibrated for 30 to 45 minutes; then, their contractility was tested with a KCl–physiological saline solution (40 mmol/L), followed by 2 washout periods. They were further incubated for 30 to 45 minutes with or without LNNA (100 μmol/L), then preconstricted with a half‐maximal effective concentration dose of thromboxane A2 analog U46619 (0.1 to 10 nmol/L), followed by dose‐response curves to ACh (0.1 nmol/L to 3 μmol/L). To study vascular smooth muscle cell function, we mechanically removed the endothelium by gentle rubbing with human hair and confirmed the removal by the loss of response to ACh. After incubation, single cumulative concentration‐response curves to PE (0.1 nmol/L to 3 μmol/L), followed by single cumulative concentration‐response curves to sodium nitroprusside (0.1 nmol/L to 30 μmol/L), were performed.
Quantification of TGs in Mouse Livers
FFAs from the liver were quantified as previously reported32–33 including tissue lipid extraction34 and separation into TG and phospholipid classes by using an aminoisopropyl column (Varian).35 FFAs were trans‐methylated according to a modified protocol previously described by Lepage and Roy.36 Gas chromatography–mass spectometry was operated in chemical ionization mode using ammonia as the reagent gas and was internally validated. FFAs were identified according to their retention time and m/z ratio; concentrations were determined by using calibration curves with internal and external standards.
Adipocyte Size Analysis
Isolated epididymal and mesenteric adipose tissues embedded in paraffin sections for H&E staining were visualized by using light microscopy, and ImageJ was used to measure the mean diameter of 30 to 50 adipocytes. The average of 3 separate images was used to calculate mean adipocyte diameter.
Real‐Time Quantitative Polymerase Chain Reaction
Total RNA was extracted from various tissues using the RNeasy mini‐kit (Qiagen Canada) according to the manufacturer's protocol. Reverse transcription reaction (100 ng) was performed as previously described4,37 using the Moloney murine leukemia virus reverse transcriptase (200 U; Invitrogen). Quantitative polymerase chain reaction (qPCR) was performed using the EvaGreen qPCR Mastermix (Mastermix‐LR; Applied Biological Materials Inc). Primers of target genes were designed using the Clone Manager software (Table 1). The ΔΔCT method was used for analysis of relative gene expression using cyclophilin A as the housekeeping gene. For each pair of primers used, its optimal concentration, cDNA template concentration, and annealing temperature were optimized by performing standard curves that yielded efficiencies of 100±10%.
Statistical Analysis
Results are presented as mean±SEM, and n indicates number of mice. EC50 is the half‐maximum effective concentration for each concentration‐response curve, estimated by using Graph Pad Prism 5.0 software according to the variable slope sigmoidal dose‐response curve formula:
where bottom is the Y value at the bottom plateau, top is the Y value at the top plateau, and Hillslope describes the steepness of the curve. Emax is the maximal ACh‐induced dilation at the maximal dose tested. Normality tests were first performed for all groups using the d'Agostino–Pearson omnibus test. If sample sizes were normally distributed, parametric tests were performed: the unpaired Student t test was used to compare 2 groups and the 2‐way ANOVA followed by Bonferroni posttest were performed for comparison of more than 2 groups. When groups did not follow a Gaussian distribution, nonparametric tests were performed: the Mann–Whitney U test was used to compare 2 groups and the Kruskal–Wallis test followed by the Dunn's posttest were used for comparison of more than 2 groups. When “n” was too low to test for normality, the z‐score method (Y–mean/SD) was used for each individual datum (Y) followed by normality test using the d'Agostino–Pearson test. If the transformed data sets followed a normal distribution, parametric tests were used; if data sets did not follow a normal distribution, nonparametric tests were used. In all cases, P<0.05 was considered statistically significant.
Results
Acute Addition of Angptl2 Led to Impaired Endothelial Function in the Femoral Artery, Which Was Reversed by Addition of the Antioxidant NAC
The effect of an acute addition of angptl2 on endothelial function in the femoral artery was first investigated. ACh‐induced vasodilation in 3‐ to 4‐month‐old WT mice was significantly reduced after acute addition of 50 nmol/L angptl2‐GST (Emax [%]: TBSE=99±1 and angptl2=80±7, P<0.05, n=4 to 5), as shown in Figure 2. Because angptl2 has been demonstrated to have pro‐oxidative properties in cancer cells,26 we next asked whether an antioxidant could reverse the detrimental effects of angptl2 on endothelial function. Indeed, endothelial dysfunction caused by the acute angptl2 addition was completely reversed by NAC (10 μmol/L; Emax [%]: angptl2=80±7 and angptl2+NAC=98±1, P<0.05, n=4 to 5) with no overall effects by NAC alone without angptl2 addition (Emax [%]: TBSE=99±1 and TBSE+NAC=95±3, n=4 to 5). These data suggest that angptl2 induces deleterious effects on endothelial function, at least partially via its pro‐oxidative properties.
Figure 2.
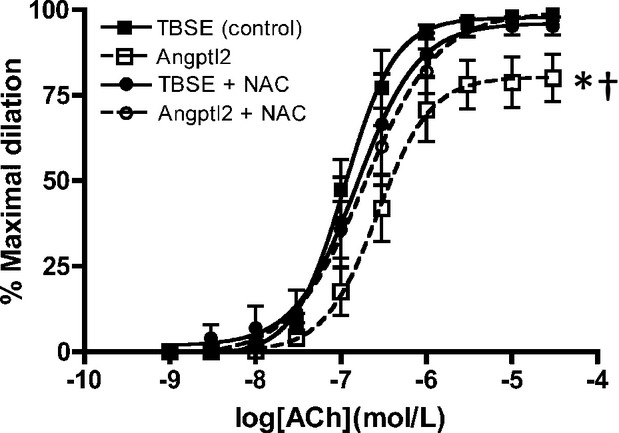
Vascular reactivity of pressurized femoral arteries measured by ACh‐mediated dilation in 3‐ to 4‐month‐old WT (n=4 to 5) with addition of Tris‐buffered saline EDTA (control) or angptl2‐Glutathion S‐transferase (50 nmol/L) with or without antioxidant NAC (10 μmol/L). The z‐score method followed by the d'Agostino–Pearson omnibus test was used to test normality of data sets, after which the 1‐way ANOVA followed by the Bonferonni posttest were used. *P<0.05 vs control; †P<0.05 vs + NAC. ACh indicates acetylcholine; Angptl2, angiopoietin‐like‐2; GST, Glutathion S‐transferase; NAC, N‐acetylcysteine; TBSE, Tris‐buffered saline EDTA; WT, wild‐type.
Phenotype of Angptl2 KD Mice
To further study the pathophysiological role of angptl2, we generated an angptl2 KD mouse model. Angptl2 KD mice were born alive following a Mendelian pattern and were grossly comparable to WT littermates. Verification of angptl2 depletion was determined by quantification by qPCR and Western blot (Figure 1B and 1C). Young mice from 3 to 4 months of age showed similar fasting plasma parameters, with higher (P=0.04; n=13) insulin levels in KD mice (Table 2) but with normal glucose and insulin tolerance (data not shown). Surprisingly, KD mice (n=28) showed significantly slower heart rate compared with WT mice (n=24) at this age, while systolic and diastolic blood pressures were similar for the 2 strains (Table 3).
Table 2.
Parameters of Fasting Plasma in 3‐ to 4‐Month‐Old WT and Angptl2 KD Mice
| WT | KD | |
|---|---|---|
| Weight, g | 24.7±0.4 (10) | 23.4±1.1 (8) |
| Glucose, mmol/L | 10.7±1.4 (10) | 13.4±0.9 (8) |
| Triglycerides, mmol/L | 0.63±0.09 (10) | 0.52±0.06 (8) |
| Adiponectin, μg/mL | 7.3±0.2 (8) | 7.4±0.4 (7) |
| Leptin, ng/mL | 6.3±2.2 (8) | 5.7±0.9 (8) |
| FFAs, mmol/L | 0.55±0.05 (10) | 0.51±0.06 (8) |
| Cholesterol—total, mmol/L | 2.85±0.17 (10) | 2.78±0.17 (8) |
| Cholesterol—HDL, mmol/L | 2.65±0.13 (10) | 2.60±0.13 (8) |
| Cholesterol—LDL, mmol/L | 0.12±0.02 (6) | 0.15±0.03 (5) |
| Cholesterol―total/HDL | 1.08±0.01 (10) | 1.06±0.01 (8) |
| LDL/HDL | 0.04±0.01 (6) | 0.05±0.01 (5) |
| Insulin, ng/mL | 0.18±0.04 (13) | 0.29±0.04 (13)‡ |
Data presented as mean±SEM of (n) mice. The Mann–Whitney U test was used. FFA indicates free fatty acid; HDL, high‐density lipoprotein; KD, knockdown; LDL, low‐density lipoprotein; WT, wild‐type.
‡P<0.05 vs WT.
Table 3.
Measurements of Heart Rate and Blood Pressures by Tail‐Cuff Plethysmography in 3‐ to 4‐Month‐Old WT and Angptl2 KD Male Mice
| WT | KD | |
|---|---|---|
| Heart rate, bpm | 680±9 (24) | 640±11 (28)‡ |
| Systolic blood pressure, mm Hg | 153±3 (24) | 155±3 (28) |
| Diastolic blood pressure, mm Hg | 122±3 (24) | 119±3 (28) |
Data presented as mean±SEM of (n) mice. Data sets were tested for normality using the d'Agostino–Pearson normality test and the unpaired Student t test was used. KD indicates knockdown; WT, wild‐type.
‡P<0.05 vs WT.
Angptl2 KD Mice Had Better Endothelial Function in the Femoral Artery as Measured by Vasodilation to ACh
Mice from 3 to 4 months of age were used to study vascular endothelial function in the femoral artery (Figure 3). Under control conditions (Figure 3A), the maximal dilation to ACh resulting from the combination of NO, PGI2, and EDHF was slightly greater in KD (n=8) than in WT mice (n=7), as shown by the higher Emax and a tendency for a higher sensitivity indicated by their EC50 values (P=0.06, Table 4). Inhibition of NOS by LNNA reduced this response in arteries isolated from WT littermates and KD mice (Figure 3B and 3C, Table 4), while inhibition of cyclooxygenase with Indo had no impact on ACh‐induced vasodilation (Figure 3B and 3C, Table 4). In the presence of LNNA and Indo, combined activities of NO and PGI2 are abolished, revealing that of EDHF38–39; these experimental conditions revealed a lower EDHF contribution in KD mice (Figure 3B and 3C). In WT littermates, compared with LNNA alone, where both PGI2 and EDHF are present, incubation with LNNA and Indo, where only EDHF is present, did not result in additional effects (Figure 3B, Table 4), suggesting that PGI2 per se played no role in ACh‐mediated dilation in WT littermates. In contrast, in KD mice, Emax decreased significantly in the presence of LNNA combined with Indo (Figure 3C, Table 4), suggesting that PGI2 contributed significantly to ACh‐induced femoral artery dilation in KD mice but not WT littermates in the absence of NO. As there are interactions and compensations among the 3 main EDRFs40 under each individual experimental condition, PGI2 contribution to vasodilation could only be revealed following NO inhibition in KD mice. Overall, these results suggest that angptl2 KD results in better endothelial function, which was associated with greater contribution of PGI2 and less EDHF to vasodilation, at least in young and healthy mice.
Figure 3.
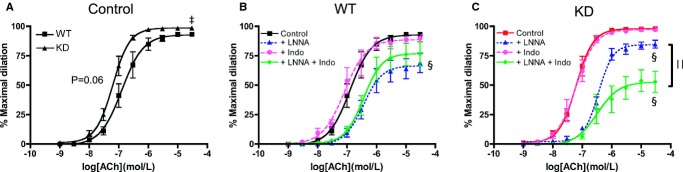
Vascular reactivity as measured by ACh‐mediated dilation in femoral arteries of WT (n=7) and angptl2 KD (n=8) mice at 3 to 4 months of age in no‐drug control condition (A) and in the presence of LNNA (100 μmol/L), indomethacin (Indo, 10 μmol/L), or their combination in arteries isolated from WT mice (B) and KD mice (C). The z‐score method followed by the d'Agostino–Pearson omnibus test was used to test normality of data sets, after which the unpaired Student t test (A) and the 1‐way ANOVA followed by the Bonferonni posttest were used (B and C). ‡P<0.05 vs WT; §P<0.05 vs control; ||P<0.05 vs +LNNA. ACh indicates acetylcholine; KD, knockdown; LNNA, Nω‐nitro‐l‐arginine; WT, wild‐type.
Table 4.
Efficacy (Emax) and Sensitivity (EC50) to Acetylcholine in Femoral Arteries Isolated From 3‐ to 4‐Month‐Old WT and Angptl2 KD Male Mice
| WT | KD | |||
|---|---|---|---|---|
| Emax (%) | EC50 | Emax (%) | EC50 | |
| Control | 93±2 (7) | 6.8±0.2 (7) | 98±1 (8)‡ | 7.2±0.1 (8) |
| +LNNA | 69±8 (7)§ | 6.3±0.2 (7) | 85±3 (8)§ | 6.4±0.1 (8)§ |
| +Indo | 90±3 (7) | 7.0±0.2 (7) | 97±1 (8)‡ | 7.2±0.1 (8) |
| +LNNA+Indo | 77±10 (7) | 6.4±0.1(7) | 53±9 (8)§∥ | 6.2±0.2 (8)§ |
Vessels were preconstricted to 30% to 50% of maximal diameter with phenylephrine (1 to 3 μmol/L). Emax are expressed as the percentage of the maximal diameter. Data presented as mean±SEM of (n) mice. The z‐score method followed by the d'Agostino–Pearson omnibus test was used to test normality of data sets, after which the unpaired Student t test (‡) and the 1‐way ANOVA followed by the Bonferonni posttest were used (§ and ∥). KD indicates knockdown; WT, wild‐type; LNNA, Nω‐nitro‐l‐arginine; Indo, indomethacin.
‡P<0.05 vs WT; §P<0.05 vs control; ∥P<0.05 vs +LNNA.
Endothelial Function in the Femoral Artery Was Preserved in KD Mice Fed an HFD
At 6 months of age, when fed an RD, the sensitivity to ACh under no‐drug control condition was, as similarly observed at 3 to 4 months of age, slightly greater in femoral arteries from KD (n=13) compared with WT (n=10) mice, as indicated by a shift to the left of the dose‐response curve (Figure 4A and 4B) and by the higher EC50 values (Table 5). The efficacies (Emax) were identical in both strains (Figure 4, Table 5). For both KD and WT mice fed an RD diet, addition of LNNA to inhibit NOS significantly decreased Emax and EC50 values similarly (Figure 4, Table 5), suggesting that NO contributed equally for both strains of mice. When fed an HFD (n=9 to 10), both strains of mice displayed similar global responses to ACh, while LNNA significantly decreased the sensitivity of ACh‐mediated dilation compared with no‐drug condition in KD mice only. In WT mice, LNNA no longer had a significant inhibition on dilation, suggestive of a lower eNOS activity in WT mice fed an HFD. In KD mice, however, LNNA‐sensitive ACh‐mediated dilations were similar under either diet, suggesting that NO‐dependent dilation sensitive to LNNA was preserved in KD mice. To further confirm preservation of NO in HFD‐fed angptl2 KD mice compared with WT mice, we loaded femoral arteries with a fluorescent dye specific for NO production, DAF‐2, as previously described.30 In agreement with our functional data, a 3‐month HFD significantly reduced ACh‐induced changes in NO‐associated fluorescence only in WT (n=6), while it was preserved in angptl2 KD mice (n=6) (Figure 4C). Artery preincubation with Indo alone or in combination with LNNA to reveal contribution of EDHF did not show differences in ACh‐mediated dilation across groups (Table 5). Previously observed contribution of PGI2 in KD mice was lost at 6 months of age under either diet regimen (Table 5). Furthermore, while vasoconstriction to PE was slightly but significantly greater in HFD‐fed WT mice compared with all other groups of mice (Figure 5A), endothelium‐independent dilation to sodium nitroprusside was similar across all groups of mice (Figure 5B). Taken together, these data suggest that in angptl2 KD mice treated with an HFD, eNOS‐derived NO production is preserved in the femoral arteries.
Figure 4.
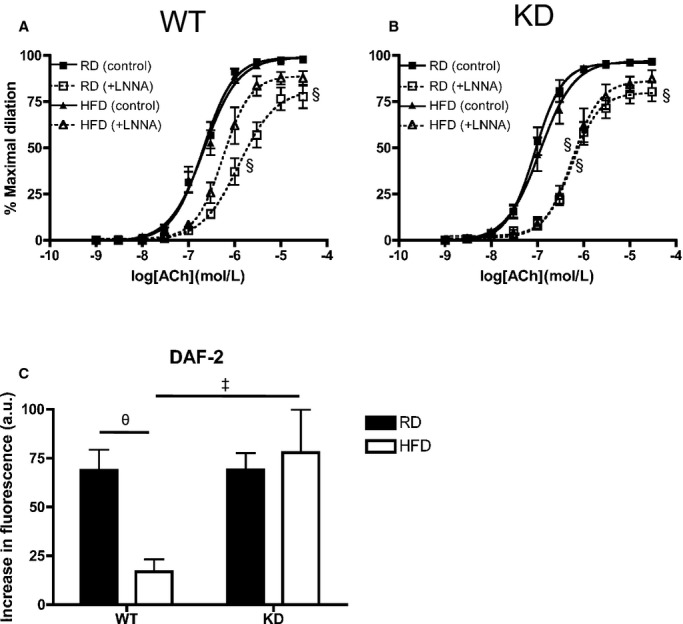
Vascular reactivity as measured by ACh‐mediated dilation in femoral arteries under no‐drug control condition and with LNNA of 6‐month‐old (A) WT (n=7 to 10) and (B) angptl2 KD (n=9 to 13) mice fed an RD or HFD. The Kruskal–Wallis test followed by the Dunn's posttest were used for data sets not normally distributed. §P<0.05 vs control. C, Production of NO was measured by loading femoral arteries with DAF‐2 with average increases in fluorescence intensities during addition of 10 μmol/L ACh (n=6). The z‐score method followed by the d'Agostino–Pearson omnibus test was used to test normality of data sets, after which the 2‐way ANOVA followed by the Bonferonni posttest were used. ‡P<0.05 vs WT; θP<0.05 vs RD. ACh indicates acetylcholine; DAF‐2, 4,5‐diaminoflorescein diacetate; HFD, high‐fat diet; KD, knockdown; LNNA, Nω‐nitro‐l‐arginine; NO, nitric oxide; RD, regular diet; WT, wild‐type.
Table 5.
Efficacy (Emax) and Sensitivity (EC50) to Acetylcholine in Femoral Arteries Isolated From 6‐Month‐Old WT and Angptl2 KD Male Mice Fed a Regular (RD) or High‐Fat Diet (HFD)
| WT+RD | WT+HFD | KD+RD | KD+HFD | |||||
|---|---|---|---|---|---|---|---|---|
| Emax (%) | EC50 | Emax (%) | EC50 | Emax (%) | EC50 | Emax (%) | EC50 | |
| Control | 98±1 (10) | 6.7±0.1 (10) | 98±1 (9) | 6.7±0.1 (9) | 97±1 (13) | 7.0±0.1 (13)‡ | 96±1 (10) | 6.9±0.1 (10) |
| +LNNA | 78±6 (9)§ | 5.9±0.1 (9)§ | 88±4 (7) | 6.3±0.1 (7) | 80±5 (12)§ | 6.2±0.1 (12)§ | 87±5 (9) | 6.2±0.1 (9)§ |
| +Indo | 92±6 (10) | 6.8±0.2 (10) | 96±2 (8) | 6.8±0.1 (8) | 97±1 (13) | 6.9±0.1 (13) | 96±1 (10) | 7.0±0.1 (10) |
| +LNNA+Indo | 74±7 (10)§ | 5.9±0.1 (10)§ | 81±7 (8) | 6.2±0.1 (8)§ | 65±7 (13)§ | 5.9±0.2 (13)§ | 74±6 (9) | 6.2±0.1 (9)§ |
Vessels were preconstricted to 30% to 50% of maximal diameter with phenylephrine (1 to 3 μmol/L). Emax are expressed as the percentage of the maximal diameter. Data presented as mean±SEM of (n) mice. The Kruskal–Wallis test followed by the Dunn's posttest were used. KD indicates knockdown; WT, wild‐type; LNNA, Nω‐nitro‐l‐arginine; Indo, indomethacin.
‡P<0.05 vs WT+RD; §P<0.05 vs control (within respective groups).
Figure 5.
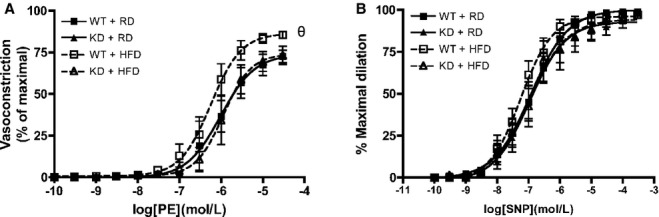
Vascular smooth muscle cell function was assessed in femoral arteries from WT and angptl2 KD mice, by (A) vasoconstriction to phenylephrine (PE), and (B) dilation to sodium nitroprusside (SNP) in the absence of the endothelium. Data are mean±SEM of n=4 to 6 mice, and compared using the Mann–Whitney U test. θP<0.05 vs RD. KD indicates knockdown; RD, regular diet; WT, wild‐type.
In our experimental models, the HFD did not change DHE staining, a marker of oxidative stress, in femoral arteries from both strains of mice (Figure 6).
Figure 6.
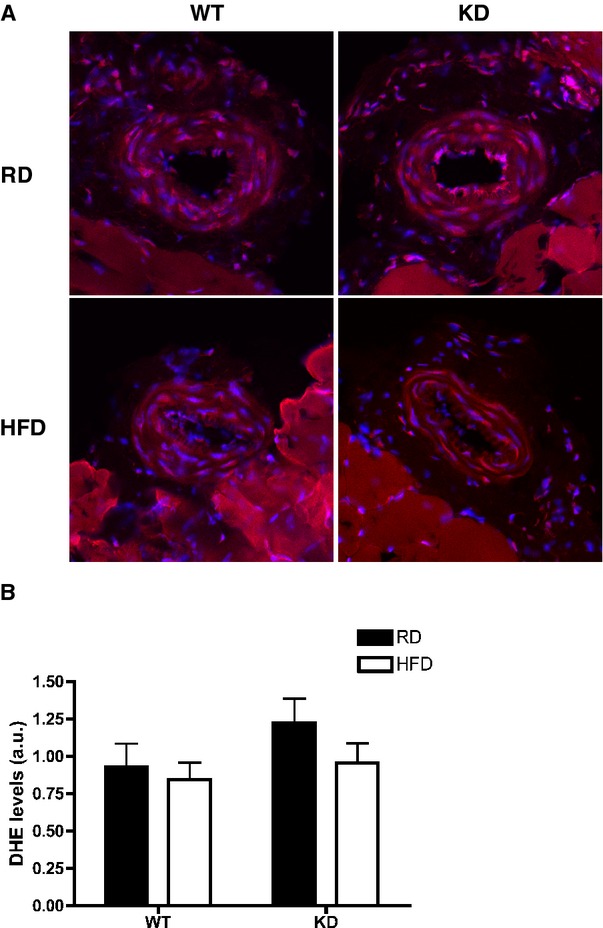
A, DHE staining in femoral arteries of WT and KD mice fed an RD or HFD, and (B) quantifications of DHE intensities in femoral arteries; n=3 to 5. DHE indicates dihydroethidium; HFD, high‐fat diet; KD, knockdown; RD, regular diet; WT, wild‐type.
An HFD Recruited a Compensatory NO Pathway in the Mesenteric Arteries of WT, Which Was Absent in KD mice
In mesenteric arteries, global endothelial function measured by ACh‐mediated vasorelaxation was similar for the 2 strains of mice (Figure 7A). After an HFD, global endothelial function remained similar as well (Figure 7B). Interestingly, deciphering out contribution of NO using LNNA showed that under the RD regimen, there was no significant functional consequence of inhibiting NO with LNNA in WT mice (n=6; Figure 7C), confirming, as reported previously,41 that in these mesenteric resistance arteries, EDHF is most likely the main vasodilator. When fed an HFD, however, LNNA significantly reduced ACh‐induced relaxation (Figure 7D, Table 6) in WT mice (n=6), suggesting a decrease in EDHF‐mediated relaxation but compensated by NO. In contrast, LNNA (n=8) unexpectedly reduced ACh‐induced relaxation in angptl2 KD mice fed an RD (Figure 7E, Table 6), suggesting that both NO and EDHF contribute to the relaxation. The effect of LNNA (n=5) was, however, absent in KD mice fed an HFD (Figure 7F, Table 6), suggesting that the contribution of EDHF was preserved in mesenteric arteries of KD mice. Of note, in both strains, Indo did not modify ACh‐induced relaxation (data not shown). Unlike in the femoral artery, vasoconstriction to PE was similar among all groups of mice (Figure 8A); furthermore, endothelium‐independent relaxation to sodium nitroprusside was not different between groups (Figure 8B). Combined, these data suggest that in mesenteric arteries from angptl2 KD mice treated with an HFD, the EDHF contribution is preserved, while in WT mice NO compensates for the reduced contribution of EDHF.
Figure 7.
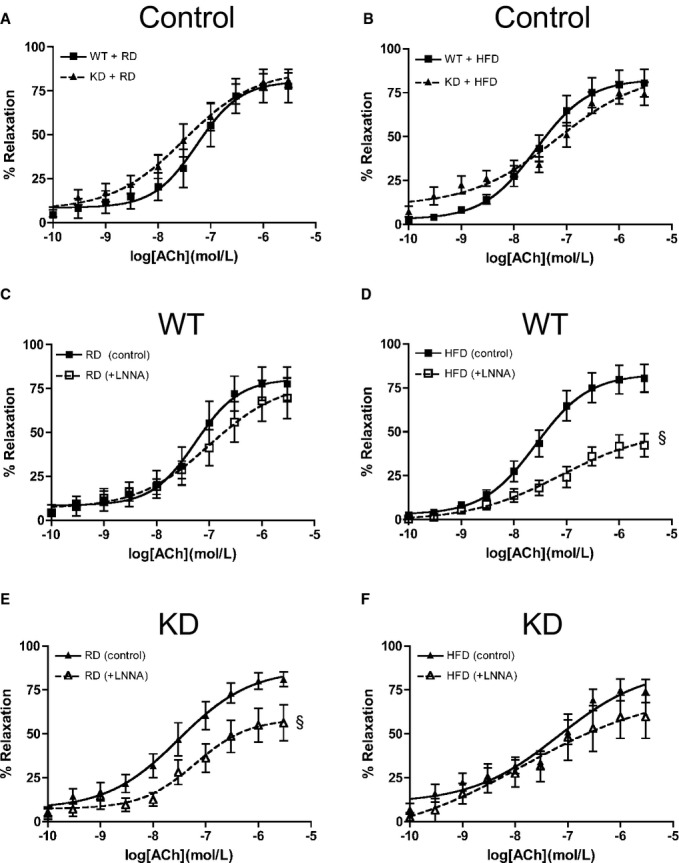
ACh‐mediated relaxation in mesenteric arteries of 6‐month‐old mice fed an (A) RD, (B) HFD under no‐drug control condition and with LNNA in WT (n=6) fed an (C) RD or (D) HFD, and in KD (n=6 to 10) fed an (E) RD or (F) HFD. The z‐score method followed by the d'Agostino–Pearson omnibus test was used to test normality of data sets, after which the 2‐way ANOVA followed by the Bonferonni posttest were used. §P<0.05 vs control. ACh indicates acetylcholine; HFD, high‐fat diet; KD, knockdown; LNNA, Nω‐nitro‐l‐arginine; RD, regular diet; WT, wild‐type.
Table 6.
Efficacy (Emax) and Sensitivity (EC50) to Acetylcholine in Mesenteric Arteries Isolated From 6‐Month‐Old WT and angptl2 KD Male Mice Fed a Regular (RD) or High‐Fat Diet (HFD)
| WT+RD | WT+HFD | KD+RD | KD+HFD | |||||
|---|---|---|---|---|---|---|---|---|
| Emax (%) | EC50 | Emax (%) | EC50 | Emax (%) | EC50 | Emax (%) | EC50 | |
| Control | 78±10 (6) | 7.3±0.1 (6) | 80±8 (6) | 7.6±0.2 (6) | 81±4 (10) | 7.5±0.3 (8) | 74±7 (6) | 7.2±0.2 (5) |
| +LNNA | 70±12 (6) | 7.0±0.3 (3) | 42±7 (6)§ | 7.4±0.3 (6) | 56±10 (8)§ | 7.4±0.4 (7) | 60±12 (5) | 8.1±0.4 (5) |
Vessels were preconstricted with thromboxane A2 analog U46619 (0.1 to 10 μmol/L). Emax values are expressed as the percentage of the maximal diameter. Data presented as mean±SEM of (n) mice. The z‐score method followed by the d'Agostino–Pearson omnibus test was used to test normality of data sets, after which the 2‐way ANOVA followed by the Bonferonni posttest were used. KD indicates knockdown; WT, wild‐type; LNNA, Nω‐nitro‐l‐arginine.
§P<0.05 vs control (within respective groups).
Figure 8.
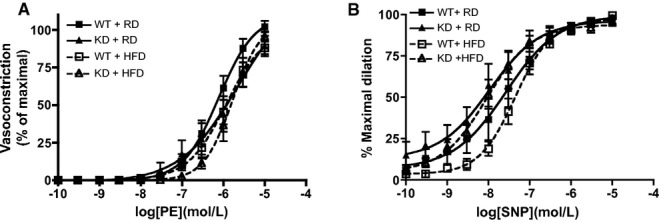
Vascular smooth muscle cell function was assessed in mesenteric arteries from WT and angptl2 KD mice, by (A) vasoconstriction to phenylephrine (PE) and (B) dilation to sodium nitroprusside (SNP) in the absence of the endothelium. Data are mean±SEM of n=4 to 6 mice. KD indicates knockdown; WT, wild‐type.
KD Mice Had a Better Lipid Profile Than WT Mice Fed an HFD
We examined the phenotypic profiles of WT and KD mice after a 3‐month RD or HFD diet treatment (Table 7). Similar to mice 3 to 4 months of age, 6‐month‐old KD mice fed the RD had similar body weight compared with age‐matched WT. After being fed the HFD for 3 months, both WT and KD mice significantly gained weight but KD mice weighed less than WT mice (Table 7). During the 3‐month diet treatment, food intake was also similar in the 2 strains of mice fed either diet (data not shown). However, after the HFD, circulating leptin level rose significantly only in WT mice, while fasting plasma glucose, TGs, adiponectin, FFAs, and cholesterol (HDL, total cholesterol) were similar for the 2 strains (Table 7). Although total cholesterol and HDL levels increased in WT and KD mice after HFD, LDL levels increased only in WT, but not in KD mice, so that total cholesterol‐to‐HDL and LDL‐to‐HDL ratios increased significantly only in WT mice (Table 7). Fasting insulin in WT mice significantly elevated after HFD, while they were not different between strains. In addition, serum angptl2 was higher in WT mice fed an HFD compared with RD (149±18 versus 77±7 ng/mL, n=3 to 4, P=0.057, Mann–Whitney U test). These results indicate that after exposure to an HFD, the lipid profile was better in angptl2 KD compared with WT mice.
Table 7.
Parameters of Fasting Plasma in 6‐Month‐Old WT and Angptl2 KD Mice Fed a Regular Diet (RD) or a 3‐Month High‐Fat Diet (HFD)
| WT | KD | |||
|---|---|---|---|---|
| RD | HFD | RD | HFD | |
| Weight, g | 26.3±0.9 (10) | 33.1±1.4 (9)θ | 24.2±0.5 (13) | 29.0±1.6 (11)‡θ |
| Glucose, mmol/L | 17.0±1.1 (10) | 17.4±1.7 (9) | 13.5±1.1 (12) | 17.1±1.5 (11) |
| Triglycerides, mmol/L | 0.45±0.04 (10) | 0.49±0.05 (9) | 0.41±0.03 (12) | 0.49±0.03 (11) |
| Adiponectin, μg/mL | 8.5±0.4 (5) | 10.1±0.3 (5) | 9.7±1.1 (5) | 8.5±0.7 (5) |
| Leptin, ng/mL | 2.1±0.7 (5) | 20.2±2.1 (5)θ | 2.3±0.5 (5) | 7.5±2.3 (5) |
| FFA, mmol/L | 0.39±0.05 (7) | 0.50±0.05 (5) | 0.39±0.03 (7) | 0.44±0.06 (7) |
| Cholesterol—total, mmol/L | 2.7±0.1 (10) | 3.9±0.2 (9)θ | 2.7±0.2 (12) | 3.9±0.3 (11)θ |
| Cholesterol—HDL, mmol/L | 2.5±0.1 (10) | 3.3±0.1 (9)θ | 2.4±0.2 (12) | 3.5±0.2 (11)θ |
| Cholesterol—LDL, mmol/L | 0.15±0.02 (9) | 0.41±0.11 (8)θ | 0.13±0.01 (12) | 0.25±0.05 (11) |
| Cholesterol—total/HDL | 1.07±0.02 (10) | 1.17±0.03 (9)θ | 1.12±0.02 (12) | 1.12±0.01 (11) |
| LDL/HDL | 0.06±0.01 (9) | 0.12±0.03 (8)θ | 0.06±0.01 (11) | 0.08±0.02 (11) |
| Insulin, ng/mL | 0.10±0.02 (9) | 0.27±0.03 (9)θ | 0.14±0.02 (12) | 0.30±0.08 (10) |
Data presented as mean±SEM of (n) mice. The Kruskal–Wallis test followed by the Dunn's posttest were used for data sets not normally distributed; 2‐way ANOVA followed by the Bonferroni posttest was used to compare normally distributed data sets. FFA indicates free fatty acid; HDL, high‐density lipoprotein; KD, knockdown; LDL, low‐density lipoprotein; WT, wild‐type.
θP<0.05 vs RD (within the same strain); ‡P<0.05 vs WT (within the same treatment).
KD Mice Had Similar Blood Pressures as WT Mice but a Lower Resting Heart Rate
While systolic and diastolic blood pressures did not differ between 2 strains of mice and were not affected by the HFD (Table 8), the previously mentioned lower basal heart rate in KD mice at 3 months of age (Table 3) was maintained (n=7 to 10) at 6 months of age for the RD‐fed group (Table 8).
Table 8.
Measurements of Heart Rate and Blood Pressures by Tail‐Cuff Plethysmography in 6‐Month‐Old WT and angptl2 KD Mice Fed a Regular Diet (RD) or a 3‐Month High‐Fat Diet (HFD)
| WT | KD | |||
|---|---|---|---|---|
| RD | HFD | RD | HFD | |
| Heart rate, bpm | 719±16 (10) | 760±11 (6) | 624±29 (7)‡ | 713±18 (8) |
| Systolic blood pressure, mm Hg | 145±6 (10) | 146±5 (6) | 146±2 (7) | 153±5 (8) |
| Diastolic blood pressure, mm Hg | 111±7 (10) | 116±6 (6) | 113±3 (7) | 123±5 (8) |
Data presented as mean±SEM of (n) mice. The Mann–Whitney U test was used. KD indicates knockdown; WT, wild‐type.
‡P<0.05 vs WT.
Lower Liver TG Levels and Prevention of Upregulation of TNFα Gene Expression in KD Mice Fed an HFD
Because we observed a better lipid profile on an HFD treatment in KD mice, we examined the phenotype of the liver, a major organ involved in lipid handling. Liver sections of KD mice (n=4) fed an HFD showed less lipid droplet accumulation compared with WT mice (n=5) fed an HFD (Figure 9A). This was associated with significantly lower levels of liver TGs measured with the use of HPLC in KD mice (n=4) fed an HFD compared with WT mice (n=5) (Figure 9B). Phospholipid levels remained similar among all groups (data not shown). Expressions of hepatic genes were examined using qPCR (Figure 9C through 9F): notably, hepatic angptl2 gene expression was significantly greater after HFD in the WT mice (n=7; Figure 9C). In terms of inflammatory markers, TNFα mRNA levels significantly increased in HFD‐fed WT mice (n=7) but did not change in KD mice (n=6; Figure 9D), while transforming growth factor‐β increased similarly after HFD (Figure 9E) and interleukin‐6 gene expression was unchanged and similar between the 2 strains (Figure 9F). The expression of essential genes coding for proteins involved in the regulation of lipid metabolism was not different between groups (Figure 10). Collectively, these data suggest that an HFD promoted inflammation that was associated with increased TG levels in the liver of WT mice, which was less severe in angptl2 KD mice.
Figure 9.
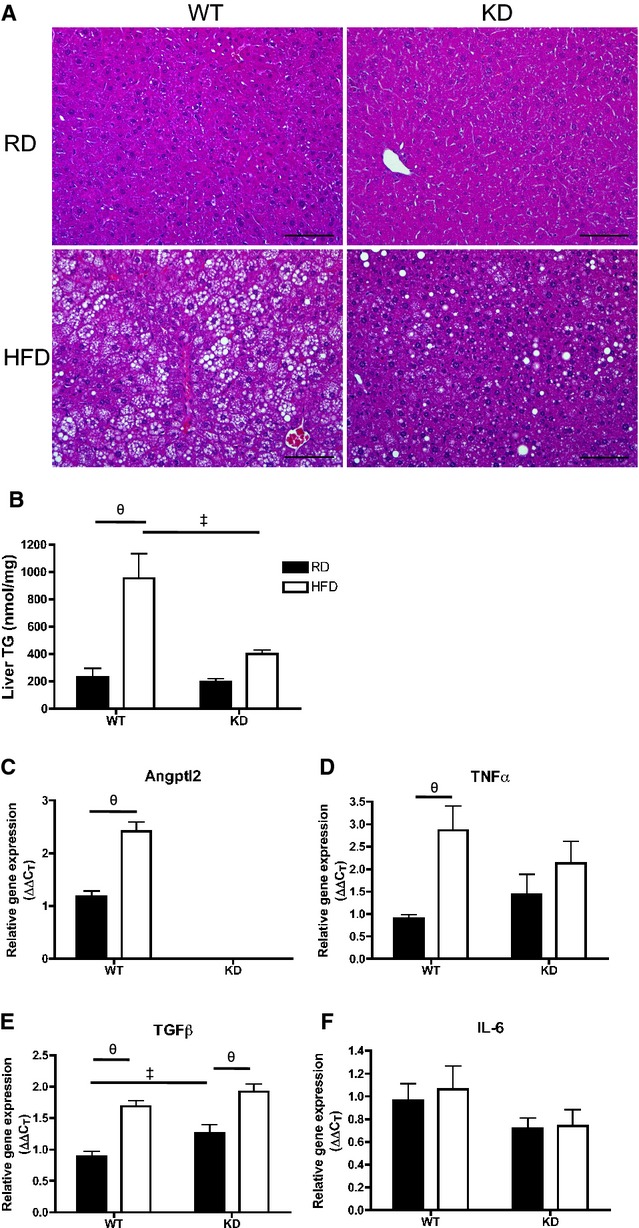
A, H&E‐stained liver sections (scale bar=100 μm) and (B) quantification of triglyceride (TG) content in liver of 6‐month‐old WT and angptl2 KD mice fed a regular diet (RD) or a 3‐month high‐fat diet (HFD); n=4 to 5. C through F, Quantitative RT‐PCR of mRNAs encoding for angptl2 and various inflammatory markers in liver of WT or angptl2 KD mice fed an RD or HFD; n=6 to 7. The z‐score method followed by the d'Agostino–Pearson omnibus test was used to test normality of data sets, after which the 2‐way ANOVA followed by the Bonferonni posttest were used. ‡P<0.05 vs WT; θP<0.05 vs RD. KD indicates knockdown; RT‐PCR, real‐time quantitative polymerase chain reaction; TGF, transforming growth factor; WT, wild‐type.
Figure 10.

A through L, Expression of genes coding for proteins involved in lipid metabolism regulation in the liver by qPCR; n=6 to 7. Data sets were tested for normality using the z‐score method and the d'Agostino–Pearson omnibus test, after which 2‐way ANOVA followed by the Bonferroni posttest was used. All except I passed the normality test, where the Krustal–Wallis followed by Dunn's posttest were used. ‡P<0.05 vs WT; θP<0.05 vs RD. FXR indicates farnesoid X receptor; HFD, high‐fat diet; HMG‐CoA, 3‐hydroxy‐3‐methylglutaryl coenzyme‐A; HSL, hormone‐sensitive lipase; KD, knockdown; LPL, lipoprotein lipase; PPAR, peroxisome proliferator‐activated receptor; qPCR, quantitative polymerase chain reaction; RD, regular diet; SREBP, sterol regulatory element binding protein.
Smaller Adipocyte Size in Fat Depots and Prevention of Inflammatory Gene Expression in Epididymal White Adipose Tissue of HFD‐fed KD Mice
Efficiency of fat storage in adipose tissue is highly related to lipid profiling.42 Under basal conditions when fed an RD, adipocyte cell size as measured by cell diameter (μm) in mesenteric white adipose tissue (mWAT) (Figure 11A and 11B) and epididymal WAT (eWAT) (Figure 11C and 11D) was similar between the 2 strains (n=3 to 6). Remarkably, when fed an HFD, adipocytes in mWAT and eWAT both significantly increased in size only in WT (n=5) (Figure 11A through 11D). Analysis by qPCR in eWAT (Figure 11E through 11J) revealed that unlike in the liver, angptl2 mRNA levels did not change after an HFD in WT mice (n=7; Figure 11E). Lipoprotein lipase (LPL) mRNA expression, although not different between RD and HFD for both strains, was significantly lower in HFD‐fed KD compared with WT mice (n=7; Figure 11F). Analysis of LPL activity, however, revealed no differences between the strains (data not shown). Interestingly, expressions of adiponectin mRNA decreased significantly after an HFD in WT (n=7) but remained unchanged in KD mice (n=7; Figure 11G), while leptin mRNA significantly increased only in eWAT of WT mice (n=7; Figure 11H) and corresponded to a similar pattern found in plasma leptin levels (Table 7). Similarly, proinflammatory gene markers transforming growth factor‐β and TNFα gene expressions were significantly increased in HFD‐fed WT mice (n=7), but not in KD mice (Figure 11I and 11J). In eWAT, expression of essential genes coding for proteins involved in the regulation of lipid metabolism was not different between groups (Figure 12). Altogether, these data suggest that the inflammation induced by the metabolic stress of an HFD is prevented in adipocytes of angptl2 KD mice.
Figure 11.
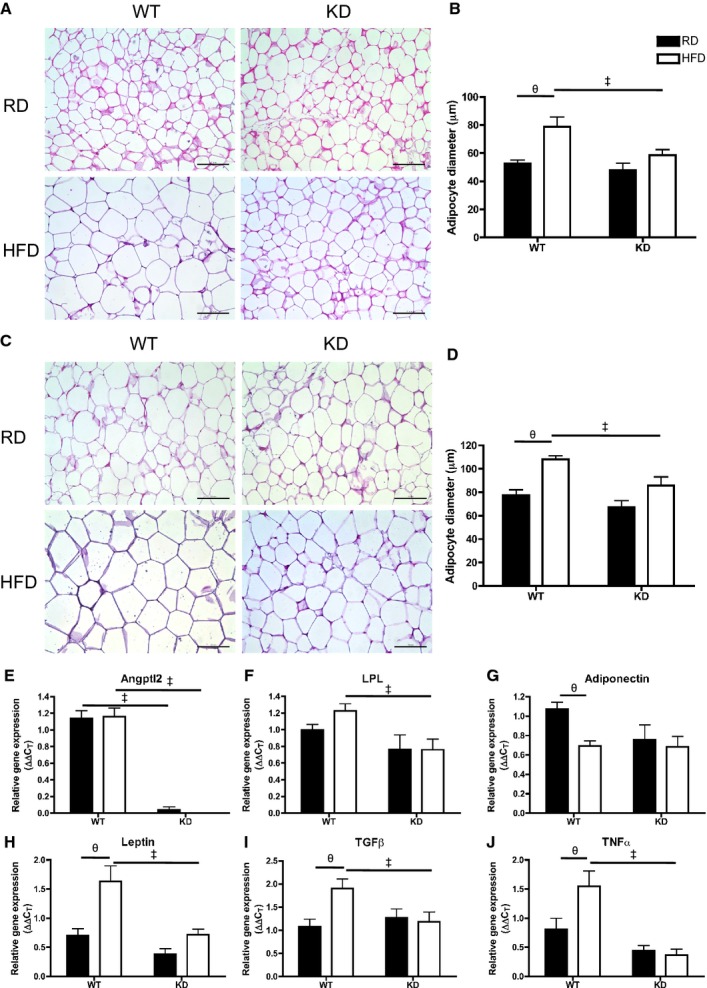
A, Hematoxylin‐eosin–stained mesenteric white adipose tissue (mWAT) in different groups (scale bar=100 μm) and (B) quantification of adipocyte size of diameter measurements (average of 3 quantifying analyses was used per animal) in mWAT; n=3 to 5. C, Hematoxylin‐eosin–stained epididymal WAT (eWAT) in different groups (scale bar=100 μm) and (D) quantification of adipocyte diameters in eWAT; n=3 to 6. E through J, Gene expression analysis by qPCR in eWAT of WT and KD mice fed an RD or HFD; n=6 to 7. The z‐score method followed by the d'Agostino–Pearson omnibus test was used to test normality of data sets, after which the 2‐way ANOVA followed by the Bonferonni posttest were used, except in E, where the Kruskal–Wallis followed by Dunn's posttest were used as it did not pass normality test. ‡P<0.05 vs WT; θP<0.05 vs RD. HFD indicates high‐fat diet; KD, knockdown; qPCR, quantitative polymerase chain reaction; RD, regular diet; WT, wild‐type.
Figure 12.
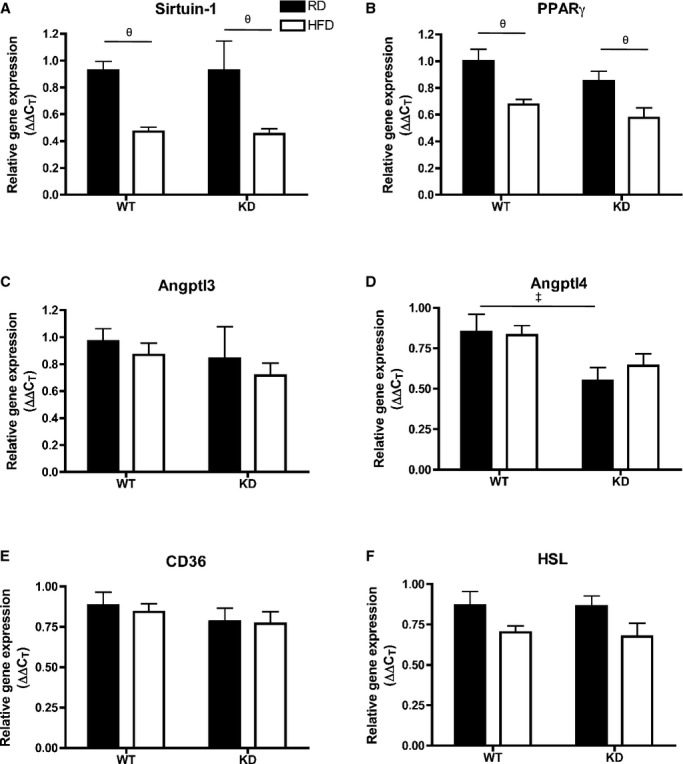
A through F, Expression of genes coding for proteins involved in lipid metabolism regulation in the eWAT by qPCR; n=6 to 7. Data sets were tested for normality using the z‐score method and the d'Agostino–Pearson omnibus test, after which 2‐way ANOVA followed by Bonferroni's posttest were used. ‡P<0.05 vs WT; θP<0.05 vs RD. eWAT indicates epididymal white adipose tissue; HSL, hormone‐sensitive lipase; KD, knockdown; PPAR, peroxisome proliferator‐activated receptor; qPCR, quantitative polymerase chain reaction; RD, regular diet; WT, wild‐type.
Discussion
The novel findings in this study are that KD of angptl2 results in (1) better femoral endothelial function via NO/PGI2 recruitment in young mice, (2) preserved endothelial dilatory function after HFD associated with maintained NO release in the femoral artery (a conductance artery) and EDHF contribution in the mesenteric artery (a resistance artery), (3) a better lipid profile when exposed to the metabolic challenge of an HFD, and (4) a lower inflammatory status of the liver and eWAT. To the best of our knowledge, this is the first report of the impact of angptl2 in the contribution of the various EDRF and their resistance against a stress induced by an HFD.
In the first part of the study, we tested the acute effects of angptl2 on endothelial function. We found that acute addition of angptl2‐GST significantly reduced vasodilation in femoral arteries from WT mice, an effect that was reversed with the addition of antioxidant NAC, implicating the pro‐oxidative role of angptl2 on deteriorating endothelial function, at least acutely. The endothelium, and ultimately vascular function, is highly sensitive to increased oxidative stress, with NO bioavailability and EDHF activity being major targets.43 This is the first demonstration of the prooxidative effect of angptl2 on vascular cells and is in accordance with the recent report by Aoi et al that used cancer cells.26
The next logical step was to then examine the role of angptl2 in regulating endothelial function chronically. In characterizing the endothelial function in femoral arteries in which NO is the main EDRF, we found that, in addition to NO, PGI2 unexpectedly contributed to the vasodilation in young healthy KD mice, which may reflect a remodeling of the EDRF induced by angptl2 KD. It is known that NO and PGI2 produced by the endothelium play a protective and antiproliferative role as potent inhibitor of platelet function and vasorelaxant.44 In addition, our data reveal that the contribution of EDHF is greater in femoral arteries isolated from young WT mice compared with KD mice. We previously reported that with age, the contribution of EDHF increases, compensating for the decline in eNOS activity and/or NO bioavailability.21 The current results, therefore, suggest that the lack of angptl2 expression may protect endothelial function by influencing the relative contribution of the 3 main EDRFs—NO, PGI2, and EDHF.
In the second part of the study, we compared the effects of an HFD on endothelial function between WT and KD mice. The current observations are in line with recently reported results demonstrating HFD‐fed angptl2 knockout mice with attenuated endothelial dysfunction in isolated descending thoracic aortas,5 while endothelial cell–derived angptl2 contributed to eNOS inactivation.5 In the current work, we also reveal the preservation of NO‐mediated vasodilation in HFD‐fed KD, but not WT mice, as confirmed by monitoring ACh‐induced NO release using DAF‐2 in femoral arteries. These results complement what has been recently found in aortas of HFD‐fed angptl2 knockout mice, where levels of phospho‐eNOS was higher than in WT mice.5 However, the exact mechanism by which NO‐mediated dilation is maintained remains unknown. Repeatedly, literature has demonstrated endothelial dysfunction characterized by decreased NO bioavailability due to increased oxidative stress.18,45 Because we observed complete rescue of endothelial function with NAC after an acute addition of angptl2, we asked if lower oxidative stress levels in HFD‐fed KD mice could explain our functional data regarding NO. In the present study, however, oxidative stress evaluated by DHE staining did not reveal differences in the femoral artery. Presumably, a 3‐month HFD was not as strong a stress on the femoral artery compared with an acute addition of a pharmacological dose of angptl2 directly in the vessel bath. In addition, it was noted that an HFD increased efficacy to PE in denuded femoral arteries from WT, as has been observed by others,46 but not in KD mice. Despite that difference, low doses of PE were used depending on individual arteries to preconstrict the arteries at 30% to 50% maximal diameter, suggesting that the observed differences in vascular function were not due to different efficacies to PE.
Because the vasodilatory function of the endothelium displays high heterogeneity among various vascular beds, where responses and adaptation to stress also show great diversity, we examined, in parallel, endothelial function in mesenteric resistance arteries. After an RD, 6‐month‐old mice did not show differences in global endothelial function. In the mesenteric resistance arteries, it has been reported that the main vasodilator is EDHF,47 which is in accordance with the current results in WT mice where relaxations were insensitive to NOS inhibition. In contrast, inhibiting NOS with LNNA significantly reduced ACh‐induced relaxation in RD‐fed angptl2 KD mice, suggesting that both EDHF and NO synergistically contributed to relaxation. This could reflect an impact of knocking down angptl2 on EDRF signaling in mesenteric arteries, similar to the remodeling of EDRF observed in femoral arteries from young KD mice in which PGI2 synergizes with the main EDRF NO.
In mesenteric arteries from WT mice fed an HFD, the contribution of EDHF to the relaxation was significantly reduced but compensated by NO. We previously showed that EDHF was a factor sensitive to mild dyslipidemia‐associated oxidative stress21; therefore, it is possible that in WT mice challenged by a 3‐month HFD, the NOS systems compensates for this impaired EDHF activity at the early stage of obesity. In KD mice fed an HFD, however, LNNA no longer reduced ACh‐induced relaxation of isolated mesenteric arteries, suggesting that the EDHF component was preserved. Altogether, in normal WT mice, our results demonstrate that regardless of the arterial bed, the main EDRF is sensitive to the metabolic stress induced by an HFD, and it is efficiently compensated for by a secondary EDRF, maintaining global endothelial function. In the long term, as in aging and chronic obesity, however, adaptive mechanisms may become overwhelmed, leading to permanent endothelial dysfunction, the primary step toward atherosclerosis. With low angptl2 levels, as in the KD mice, the endothelium is more resistant to the stress induced by the HFD, maintaining the functionality of the respective main EDRF. By extrapolation, lowering angptl2 may delay endothelial dysfunction.
We further characterized the impact of knocking down angptl2 on the metabolic stress induced by an HFD. In 2 recent studies focusing on insulin resistance and atherosclerosis, authors did not report differences between the lipid profiles of HFD‐fed WT and angptl2 knockout mice.3,5 Although we previously reported that infusion of purified recombinant angptl2 in young 3‐month‐old severely dyslipidemic (LDLR−/−;hApoB+/+) mice further increased LDL cholesterol levels and accelerated atherogenesis,4 we did not expect the difference in lipid handling between KD and WT mice fed an HFD reported in the present study. Indeed, after a 3‐month HFD, total cholesterol‐to‐HDL and LDL‐to‐HDL ratios remained unchanged in KD mice, which is in stark contrast with the expected increase in cholesterol levels3 and with that measured in WT mice. The favorable lipid profile of KD mice fed an HFD was associated with significantly lower levels of TG in the liver, which is in accordance with the results of Tabata et al.3 In addition, we found that KD mice exhibited a smaller degree of adipocyte hypertrophy in both eWAT and mWAT. There were lower levels of TNFα and transforming growth factor‐β mRNA in eWAT from HFD‐fed KD mice, likely a consequence of reduced fat accumulation associated with the lack of expression of angptl2,3 suggesting lower levels of inflammation in adipose tissues of KD mice. In line with this, it has been shown that impaired excess fat storage in adipocytes is closely linked to ectopic fat deposition,48 as observed in the increased hepatic lipid accumulation in HFD‐fed WT but not in KD mice. Unlike Tabata et al,3 however, we did not observe lower fasting insulin levels in KD mice. Discrepancies may be explained by the modality of genetic inactivation used, being a knockout and ours a KD model. Taken together, the more favorable lipid profile, lower fat accumulation, and proinflammatory gene expression may explain why the endothelium‐dependent dilatory function of arteries isolated from KD mice was insensitive to the HFD in contrast to arteries from WT mice.
An unexpected finding in this study was that angptl2 KD mice had lower basal heart rate than WT mice, which was consistent from 3 to 6 months of age. With the HFD, heart rate increased in KD mice but still tended to be lower than that in WT mice. This observation may contribute as well to the better endothelial function in KD mice. Indeed, studies in the middle‐aged and elderly have shown that an elevated resting heart rate correlated with subclinical inflammation49 and lowering basal heart rate in atherosclerosis‐prone mice in different studies delayed endothelial dysfunction associated with reduced oxidative stress.50–51 The beneficial effects of a lower heart rate on the endothelium is suggested to be a combination of sustained shear stress and lower cyclic mechanical stress, resulting in reduced damage to the endothelium.52
Heart rate, blood pressure, and energy expenditure have been shown to be regulated by leptin through central coactivation of the sympathetic nervous system and renin‐angiotensin system (see review in Mark 53). In our experimental models, leptin levels did not increase in the blood of KD mice fed an HFD. Therefore, a lack of increase in leptin during HFD in KD mice could participate in limiting the observed changes in heart rate and metabolic phenotype. Despite contrasting leptin levels, food intake during the 3‐month diet treatment was not significantly different between the 2 strains of mice. Interestingly, a recent study reported a possible link between adipocyte‐derived adiponectin, which shares established interactions with leptin in settings of obesity,54 and angptl2.8 In the current study, however, plasma levels of adiponectin were similar among all groups of mice. Taken together, angptl2 may interact with leptin pathways, but this hypothesis needs to be further tested.
To conclude, accumulating reports are starting to highlight the importance of angptl2 involvement in a plethora of pathologies ranging from obesity and cancer to atherosclerosis in the most recent reports.4–5,11 A common phenomenon underlying the aforementioned pathologies is the presence of an inflammatory environment. Inflammation in the endothelium favors homeostatic imbalances between vasodilators and vasoconstrictors, ultimately leading to endothelial dysfunction. Our data reveal better endothelial function in the arteries of angptl2 KD mice with preserved NO‐mediated dilation in femoral arteries and preserved EDHF‐dilation in mesenteric arteries against a 3‐month HFD‐induced hypercholesterolemia and fat accumulation in the liver and adipose tissues. Because targeting endothelial dysfunction is a rational therapeutic approach in treating patients with cardiovascular disease,55 further understanding the role of angptl2 in endothelial dysfunction associated with increased inflammation may reveal a new possible target to treatment and prevention of a range of cardiovascular disorders.
Sources of Funding
This work was supported by the Canadian Institutes of Health Research (MOP 14496 and 133649 to Dr Thorin) and the Foundation of the Montreal Heart Institute (Dr Thorin). Carol Yu is supported by the Postgraduate Scholarship from the Natural Sciences and Engineering Research Council of Canada. Dr Martel is supported by the Postgraduate Scholarship from the Fonds de la Recherche en Santé du Québec.
Disclosures
None.
Acknowledgments
We would like to thank Maya Mamarbachi and Marie‐Élaine Clavet‐Lanthier for their technical support in validating the genotype of the mice and processing and staining of tissue samples, respectively.
References
- 1.Reaven GM. Banting Lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988; 1988:1595-1607. [DOI] [PubMed] [Google Scholar]
- 2.Krauss RM, Winston M, Fletcher RN, Grundy SM. Obesity: impact of cardiovascular disease. Circulation. 1998; 98:1472-1476. [PubMed] [Google Scholar]
- 3.Tabata M, Kadomatsu T, Fukuhara S, Miyata K, Ito Y, Endo M, Urano T, Zhu HJ, Tsukano H, Tazume H, Kaikita K, Miyashita K, Iwawaki T, Shimabukuro M, Sakaguchi K, Ito T, Nakagata N, Yamada T, Katagiri H, Kasuga M, Ando Y, Ogawa H, Mochizuki N, Itoh H, Suda T, Oike Y. Angiopoietin‐like protein 2 promotes chronic adipose tissue inflammation and obesity‐related systemic insulin resistance. Cell Metab. 2009; 10:178-188. [DOI] [PubMed] [Google Scholar]
- 4.Farhat N, Thorin‐Trescases N, Mamarbachi M, Villeneuve L, Yu C, Martel C, Duquette N, Gayda M, Nigam A, Juneau M, Allen BG, Thorin E. Angiopoietin‐like 2 promotes atherogenesis in mice. J Am Heart Assoc. 2013; 2:e00020110.1161/JAHA.113.000201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horio E, Kadomatsu T, Miyata K, Arai Y, Hosokawa K, Doi Y, Ninomiya T, Horiguchi H, Endo M, Tabata M, Tazume H, Tian Z, Takahashi O, Terada K, Takeya M, Hao H, Hirose N, Minami T, Suda T, Kiyohara Y, Ogawa H, Kaikita K, Oike Y. Role of endothelial cell‐derived Angptl2 in vascular inflammation leading to endothelial dysfunction and atherosclerosis progression. Arterioscler Thromb Vasc Biol. 2014; 34:790-800. [DOI] [PubMed] [Google Scholar]
- 6.Doi Y, Ninomiya T, Hirakawa Y, Takahashi O, Mukai N, Hata J, Iwase M, Kitazono T, Oike Y, Kiyohara Y. Angiopoietin‐like protein 2 and risk of type 2 diabetes in a general Japanese population: the Hisayama study. Diabetes Care. 2012; 36:98-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tazume H, Miyata K, Tian Z, Endo M, Horiguchi H, Takahashi O, Horio E, Tsukano H, Kadomatsu T, Nakashima Y, Kunitomo R, Kaneko Y, Moriyama S, Sakaguchi H, Okamoto K, Hara M, Yoshinaga T, Yoshimura K, Aoki H, Araki K, Hao H, Kawasuji M, Oike Y. Macrophage‐derived angiopoietin‐like protein 2 accelerates development of abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2012; 32:1400-1409. [DOI] [PubMed] [Google Scholar]
- 8.Tian Z, Miyata K, Tazume H, Sakaguchi H, Kadomatsu T, Horio E, Takahashi O, Komohara Y, Araki K, Hirata Y, Tabata M, Takanashi S, Takeya M, Hao H, Shimabukuro M, Sata M, Kawasuji M, Oike Y. Perivascular adipose tissue‐secreted angiopoietin‐like protein 2 (Angptl2) accelerates neointimal hyperplasia after endovascular injury. J Mol Cell Cardiol. 2013; 57:1-12. [DOI] [PubMed] [Google Scholar]
- 9.Okada T, Tsukano H, Endo M, Tabata M, Miyata K, Kadomatsu T, Miyashita K, Semba K, Nakamura E, Tsukano M, Mizuta H, Oike Y. Synoviocyte‐derived angiopoietin‐like protein 2 contributes to synovial chronic inflammation in rheumatoid arthritis. Am J Pathol. 2010; 176:2309-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogata A, Endo M, Aoi J, Takahashi O, Kadomatsu T, Miyata K, Tian Z, Jinnin M, Fukushima S, Ihn H, Oike Y. The role of angiopoietin‐like protein 2 in pathogenesis of dermatomyositis. Biochem Biophys Res Commun. 2012; 418:494-499. [DOI] [PubMed] [Google Scholar]
- 11.Aoi J, Endo M, Kadomatsu T, Miyata K, Nakano M, Horiguchi H, Ogata A, Odagiri H, Yano M, Araki K, Jinnin M, Ito T, Hirakawa S, Ihn H, Oike Y. Angiopoietin‐like protein 2 is an important facilitator of inflammatory carcinogenesis and metastasis. Cancer Res. 2011; 71:7502-7512. [DOI] [PubMed] [Google Scholar]
- 12.Farhat N, Thorin‐Trescases N, Voghel G, Villeneuve L, Mamarbachi M, Perrault LP, Carrier M, Thorin E. Stress‐induced senescence predominates in endothelial cells isolated from atherosclerotic chronic smokers. Can J Physiol Pharmacol. 2008; 86:761-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitazawa M, Nagano M, Masumoto KH, Shigeyoshi Y, Natsume T, Hashimoto S. Angiopoietin‐like 2, a circadian gene, improves type 2 diabetes through potentiation of insulin sensitivity in mice adipocytes. Endocrinology. 2011; 152:2558-2567. [DOI] [PubMed] [Google Scholar]
- 14.Muramoto A, Tsushita K, Kato A, Ozaki N, Tabata M, Endo M, Oike Y, Oiso Y. Angiopoietin‐like protein 2 sensitively responds to weight reduction induced by lifestyle intervention on overweight Japanese men. Nutr Diabetes. 2011; 1:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980; 288:373-376. [DOI] [PubMed] [Google Scholar]
- 16.Moncada S, Erusalimsky JD. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat Rev Mol Cell Biol. 2002; 3:214-220. [DOI] [PubMed] [Google Scholar]
- 17.Komori K, Vanhoutte PM. Endothelium‐derived hyperpolarizing factor. Blood Vessels. 1990; 27:238-245. [DOI] [PubMed] [Google Scholar]
- 18.Durand MJ, Gutterman DD. Diversity in mechanisms of endothelium‐dependent vasodilation in health and disease. Microcirculation. 2013; 20:239-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winquist RJ, Bunting PB, Baskin EP, Wallace AA. Decreased endothelium‐dependent relaxation in New Zealand genetic hypertensive rats. J Hypertens. 1984; 2:541-545. [DOI] [PubMed] [Google Scholar]
- 20.Lockette W, Otsuka Y, Carretero O. The loss of endothelium‐dependent vascular relaxation in hypertension. Hypertension. 1986; 8:II61-II66. [DOI] [PubMed] [Google Scholar]
- 21.Krummen S, Drouin A, Gendron ME, Falck JR, Thorin E. ROS‐sensitive cytochrome P450 activity maintains endothelial dilatation in ageing but is transitory in dyslipidaemic mice. Br J Pharmacol. 2006; 147:897-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Najibi S, Cowan CL, Palacino JJ, Cohen RA. Enhanced role of potassium channels in relaxations to acetylcholine in hypercholesterolemic rabbit carotid artery. Am J Physiol. 1994; 266:H2061-H2067. [DOI] [PubMed] [Google Scholar]
- 23.Huang A, Sun D, Smith CJ, Connetta JA, Shesely EG, Koller A, Kaley G. In eNOS knockout mice skeletal muscle arteriolar dilation to acetylcholine is mediated by EDHF. Am J Physiol Heart Circ Physiol. 2000; 278:H762-H768. [DOI] [PubMed] [Google Scholar]
- 24.Huang A, Sun D, Carroll MA, Jiang H, Smith CJ, Connetta JA, Falck JR, Shesely EG, Koller A, Kaley G. EDHF mediates flow‐induced dilation in skeletal muscle arterioles of female eNOS‐KO mice. Am J Physiol Heart Circ Physiol. 2001; 280:H2462-H2469. [DOI] [PubMed] [Google Scholar]
- 25.Sun D, Huang A, Smith CJ, Stackpole CJ, Connetta JA, Shesely EG, Koller A, Kaley G. Enhanced release of prostaglandins contributes to flow‐induced arteriolar dilation in eNOS knockout mice. Circ Res. 1999; 85:288-293. [DOI] [PubMed] [Google Scholar]
- 26.Aoi J, Endo M, Kadomatsu T, Miyata K, Ogata A, Horiguchi H, Odagiri H, Masuda T, Fukushima S, Jinnin M, Hirakawa S, Sawa T, Akaike T, Ihn H, Oike Y. Angiopoietin‐like protein 2 accelerates carcinogenesis by activating chronic inflammation and oxidative stress. Mol Cancer Res. 2014; 12:239-249. [DOI] [PubMed] [Google Scholar]
- 27.Damjanovic M, Barton M. Fat intake and cardiovascular response. Curr Hypertens Rep. 2008; 10:25-31. [DOI] [PubMed] [Google Scholar]
- 28.Bolduc V, Baraghis E, Duquette N, Thorin‐Trescases N, Lambert J, Lesage F, Thorin E. Catechin prevents severe dyslipidemia‐associated changes in wall biomechanics of cerebral arteries in LDLr−/−:hApoB+/+ mice and improves cerebral blood flow. Am J Physiol Heart Circ Physiol. 2012; 302:H1330-H1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorin E, Shreeve SM, Thorin‐Trescases N, Bevan JA. Reversal of endothelin‐1 release by stimulation of endothelial alpha2‐adrenoceptor contributes to cerebral vasorelaxation. Hypertension. 1997; 30:830-836. [DOI] [PubMed] [Google Scholar]
- 30.Drouin A, Thorin‐Trescases N, Hamel E, Falck JR, Thorin E. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. 2007; 73:73-81. [DOI] [PubMed] [Google Scholar]
- 31.Drouin A, Farhat N, Bolduc V, Thorin‐Trescases N, Gillis MA, Villeneuve L, Nguyen A, Thorin E. Up‐regulation of thromboxane A(2) impairs cerebrovascular eNOS function in aging atherosclerotic mice. Pflugers Arch. 2011; 462:371-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khairallah RJ, Khairallah M, Gelinas R, Bouchard B, Young ME, Allen BG, Lopaschuk GD, Deschepper CF, Des Rosiers C. Cyclic GMP signaling in cardiomyocytes modulates fatty acid trafficking and prevents triglyceride accumulation. J Mol Cell Cardiol. 2008; 45:230-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gelinas R, Thompson‐Legault J, Bouchard B, Daneault C, Mansour A, Gillis MA, Charron G, Gavino V, Labarthe F, Des Rosiers C. Prolonged QT interval and lipid alterations beyond beta‐oxidation in very long‐chain acyl‐CoA dehydrogenase null mouse hearts. Am J Physiol Heart Circ Physiol. 2011; 301:H813-H823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957; 226:497-509. [PubMed] [Google Scholar]
- 35.Ruiz J, Antequera T, Andres AI, Petron M, Muriel E. Improvement of a solid phase extraction method for analysis of lipid fractions in muscle foods. Anal Chim Acta. 2004; 520:201-205. [Google Scholar]
- 36.Lepage G, Roy CC. Direct transesterification of all classes of lipids in a one‐step reaction. J Lipid Res. 1986; 27:114-120. [PubMed] [Google Scholar]
- 37.Gendron ME, Theoret JF, Mamarbachi AM, Drouin A, Nguyen A, Bolduc V, Thorin‐Trescases N, Merhi Y, Thorin E. Late chronic catechin antioxidant treatment is deleterious to the endothelial function in aging mice with established atherosclerosis. Am J Physiol Heart Circ Physiol. 2010; 298:H2062-H2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adeagbo AS, Triggle CR. Varying extracellular [K+]: a functional approach to separating EDHF‐ and EDNO‐related mechanisms in perfused rat mesenteric arterial bed. J Cardiovasc Pharmacol. 1993; 21:423-429. [PubMed] [Google Scholar]
- 39.Thorin E, Huang PL, Fishman MC, Bevan JA. Nitric oxide inhibits alpha2‐adrenoceptor‐mediated endothelium‐dependent vasodilation. Circ Res. 1998; 82:1323-1329. [DOI] [PubMed] [Google Scholar]
- 40.Shimokawa H, Flavahan NA, Lorenz RR, Vanhoutte PM. Prostacyclin releases endothelium‐derived relaxing factor and potentiates its action in coronary arteries of the pig. Br J Pharmacol. 1988; 95:1197-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wigg SJ, Tare M, Tonta MA, O'Brien RC, Meredith IT, Parkington HC. Comparison of effects of diabetes mellitus on an EDHF‐dependent and an EDHF‐independent artery. Am J Physiol Heart Circ Physiol. 2001; 281:H232-H240. [DOI] [PubMed] [Google Scholar]
- 42.Scherer PE. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes. 2006; 55:1537-1545. [DOI] [PubMed] [Google Scholar]
- 43.Feletou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture). Am J Physiol Heart Circ Physiol. 2006; 291:H985-H1002. [DOI] [PubMed] [Google Scholar]
- 44.de Leval X, Hanson J, David JL, Masereel B, Pirotte B, Dogne JM. New developments on thromboxane and prostacyclin modulators part II: prostacyclin modulators. Curr Med Chem. 2004; 11:1243-1252. [DOI] [PubMed] [Google Scholar]
- 45.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990; 87:1620-1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molnar J, Yu S, Mzhavia N, Pau C, Chereshnev I, Dansky HM. Diabetes induces endothelial dysfunction but does not increase neointimal formation in high‐fat diet fed C57BL/6J mice. Circ Res. 2005; 96:1178-1184. [DOI] [PubMed] [Google Scholar]
- 47.Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H, Takeshita A. Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in mice. J Clin Invest. 2000; 106:1521-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Unger RH, Orci L. Lipotoxic diseases of nonadipose tissues in obesity. Int J Obes Relat Metab Disord. 2000; 24suppl 4:S28-S32. [DOI] [PubMed] [Google Scholar]
- 49.Sajadieh A, Nielsen OW, Rasmussen V, Hein HO, Abedini S, Hansen JF. Increased heart rate and reduced heart‐rate variability are associated with subclinical inflammation in middle‐aged and elderly subjects with no apparent heart disease. Eur Heart J. 2004; 25:363-370. [DOI] [PubMed] [Google Scholar]
- 50.Custodis F, Baumhakel M, Schlimmer N, List F, Gensch C, Bohm M, Laufs U. Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E‐deficient mice. Circulation. 2008; 117:2377-2387. [DOI] [PubMed] [Google Scholar]
- 51.Drouin A, Gendron ME, Thorin E, Gillis MA, Mahlberg‐Gaudin F, Tardif JC. Chronic heart rate reduction by ivabradine prevents endothelial dysfunction in dyslipidaemic mice. Br J Pharmacol. 2008; 154:749-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thorin E, Thorin‐Trescases N. Vascular endothelial ageing, heartbeat after heartbeat. Cardiovasc Res. 2009; 84:24-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mark AL. Selective leptin resistance revisited. Am J Physiol Regul Integr Comp Physiol. 2013; 305:R566-R581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koerner A, Kratzsch J, Kiess W. Adipocytokines: leptin–the classical, resistin–the controversical, adiponectin–the promising, and more to come. Best Pract Res Clin Endocrinol Metab. 2005; 19:525-546. [DOI] [PubMed] [Google Scholar]
- 55.Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003; 23:168-175. [DOI] [PubMed] [Google Scholar]