

Abstract
Background
Genetics can be used to predict drug effects and generate hypotheses around alternative indications. To support Losmapimod, a p38 mitogen‐activated protein kinase inhibitor in development for acute coronary syndrome, we characterized gene variation in MAPK11/14 genes by exome sequencing and follow‐up genotyping or imputation in participants well‐phenotyped for cardiovascular and metabolic traits.
Methods and Results
Investigation of genetic variation in MAPK11 and MAPK14 genes using additive genetic models in linear or logistic regression with cardiovascular, metabolic, and biomarker phenotypes highlighted an association of RS2859144 in MAPK14 with myeloperoxidase in a dyslipidemic population (Genetic Epidemiology of Metabolic Syndrome Study), P=2.3×10−6). This variant (or proxy) was consistently associated with myeloperoxidase in the Framingham Heart Study and Cardiovascular Health Study studies (replication meta‐P=0.003), leading to a meta‐P value of 9.96×10−7 in the 3 dyslipidemic groups. The variant or its proxy was then profiled in additional population‐based cohorts (up to a total of 58 930 subjects) including Cohorte Lausannoise, Ely, Fenland, European Prospective Investigation of Cancer, London Life Sciences Prospective Population Study, and the Genetics of Obesity Associations study obesity case–control for up to 40 cardiovascular and metabolic traits. Overall analysis identified the same single nucleotide polymorphisms to be nominally associated consistently with glomerular filtration rate (P=0.002) and risk of obesity (body mass index ≥30 kg/m2, P=0.004).
Conclusions
As myeloperoxidase is a prognostic marker of coronary events, the MAPK14 variant may provide a mechanistic link between p38 map kinase and these events, providing information consistent with current indication of Losmapimod for acute coronary syndrome. If replicated, the association with glomerular filtration rate, along with previous biological findings, also provides support for kidney diseases as alternative indications.
Keywords: acute coronary syndrome, drug target gene, exome sequencing, myeloperoxidase, rare variation
Introduction
Previously, Losmapimod (a fast‐acting p38 MAPK‐α and MAPK‐β inhibitor) and related compounds had been applied toward a number of indications, such as rheumatoid arthritis and depression, with failure to obtain proof‐of‐concept. These chronic disease settings may not take advantage of the rapid stress‐mediated response inherent in p38 mitogen‐activated protein kinase (MAPK) activity. MAPK is an intracellular kinase that functions as an important mediator of the inflammatory signaling cascade that leads to activation of cytokine production, more easily observed during acute events, such as acute coronary syndrome. In the setting of statin therapy, Losmapimod significantly attenuated the postpercutaneous intervention inflammatory response, as measured by high‐sensitivity C‐reactive protein (CRP).1 In a phase 2 study, the effects of Losmapimod on safety, infarct size, and cardiac function were evaluated.2 Encouraging results3 have led to the progression of Losmapimod to a phase 3 outcome trial for acute coronary syndromes.
The genes that encode MAPK‐α and ‐β (MAPK14 and MAPK11, respectively) were included in a large sequencing experiment examining the exons and flanking regions of 202 drug target genes in more than 14 000 participants phenotyped for a wide range of diseases and medically relevant traits.4 The Nelson et al sequencing study reported common and rare variation in 202 genes including MAPK14 and MAPK11, drug target genes for Losmapimod, and tested associations of common variants and rare coding variants, in aggregate, with 12 diseases.4 No common diseases were found to be associated with MAPK14 or MAPK11 in any published study including Nelson et al.4 The present work focuses on profiling variants within the MAPK11 and MAPK14 genes for association with cardiovascular and metabolic phenotypes and related biomarkers. In‐depth phenotype information coupled with a complete picture of common and rare genetic variation available from the sequencing study provides an opportunity to use genetics as an instrument to better understand the role of MAPK‐α/β in common diseases (including acute coronary syndrome) and its relationship to related biomarkers. It is anticipated that the low‐frequency range (0.1% to 5% minor allele frequency [MAF]) could contain functional variation with larger effect sizes than that observed with common variation (>5% MAF).5 Should such variants be found that mimic on‐target effects, they may make useful tools for predicting drug effects and suggesting alternative indications for MAPK‐α/β modulators.
Variants identified by Nelson et al and present in the Genetic Epidemiology of Metabolic Syndrome Study (GEMS)6 and Cohorte Lausannoise (CoLaus) study7 were profiled for association with cardiovascular and metabolic phenotypes and related biomarkers (38 and 40 traits, respectively). Analyses of these traits were performed on all sequenced subjects for GEMS (n=1576) and CoLaus (n=2086) and within dyslipidemic subjects for GEMS (n=787). The variants identified as associated in these initial analyses were evaluated in a small replication study (only myeloperoxidase [MPO] and glomerular filtration rate [GFR] results were obtained) within the Cardiovascular Health Study (CHS) and Framingham Heart Study (FHS). The same variants were then analyzed more broadly for association with 40 cardiovascular and metabolic traits in a meta‐analysis in an expanded set including CoLaus, Life Sciences Prospective Population Study (LOLIPOP), European Prospective Investigation of Cancer (EPIC)‐Norfolk, Ely, Fenland, and Genetics of Obesity Associations (GenOA) studies (Table 1 provides a summary of samples analyzed and Figure 1 a study flow diagram) to provide a cardiovascular and metabolic profile. We summarize the results and describe how they may relate to clinical trial results.
Table 1.
Summary of the Sample Characteristics and Phenotypes Analyses for Each Sample
| GEMS Dyslipidemia | GEMS Normolipidemia | CHS | FHS | GenOA Obesity Cases | GenOA Obesity Controls | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | 787 | 792 | 3897 | 2819 | 860 | 947 | ||||||
| Categorical variable, N (%) | ||||||||||||
| Men | 467 (59%) | 459 (58%) | 1707 (44%) | 1308 (46%) | 241 (28%) | 376 (40%) | ||||||
| Statins | 339 (43%) | 21 (3%) | 76 (2%) | |||||||||
| Lipid med | 398 (51%) | 21 (3%) | 203 (5%) | 584 (21%) | ||||||||
| Disease status, N (%) | ||||||||||||
| CAD | 188 (24%) | 18 (2%) | 744 (19%) | |||||||||
| Dyslipidemia | 787 (100%) | 0 (0%) | ||||||||||
| Obesity | 284 (61%) | 184 (39%) | 1083 (28%) | 852 (30%) | 860 (100%) | 947 (0%) | ||||||
| T2D | 0 (0%) | 0 (0%) | 1104 (28%) | 366 (13%) | ||||||||
| Continuous variable (N, Mean (SD)) | ||||||||||||
| Age, y | 775 | 49 (9) | 781 | 55.5 (9.3) | 3897 | 72.8 (5.6) | 2819 | 61 (9) | 860 | 49.9 (10.7) | 947 | 44.7 (15.1) |
| Hypertension | ||||||||||||
| DBP, mm Hg | 771 | 82 (11) | 780 | 81 (11) | 3886 | 70 (11) | 2819 | 74 (10) | 857 | 82 (10) | 947 | 74 (9) |
| SBP, mm Hg | 771 | 132 (17) | 780 | 132 (18) | 3890 | 135 (21) | 2819 | 127 (19) | 858 | 135 (18) | 947 | 121 (15) |
| Inflammation | ||||||||||||
| Adiponectin, μg/mL | 743 | 5.5 (3.9) | 757 | 8.1 (5.2) | 2352 | 10 (6.3) | ||||||
| CRPU, mg/L | 743 | 6 (10) | 757 | 5 (8) | ||||||||
| Fibrinogen, g/L | 605 | 2.6 (0.62) | 689 | 2.55 (0.64) | 3874 | 3.2 (0.65) | 2808 | 3.8 (0.74) | ||||
| IL‐1b, pg/mL | 603 | 47.9 (115) | 689 | 47.5 (103) | ||||||||
| IL‐6, pg/mL | 603 | 45 (72) | 689 | 51 (64) | 2810 | 4 (4.8) | ||||||
| IL‐8, pg/mL | 603 | 38.2 (76.6) | 689 | 48.4 (80.3) | ||||||||
| Leptin, ng/mL | 685 | 16.1 (11.7) | 696 | 14.5 (12.2) | ||||||||
| MPO, ng/mL | 605 | 52 (104) | 690 | 43 (51.7) | 3044 | 49 (49) | 2722 | 48 (31) | ||||
| TNFA pg/mL | 717 | 88 (132) | 821 | 112 (425) | 2144 | 145 (120) | ||||||
| Insulin sensitivity | ||||||||||||
| 2‐h glucose, mmol/L | 958 | 7.2 (2.7) | ||||||||||
| Glucose, mmol/L | 683 | 5.2 (0.6) | 752 | 5.1 (0.5) | 3897 | 6.1 (1.8) | 2771 | 5.8 (1.4) | 858 | 5.8 (2.0) | 945 | 4.7 (0.5) |
| HOMA‐B | 2730 | 142 (376) | ||||||||||
| HOMA‐IR | 651 | 3.1 (2.5) | 728 | 1.8 (1.3) | 2730 | 4.11 (3.65) | 847 | 2.98 (2.7) | 941 | 0.78 (0.41) | ||
| Insulin, mIU/mL | 743 | 12.9 (9.6) | 753 | 7.7 (5.8) | 3866 | 16.6 (23) | 2730 | 15.24 (10.6) | 848 | 12.4 (9.7) | 941 | 4.38 (2.3) |
| Kidney function | ||||||||||||
| Creatinine, μmol/L | 3897 | 84 (26.7) | ||||||||||
| CRU, mmol/day | ||||||||||||
| GFR | 2819 | 85 (18.9) | ||||||||||
| MACR | ||||||||||||
| MALB | ||||||||||||
| Lipid | ||||||||||||
| APOB, g/L | 743 | 1.2 (0.3) | 756 | 1.04 (0.24) | ||||||||
| CHOL, mmol/L | 775 | 5.7 (1.2) | 781 | 5.5 (0.9) | 3895 | 212 (39) | 2819 | 5.2 (0.9) | 859 | 5.3 (1.02) | 945 | 5.1 (0.97) |
| HDL, mmol/L | 775 | 0.9 (0.2) | 781 | 1.6 (0.3) | 3890 | 1.4 (0.4) | 2818 | 1.4 (0.4) | 854 | 1.27 (0.36) | 945 | 1.63 (0.42) |
| LDL, mmol/L | 618 | 3.4 (1.1) | 781 | 3.4 (0.9) | 3836 | 3.37 (0.9) | 3778 | 3.1 (0.84) | 837 | 3.24 (0.89) | 943 | 3.07 (0.83) |
| TRIG, mmol/L | 775 | 3.5 (2.2) | 781 | 1 (0.3) | 3895 | 1.6 (0.85) | 2819 | 1.6 (1.0) | 858 | 1.76 (1.0) | 945 | 0.94 (0.5) |
| Liver function | ||||||||||||
| ALB, g/L | ||||||||||||
| ALP, U/L | ||||||||||||
| ALT, U/L | ||||||||||||
| GGT, U/L | ||||||||||||
| Obesity | ||||||||||||
| BMI, kg/m2 | 775 | 28.7 (3.6) | 780 | 28.3 (3.7) | 3897 | 26.4 (4.5) | 2819 | 28.2 (5.3) | 857 | 40.7 (8.96) | 947 | 20.68 (2.03) |
| Body fat, % | ||||||||||||
| Hip, cm | 764 | 108 (8.1) | 778 | 108.3 (8.9) | 3885 | 102 (9.6) | 2785 | 105 (10.4) | ||||
| Waist, cm | 765 | 98.3 (10.6) | 778 | 95.8 (12.4) | 3897 | 94 (12.8) | 2789 | 100 (14.1) | 856 | 116 (20.3) | 946 | 78.6 (7.5) |
| Weight, kg | 775 | 84.7 (13.6) | 781 | 83.2 (14) | 3897 | 159 (31.5) | 2818 | 79.5 (17.5) | 858 | 114 (29) | 947 | 61.1 (10.2) |
| Waist/hip ratio | 3879 | 0.92 (0.1) | 2782 | 0.95 (0.08) | ||||||||
| Others | ||||||||||||
| Calcium, mmol/L | ||||||||||||
| Uric acid, μmol/L | 3897 | 339 (89) | ||||||||||
| CoLaus | LOLIPOPW | EPIC | Fenland | ELY | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | 5846 | 6565 | 20 370 | 6379 | 1722 | |||||
| Categorical variable, N (%) | ||||||||||
| Men | 2778 (48%) | 4264 (65%) | 9604 (47%) | 2938 (46%) | 792 (46%) | |||||
| Statins | 498 (9%) | 1106 (17%) | ||||||||
| Lipid med | 308 (2%) | 173 (3%) | 133 (8%) | |||||||
| Disease status, N (%) | ||||||||||
| CAD | 1369 (7%) | |||||||||
| Dyslipidemia | 520 (9%) | 729 (11%) | 2258 (11%) | 748 (12%) | 208 (12%) | |||||
| Obesity | 904 (15%) | 1736 (26%) | 2974 (15%) | 1238 (19%) | 380 (22%) | |||||
| T2D | 385 (7%) | 511 (8%) | 644 (3%) | 81 (1%) | 93 (5%) | |||||
| Continuous variable (N, Mean (SD)) | ||||||||||
| Age, y | 5846 | 53 (10.7) | 6565 | 52.9 (11.4) | 20,364 | 59.3 (9.2) | 6378 | 46.7 (7.3) | 1722 | 61.1 (9.1) |
| Hypertension | ||||||||||
| DBP, mm Hg | 5845 | 79.3 (11.0) | 6563 | 79.6 (10.6) | 17 532 | 82.3 (11.0) | 6373 | 75.6 (10.1) | 1714 | 78.7 (10.3) |
| SBP, mm Hg | 5845 | 128 (18.1) | 6563 | 131 (19.4) | 17 532 | 135 (18.1) | 6374 | 123 (15.3) | 1714 | 132 (16.5) |
| Inflammation | ||||||||||
| Adiponectin, μg/mL | 5773 | 10 (8) | 4944 | 6.5 (3.4) | 763 | 7.5 (3.7) | ||||
| CRPU, mg/L | 5636 | 2.5 (3.5) | 6236 | 4.2 (7.9) | 13 616 | 2.9 (4.3) | 4847 | 2.9 (4.5) | 655 | 1.9 (3.4) |
| Fibrinogen, g/L | 16 169 | 2.9 (0.8) | ||||||||
| IL‐1b, pg/mL | 3599 | 6.3 (31) | ||||||||
| IL‐6, pg/mL | 5340 | 10 (107) | 395 | 1.9 (3.2) | ||||||
| IL‐8, pg/mL | 1152 | 36 (201) | ||||||||
| Leptin, ng/mL | 3558 | 13.6 (10.8) | 4941 | 15.8 (16.6) | 1708 | 22.1 (22.8) | ||||
| MPO, ng/mL | 2534 | 102 (79) | ||||||||
| TNFA, pg/mL | 5730 | 6.4 (54.3) | ||||||||
| Insulin sensitivity | ||||||||||
| 2‐h glucose, mmol/L | 510 | 5.9 (2.6) | 6201 | 5.3 (1.6) | 1589 | 6.4 (2.4) | ||||
| Glucose, mmol/L | 5636 | 5.5 (1.1) | 6565 | 5.4 (1.7) | 6319 | 4.8 (0.5) | 1717 | 5.1 (0.8) | ||
| HOM‐AB | 5000 | 92 (72) | 6257 | 119 (157) | 6235 | 124 (84) | 1707 | 136 (90) | ||
| HOMA‐IR | 5000 | 2.3 (2.3) | 6257 | 2.7 (3.8) | 6238 | 1.72 (1.4) | 1707 | 2.25 (1.63) | ||
| Insulin, mIU/mL | 5171 | 8.8 (6.2) | 6257 | 10.2 (10.3) | 6261 | 6.7 (4.7) | 1709 | 8.4 (5.2) | ||
| Kidney function | ||||||||||
| Creatinine, μmol/L | 5635 | 80 (22.2) | 6564 | 90.6 (18.6) | 13 654 | 86.3 (18.2) | 6274 | 76.2 (14.7) | 792 | 84.5 (15.9) |
| CRU, mmol/day | 5508 | 13.3 (6.7) | 6360 | 10.5 (7.7) | ||||||
| GFR | 5635 | 78.7 (15.7) | 6562 | 92.4 (27) | 13 654 | 75.8 (21.4) | ||||
| MACR | 5507 | 1.6 (7.0) | 4637 | 1.3 (3.4) | ||||||
| MALB | 5507 | 18.9 (80.1) | 4637 | 12.9 (23) | ||||||
| Lipid | ||||||||||
| APOB, g/L | 5788 | 1.74 (1.34) | 13 469 | 1 (0.2) | 4942 | 1 (0.2) | ||||
| CHOL, mmol/L | 5636 | 5.6 (1) | 6565 | 5.4 (1.1) | 17 005 | 6.2 (1.2) | 6331 | 5.4 (1) | 1719 | 5.6 (1.1) |
| HDL, mmol/L | 5636 | 1.6 (0.4) | 6565 | 1.4 (0.4) | 16 320 | 1.4 (0.4) | 6331 | 1.5 (0.4) | 1717 | 1.5 (0.4) |
| LDL, mmol/L | 5554 | 3.3 (0.9) | 6421 | 3.3 (0.9) | 16 321 | 4.0 (1.0) | 6281 | 3.4 (0.9) | 1715 | 3.5 (0.9) |
| TRIG, mmol/L | 5636 | 1.4 (1.1) | 6564 | 1.5 (1.2) | 17 003 | 1.93 (1.1) | 6331 | 1.2 (0.8) | 1717 | 1.4 (0.8) |
| Liver function | ||||||||||
| ALB, g/L | 5636 | 44.2 (2.5) | 6565 | 43.5 (2.8) | 13 780 | 41.5 (5.7) | 6330 | 42.1 (2.6) | 497 | 43.3 (2.9) |
| ALP, U/L | 5636 | 63.6 (20.5) | 6558 | 77.4 (31.1) | 13 826 | 60.3 (20.3) | 6324 | 80.8 (21.3) | ||
| ALT, U/L | 5636 | 27.8 (19.6) | 6563 | 28.7 (19.6) | 6328 | 28.7 (15.9) | ||||
| GGT, U/L | 5636 | 33.3 (59.5) | 6565 | 43.2 (68.1) | 13 869 | 32.8 (28.6) | 6329 | 34.2 (25.5) | ||
| Obesity | ||||||||||
| BMI, kg/m2 | 5844 | 25.8 (4.6) | 6563 | 27.6 (5.1) | 17 540 | 26.1 (3.9) | 5836 | 26.9 (4.8) | 1719 | 27.2 (4.7) |
| Body fat, % | 5792 | 29.4 (9.2) | 6290 | 30.5 (8.9) | 11 534 | 31.9 (11.1) | 5761 | 30.3 (8.8) | 1698 | 33.7 (8.9) |
| Hip, cm | 5842 | 101.7 (9.3) | 6550 | 103.3 (9.5) | 5832 | 103.6 (9.2) | 1707 | 106.1 (10) | ||
| Waist, cm | 5845 | 89.3 (13.4) | 6550 | 95.5 (13.8) | 17 553 | 88.0 (12.4) | 5835 | 91 (13.3) | 1709 | 93 (13.2) |
| Weight, kg | 5845 | 73.6 (15.2) | 6563 | 80.7 (16.9) | 5836 | 78.1 (16.3) | 1720 | 76.4 (15) | ||
| Waist hip ratio | 5842 | 0.9 (0.1) | 6549 | 0.9 (0.1) | 17 536 | 0.86 (0.1) | 1707 | 0.88 (0.1) | ||
| Others | ||||||||||
| Calcium, mmol/L | 5636 | 2.2 (0.1) | 6564 | 2.3 (0.1) | 6324 | 2.2 (0.1) | 792 | 2.1 (0.1) | ||
| Uric acid, μmol/L | 5636 | 314 (85) | 6556 | 314 (89) | 13 708 | 295 (81) | ||||
ALB indicates albumin; ALT, alanine aminotransferase; APOB, apolipoprotein B; BMI, body mass index; CAD, coronary artery disease; CHOL, cholesterol; CHS, Cardiovascular Health Study; CoLaus, Cohorte Lausannoise; CRPU, ultrasensitive C‐reactive protein; CRU, urinary creatinine; DBP, diastolic blood pressure; EPIC, European Prospective Investigation of Cancer; FHS, Framingham Heart Study; GEMS, Genetic Epidemiology of Metabolic Syndrome Study; GenOA, Genetics of Obesity Associations; GFR, glomerular filtration rate; GGT, γ‐glutamyl transferase; glomerular filtration rate; HDL, high‐density lipoprotein; HOMA‐B, homeostatic model assessment – beta‐cell; HOMA‐IR, homeostatic model assessments – insulin resistance; IL, interleukin; LDL, low‐density lipoprotein; LOLIPOPW, London Life Sciences Prospective Population Study – Whites; MACR, microalbumin‐creatinine ratio; MALB, microalbuminuria; MPO, myeloperoxidase; SBP, systolic blood pressure; T2D, type 2 diabetes; TNFA, tumor necrosis factor α; TRIG, triglycerides.
Figure 1.
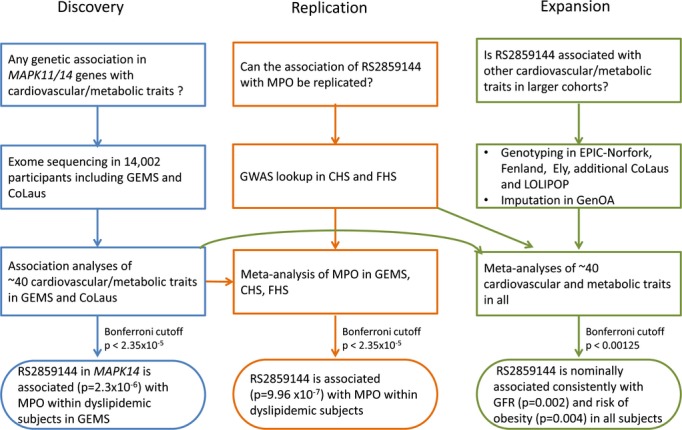
Study flow chart. CHS indicates Cardiovascular Health Study; CoLaus, Cohorte Lausannoise; FHS, Framingham Heart Study; GEMS, Genetic Epidemiology of Metabolic Syndrome Study; GenOA, Genetic Obesity Associations; GFR, glomerular filtration rate; GWAS, genome‐wide association study; LOLIPOP, London Life Sciences Prospective Population Study; MPO, myeloperoxidase.
Materials and Methods
Population Characteristics
GEMS Study
The GEMS study is a large multinational study designed to explore the genetic basis of the metabolic syndrome.6 Subjects were recruited from 2 centers in Europe (Oulu, Finland and Lausanne, Switzerland), 1 in the United States (Dallas, TX), 1 in Canada (Ottawa, Ontario), and 1 in Australia (Adelaide, South Australia). Dyslipidemic subjects were required to have the combination of an elevated plasma triglyceride (>75th percentile) and a low serum high‐density lipoprotein (HDL)‐cholesterol (<25th percentile) for their age, sex, and country threshold (age 18 to 75 years) and were nondiabetic. Unrelated normolipidemic controls were required to have plasma triglyceride lower than 50th percentile, serum HDL cholesterol >50th percentile for their age, sex, and country threshold, body mass index (BMI) >25 kg/m2, and be >40 years of age. The subjects have phenotypes for cardiovascular and metabolic traits as well as biomarkers of inflammation. Dyslipidemic subjects (n=787 subjects) and normolipidemic controls (n=792 subjects), matched by sex, age, and collection center were sequenced. In some analyses, only dyslipidemic subjects were analyzed.
Cardiovascular Health Study
The CHS is a population‐based, observational study of risk factors for clinical and subclinical cardiovascular diseases.8 The study recruited participants 65 years and older from 4 US communities (Forsyth County, North Carolina; Sacramento County, California; Washington County, Maryland; and Pittsburgh, Pennsylvania) in 2 phases: 5201 participants from 1989 to 1990, and 687 (primarily African American participants) from 1992 to 1993. CHS participants completed standardized clinical examinations and questionnaires at study baseline and at 9 annual follow‐up visits. Follow‐up for clinical events occurs every 6 months. MPO was measured from frozen plasma samples (n=3044) collected at the 1992–1993 CHS examination.
Framingham Heart Study
The FHS was initiated in 1948 and recruited, from the town of Framingham, MA, a total of 5209 participants, most from European ancestry, who have undergone biannual examinations to study cardiovascular disease and related risk factors.9 The Offspring cohort was recruited in 1971 and includes 5124 children of the original cohort and their spouses.10 Serum MPO measures was available for 2940 FHS Offspring cohort participants during the seventh cycle of examination (1998–2001).
CoLaus Study
The CoLaus study is a community‐based study of 6188 European white subjects aged 35 to 75 years. Participants were drawn from the CHUV University Hospital in Lausanne Switzerland7 and studied for cardiovascular and metabolic phenotypes.
LOLIPOP Study
The London Life Sciences Prospective Population Study (LOLIPOP) is a population‐based study of 21 915 subjects identified from the lists of 58 general practitioners in West London.11 Participants are primarily Indian Asians and European whites aged 35 to 75 years, who have been characterized for cardiovascular phenotypes. Only European whites (n=6565) were included in this analysis.
EPIC‐Norfolk
The EPIC‐Norfolk study is a cohort study investigating the relationship between diet and incident disease.12 Over 25 639 men and women aged between 45 and 74 were recruited in Norwich and the surrounding area. Subjects were characterized for cardiovascular and metabolic phenotypes. MPO was available in nonfasted baseline serum samples for 1138 incident coronary artery disease cases and 2237 controls after an 8year follow‐up.
Fenland
The Fenland Study is an ongoing, population‐based cohort study (started in 2005) designed to investigate the association between genetic and lifestyle environmental factors and the risk of obesity, insulin sensitivity, hyperglycemia, and related metabolic traits in men and women aged 30 to 55 years.13 Participants were recruited from General Practice sampling frames in the Fenland, Ely, and Cambridge areas of the Cambridgeshire Primary Care Trust in the United Kingdom. After an overnight fast, participants underwent a detailed clinical examination, and blood samples were collected. Data from up to 5105 participants were included in the current analyses.
Ely
The MRC Ely Study is a population‐based cohort randomly selected from people living in Ely and surrounding villages (East Anglia, United Kingdom), an ethnically homogeneous European ancestry population. The study design, methods, and measurements of the 3 phases have been described in detail elsewhere.14 The current analyses included individuals aged 35 to 79 years, from phase 3. Data from up to 1606 participants were included in the current analyses.
GenOA
The Genetics of Obesity Associations (GenOA) study is a case–control study with 1008 obese (BMI >30) white subjects recruited from the Ottowa obesity weight management clinic and 991 white controls (BMI <40th percentile for age and gender) from the local community.15 Obesity and type 2 diabetes status were meta‐analyzed with other studies in overall subjects.
Ethical oversight for genetic research and patient informed consent were obtained by each specific study.
Sequencing
The MAPK11 and MAPK14 genes were sequenced along with 200 other genes in 14 002 subjects.4
MPO Assay
MPO was measured in fasting serum for GEMS, FHS, and CHS and nonfasting serum for EPIC‐Norfolk. MPO was determined in GEMS by Pathway Diagnostics (Cypress, CA) using the Myeloperoxidase ELISA kit #K6631 from ALPCO Diagnostics (Salem, NH). CHS and EPIC‐Norfolk both used the CardioMPO test (PrognostiX Inc, Cleveland, OH), a Food and Drug Administration–approved sandwich enzyme–linked immunosorbent assay. Normal control values from a middle‐aged healthy population have been reported to be <640 pmol/L. The minimum detection limit (calculated using interpolation of the mean plus 2 SDs) was 30 pmol/L, and a within‐run precision of 4.8% was reported.16 In the FHS, a quantitative ELISA kit was used (Oxis, Cat. No. 21013) and read on a Molecular Devices VersaMax microplate reader. The minimum detectable dose was 0.17 ng/mL, the standard curve range was 0 to 25 ng/mL, and the intra‐assay variability was 3.15%.
Phenotypes Analyzed
Table 1 characterizes each sample set analyzed and lists all phenotypes analyzed in the initial analyses of the GEMS collection, replication analyses for the CHS and FHS, and profiling analysis for the CoLaus, LOLIPOP, EPIC‐Norfolk, Fenland Ely, and GenOA studies. Table 2 characterizes the dyslipidemic subgroups only.
Table 2.
Population Information in Dyslipidemia Subset
| GEMS | CoLaus | LOLIPOPW | CHS | FHS | EPIC | Fenland | ELY | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | 787 | 520 | 729 | 433 | 391 | 2258 | 748 | 208 | ||||||||
| Categorical variable, N (%) | ||||||||||||||||
| Men | 467 (59%) | 276 (53%) | 457 (63%) | 266 (61%) | 166 (42%) | 1018 (45%) | 328 (44%) | 98 (47%) | ||||||||
| Statins | 339 (43%) | 70 (14%) | 153 (21%) | 12 (3%) | 44 (2%) | 34 (5%) | 21 (10%) | |||||||||
| Continuous variable (N, Mean (SD)) | ||||||||||||||||
| Age, y | 775 | 49.2 (9.1) | 520 | 54.3 (10.1) | 729 | 52.7 (11.3) | 433 | 72.3 (5.3) | 391 | 61.6 (9.5) | 2258 | 59.4 (9.2) | 748 | 46.9 (7.2) | 208 | 61.5 (8.3) |
| Hypertension | ||||||||||||||||
| DBP, mm Hg | 771 | 81.7 (10.9) | 520 | 81.5 (11.5) | 729 | 81.4 (9.9) | 433 | 70.2 (10.9) | 391 | 75.2 (10.2) | 2256 | 84.7 (10.5) | 747 | 79.8 (9.8) | 208 | 82.1 (10.4) |
| SBP, mm Hg | 771 | 131.9 (17.3) | 520 | 131.9 (18.4) | 729 | 133.2 (19) | 433 | 136 (21.7) | 391 | 130.9 (18.1) | 2256 | 138.7 (17.6) | 747 | 127.3 (15.1) | 208 | 136.1 (17.4) |
| Inflammation | ||||||||||||||||
| Adiponectin, μg/mL | 743 | 5.5 (3.9) | 518 | 6.64 (4.57) | 329 | 6.58 (3.96) | 605 | 4.6 (2.2) | 95 | 6.0 (3.0) | ||||||
| CRPU, mg/L | 743 | 6 (10) | 520 | 3.7 (4.2) | 695 | 5.8 (6.6) | 1769 | 3.4 (3.9) | 605 | 4.1 (4.4) | 80 | 2.1 (2.3) | ||||
| Fibrinogen, g/L | 605 | 2.6 (0.62) | 431 | 3.27 (0.64) | 390 | 3.99 (0.76) | 2103 | 3.0 (0.8) | ||||||||
| IL‐1b, pg/mL | 603 | 47.9 (115) | 320 | 6.7 (20.3) | ||||||||||||
| IL‐6, pg/mL | 603 | 45 (72) | 496 | 18.4 (209.4) | 390 | 4.45 (4.43) | 43 | 1.5 (1.2) | ||||||||
| IL‐8, pg/mL | 603 | 38.2 (76.6) | 170 | 60.4 (281.3) | ||||||||||||
| Leptin, ng/mL | 685 | 16.1 (11.7) | 345 | 17.9 (12.3) | 605 | 25.4 (21.3) | 206 | 30.1 (26.7) | ||||||||
| MPO, ng/mL | 605 | 52 (104) | 311 | 49 (41) | 391 | 45.8 (31.7) | 352 | 102 (80) | ||||||||
| TNFA, pg/mL | 717 | 88 (132) | 512 | 5.7 (9.9) | 305 | 145.5 (120.5) | ||||||||||
| Insulin sensitivity | ||||||||||||||||
| 2h glucose, mmol/L | 45 | 6.9 (3.8) | 138 | 8.7 (3.5) | 728 | 6.2 (1.9) | 190 | 7.9 (2.9) | ||||||||
| Glucose, mmol/L | 683 | 5.2 (0.6) | 520 | 6.1 (1.9) | 729 | 6.2 (2.6) | 433 | 130 (52.2) | 384 | 6.7 (2.4) | 746 | 5.1 (0.6) | 208 | 5.4 (1) | ||
| HOMA‐B | 497 | 114.8 (88.2) | 700 | 175.2 (155.7) | 378 | 169.5 (128.1) | 738 | 166.8 (99.5) | 206 | 164.7 (88.9) | ||||||
| HOMA‐IR | 651 | 3.1 (2.5) | 497 | 3.8 (4.7) | 700 | 5.3 (8) | 378 | 6.96 (6.44) | 738 | 2.9 (2) | 206 | 3.5 (2.1) | ||||
| Insulin, mIU/mL | 743 | 12.9 (9.6) | 497 | 13.1 (8.8) | 700 | 17.9 (17.9) | 428 | 27.9 (42.4) | 378 | 22.19 (16.8) | 740 | 10.7 (6.4) | 206 | 12.1 (6.2) | ||
| Kidney function | ||||||||||||||||
| Creatinine, μmol/L | 520 | 81.9 (22) | 729 | 92.4 (27.3) | 433 | 1.18 (0.40) | 1770 | 84.4 (19.3) | 743 | 77.6 (15.2) | 97 | 88.0 (17.0) | ||||
| CRU, mmol/day | 504 | 14 108 (7139) | 743 | 11.9 (8.1) | ||||||||||||
| GFR | 520 | 77.8 (16) | 729 | 103.1 (32.1) | 391 | 80.6 (20.8) | 1770 | 77.8 (22.9) | 743 | 87.2 (16.4) | ||||||
| MACR | 504 | 2231 (6959) | 603 | 1.4 (2.9) | ||||||||||||
| MALB | 504 | 27.5 (72.1) | 603 | 16.4 (30.3) | ||||||||||||
| Lipid | ||||||||||||||||
| APOB, g/L | 743 | 1.2 (0.3) | 515 | 1.97 (1.63) | 1752 | 1.05 (0.3) | 605 | 1.1 (0.2) | ||||||||
| CHOL, mmol/L | 775 | 5.7 (1.2) | 520 | 5.8 (1.1) | 729 | 5.5 (1.1) | 433 | 206.0 (37.8) | 391 | 5.26 (1.04) | 2258 | 6.5 (1.1) | 748 | 5.7 (1) | ||
| HDL, mmol/L | 775 | 0.9 (0.2) | 520 | 1.1 (0.2) | 729 | 1 (0.1) | 433 | 35.0 (4.6) | 391 | 0.9 (0.2) | 2258 | 1.0 (0.2) | 748 | 1.1 (0.2) | 208 | 1.0 (0.2) |
| LDL, mmol/L | 618 | 3.4 (1.1) | 479 | 3.6 (1.0) | 639 | 3.2 (0.9) | 392 | 3.2 (0.98) | 355 | 3.0 (0.97) | 2258 | 4.2 (1.1) | 709 | 3.6 (0.9) | 206 | 3.7 (1) |
| TRIG, mmol/L | 775 | 3.5 (2.2) | 520 | 2.8 (2.1) | 729 | 3.1 (1.9) | 433 | 3.03 (1.44) | 391 | 3.06 (1.45) | 2258 | 3.0 (0.8) | 748 | 2.3 (1) | 208 | 2.8 (1.0) |
| Liver function | ||||||||||||||||
| ALB, g/L | 520 | 43.8 (2.6) | 729 | 43.2 (2.7) | 1784 | 41.8 (5.8) | 748 | 41.6 (2.7) | 97 | 42.2 (3.3) | ||||||
| ALP, U/L | 520 | 69.2 (21.1) | 727 | 84.8 (37.4) | 1789 | 64.6 (20.6) | 747 | 85.2 (22.1) | ||||||||
| ALT, U/L | 520 | 34 (21.6) | 729 | 34.3 (25.2) | 748 | 34.9 (19.4) | ||||||||||
| GGT, U/L | 520 | 39.1 (46.4) | 729 | 56.6 (116.6) | 1793 | 38.1 (30.9) | 748 | 41.3 (28.8) | ||||||||
| Obesity | ||||||||||||||||
| BMI, kg/m2 | 775 | 28.7 (3.6) | 520 | 29.1 (4.7) | 729 | 31 (5.6) | 433 | 28.2 (4.4) | 391 | 30.5 (5.3) | 2254 | 28.2 (3.9) | 687 | 30.6 (5.4) | 208 | 29.7 (4.6) |
| Body fat, % | 515 | 32.3 (9.7) | 690 | 35.6 (8.8) | 1412 | 35.8 (11.8) | 677 | 35.1 (9) | 205 | 36.1 (8.9) | ||||||
| Hip, cm | 764 | 108 (8.1) | 520 | 106.6 (9.7) | 728 | 108.2 (10) | 430 | 104 (9.2) | 384 | 108 (10.8) | 686 | 109.7 (10.6) | 205 | 109.8 (10) | ||
| Waist, cm | 765 | 98.3 (10.6) | 520 | 98.2 (12.5) | 728 | 104.5 (13.3) | 430 | 100.3 (11.3) | 384 | 105.9 (12.5) | 2257 | 93.8 (11.5) | 687 | 100.6 (12.9) | 206 | 99.5 (11.4) |
| Weight, kg | 775 | 84.7 (13.6) | 520 | 82.5 (15.5) | 729 | 90.4 (18) | 433 | 174 (30.4) | 391 | 84.7 (17.3) | 687 | 88.6 (17.8) | 208 | 82.3 (13.8) | ||
| Waist/hip ratio | 520 | 0.9 (0.1) | 727 | 1 (0.1) | 430 | 0.96 (0.07) | 383 | 0.98 (0.068) | 2252 | 0.89 (0.09) | 686 | 0.92 (0.08) | 205 | 0.91 (0.1) | ||
| Others | ||||||||||||||||
| Calcium, mmol/L | 520 | 2.2 (0.1) | 729 | 2.3 (0.1) | 747 | 2.2 (0.1) | 97 | 2.1 (0.1) | ||||||||
| URIC, μmol/L | 520 | 353.7 (91.5) | 726 | 340.6 (86.4) | 433 | 386.6 (95.2) | 1773 | 314.5 (83.6) | ||||||||
ALB indicates albumin ; ALP, alkaline phosphatase; ALT, alanine aminotransferase; CHS, Cardiovascular Health Study; CHOL, cholesterol; CoLaus, ; CRPU, ultrasensitive C‐reactive protein; CRU, urinary creatinine; APOB, apolipoprotein B; BMI, body mass index; CRP, C‐reactive protein; DBP, diastolic blood pressure; EPIC, European Prospective Investigation of Cancer; FHS, Framingham Heart Study; GEMS, Genetic Epidemiology of Metabolic Syndrome Study; GFR, glomerular filtration rate; GGT, γ‐glutamyl transferase; HDL, high‐density lipoprotein; HOMA‐B, homeostatic model assessment – beta‐cell; HOMA‐IR, homeostatic model assessment – insulin resistance; IL, interleukin; LDL, low‐density lipoprotein; LOLIPOPW, London Life Sciences Prospective Population Study – Whites; MACR, microalbumin‐creatinine ratio; MALB, microalbuminuria; MPO, myeloperoxidase; SBP, systolic blood pressure; TNFA, tumor necrosis factor α; TRIG, triglycerides; URIC, uric acid.
Obese participants were required to have a BMI ≥30 kg/m2 and age ≥18 years. Lean controls were required to have a current BMI that is ≤40th percentile for their age and sex groups and not previously reported having had a BMI >25th percentile for age and sex for more than a 2 ‐year consecutive period and age ≥18 years. Dyslipidemic subjects were selected for high triglycerides (≥75th percentile) and low HDL cholesterol (≤25th percentile) based on age, sex, and country threshold. Normolipidemic subjects were selected for high HDL cholesterol (above median), low triglycerides (below median), and BMI >25.6
Genotyping
CHS
Genotyping for RS612049 was performed using the custom IBCv2 genotyping array that contains high single nucleotide polymorphism (SNP) marker density and LD coverage for ≈2100 genes related to cardiovascular, inflammation, hemostasis/coagulation, and metabolic phenotypes.17
FHS
RS612049, a genome wide association study marker, from the Affymetrix 500K array was available and observed to be in perfect linkage disequilibrium with RS2859144 in the Hapmap CEU samples.
CoLaus and LOLIPOP
RS2859144 was genotyped in the entire CoLaus collection and 6565 European white subjects from the LOLIPOP collection at KBiosciences using KASPar technologies (Hoddeson, UK).
EPIC, Ely, Fenland, and EPIC‐Norfolk
RS612049 was genotyped at MRC Cambridge labs using Sequenom technology (2 plexes).
GenOA
RS2859144 was assessed in this collection by imputation. Reference haplotypes (NCBI Build 36) from the sequencing study (n=3983) were imputed into cases (810) and controls (830) from the GenOA collection with Affymetrix 6.0 array data using BEAGLE18 with the default settings. RS2859144 was well imputed with an imputation r2=0.898.
Statistical Methods
Initial analysis of the variants identified by sequencing for the MAPK14 and MAPK11 genes for the GEMS and the subset of CoLaus samples was carried out using an additive genetic model with linear or logistic regression and up to 40 cardiovascular and metabolic traits. A natural log‐transformation was applied for quantitative traits with non‐normal distributions after outliers were winsorized at 99.9th percentile of the distribution. Quantitative trait analyses were based on normalized residuals after adjusting for significant covariates including dyslipidemia status, age, sex, collection site, waist and hip circumference for the GEMS study and age, sex, BMI (if applicable), smoking, physical activity, and alcohol use for the CoLaus study. Binary traits were analyzed with and without adjustment of relevant covariates. SNPs with at least 10 copies of the minor allele were analyzed individually, whereas putatively functional SNPs with minor allele frequency <0.5% were aggregated.19 There were 7 (MAPK11) plus 15 (MAPK14) variants analyzed individually in GEMS, and 6 (MAPK11) plus 13 (MAPK14) variants in CoLaus. Three aggregate tests were carried out for each gene: (1) all rare variants that lead to a change in the amino acid sequence (nonsynonymous, nonsense, readthrough, and splice site variants), (2) all rare, amino acid–changing variants that are predicted to be functional damaging by SIFT or PolyPhen, and (3) variants in (2) plus additional rare variants at a highly conserved base position (phyloP ≥2). Analyses were conducted in all subjects and in GEMS dyslipidemic only subjects). A Bonferroni‐corrected significance threshold adjusting for the number of tested variants in both genes, number of traits, and number of subgroups analyzed was set at P=0.05/(38×(22+6)×2)=2.35×10−5 for the GEMS and P=0.05/(40×(19+6))=5.0×10−5 for the CoLaus study, respectively.
Genetic association analyses of the genotyped variant (rs2859144 or proxy) for the full set of CoLaus, LOLIPOP, EPIC‐Norfolk, Ely, Fenland, CHS, and FHS studies were carried out in each study (after transformation as described previously) under an additive genetic model using linear or logistic regression, and linear mixed‐effect or generalized estimating equation models in FHS to account for familial correlation. To accommodate the uncertainty of imputed genotypes, allelic dosages in the GenOA study were analyzed as a continuous variable in linear/logistic regression. Obese‐only and dyslipidemic‐only subgroup analyses were performed to enable replication of some context‐specific study results. In the CoLaus study, 330 subjects were removed from the analysis due to genetic relatedness (PI_HAT>0.125) based on identity by descent (IBD) sharing estimated from PLINK using existing Affy500K data. Summary statistics from all studies were meta‐analyzed using the inverse variance method.20 The P value threshold for replication of the MPO association in CHS and FHS was set at P=0.05. For the expanded cardiovascular and metabolic profile, a Bonferroni‐corrected significance threshold was used for the meta‐analysis, adjusting for the traits evaluated (P=0.05/40=0.00125).
GEMS Inflammatory Markers
Methods used for measurement of plasma lipids and glucose in serum have been described previously.6 All additional biomarkers were measured by Pathway Diagnostics (Cypress, CA) using either plasma or serum. Frozen serum and multiple aliquots of EDTA plasma specimens were received at Pathway Diagnostics on dry ice. The specimens were stored at −70°C until thawed for testing. Since it was not logistically possible to perform all of the many assays for a given subject on the same day, plasma aliquots were not pooled. Instead, individual aliquots of serum and plasma were thawed as necessary and tested as efficiently as possible; that is, as many individual assays as was practical were run with the thawed aliquots within 8 hours of being thawed. While minor heterogeneity between aliquots was possible, this method minimized any risk of specimen stability that may have occurred if the specimens were stored for long periods at 2 to 8°C or via multiple freeze/thaw cycles. Once thawed, plasma was removed for each assay and diluted (if necessary, depending on the assay) using an assay‐specific buffer.
Adiponectin was measured using an ELISA assay (R&D Systems, Minneapolis, MN); CRP and insulin were measured using chemiluminescent assays (Diagnostic Products Corp, Los Angeles, CA); LDL particle‐size was measured using a LipoPrint kit from Quantimetrix Corporation (Redondo Beach, CA); leptin was quantified using an ELISA (American Lab Products Company, Windham, NH) and apo B was measured using an immunoturbidometric assay (Polymedco). PAI‐1 was evaluated using the PAI‐1 (Plasminogen Activator Inhibitor, Type 1) ELISA kit #EL504 from Dakocytomation (Carpinteria, CA). MPO was determined using the Myeloperoxidase ELISA kit #K6631 from ALPCO Diagnostics (Salem, NH). MMP9 was measured using the MMP9 (Total) Quantikine ELISA kit #DMP900 from R&D Systems (Minneapolis, MN). Oxidized LDL was evaluated in serum using the Oxidized LDL (Ab) EIA kit #04‐BI‐20032 from ALPCO Diagnostics. Multiplex testing of interleukin (IL)‐1a, IL‐1b, IL‐6, IL‐8, IL‐10, IL‐18, monocyte chemotactic protein 1 (MCP1), soluble vascular cell adhesion molecule, soluble intracellular adhesion molecule, and tumor necrosis factor α were performed using customized kits purchased from Beckman Coulter (Fullerton, CA) for determination using the Beckman Coulter A‐Square™ reader. Prothombin F1+2 and Fibrinogen were determined using the Prothombin F1+2 ELISA kit #REFOWVVII from Dade‐Behring (Newark, DE) and the Fibrinogen (Clauss) kit #0008469110 by Instrument Laboratory (Milan, Italy), respectively. Instructions provided by the manufacturer were followed for each test kit. All colorimetric determinations were made using a Tecan GENios Pro™ (Grodig, Austria) plate reader and analyzed using Magellan™ software except for the multiplex tests, which were performed using the Beckman Coulter A‐Square™ reader and Fibrinogen, which was evaluated on the ACL Advance™ (Beckman Coulter).
While citrated plasma was the preferred matrix for the determination of Prothrombin F1+2 and Fibrinogen, only EDTA plasma was available for testing. A small comparison study performed at Pathway Diagnostics of EDTA plasma versus citrated plasma from healthy volunteers was performed and yielded similar results. At the request of GSK, Prothrombin F1+2 and Fibrinogen were performed on the EDTA specimens.
Results
The resequencing (exons, intron–exon boundaries, and untranslated regions) of the MAPK14 and MAPK11 genes (7844 bp total) in 14 002 ethnically diverse subjects including well‐phenotyped subjects from the GEMS (n=1579) and CoLaus (n=2086) collections has been described previously.4 Within all 14 002 subjects, we identified 232 and 157 SNPs, including 2822 and 27 amino acid changes and splice sites, as well as 135 and 78 SNPs in the untranslated region, respectively. For MAPK14, variants included a splice variant (RS200906391) common (MAF>0.5%) in all ethnicities and an additional coding variant present only in African Americans (S64Y). Additionally, there were 2 predicted functional (PHYLOP>2) low‐frequency variants L250F (26 carriers) and D343G (35 carriers) found mainly in Europeans. The remaining predicted functional variants were very rare, mostly private mutations. For MAPK11, we observed 1 common nonsynonymous variant in all ethnicities (R275H) and only E177K was predicted to be functional (PHYLOP>2) and was not private in Europeans. Variants identified in the MAPK11 and MAPK14 genes were reported previously.4 Figure 2 summarizes variation found in 12 514 European white, 594 African American, and 567 South Asian subjects for the MAPK14 gene and Figure 3 for the MAPK11 gene.
Figure 2.
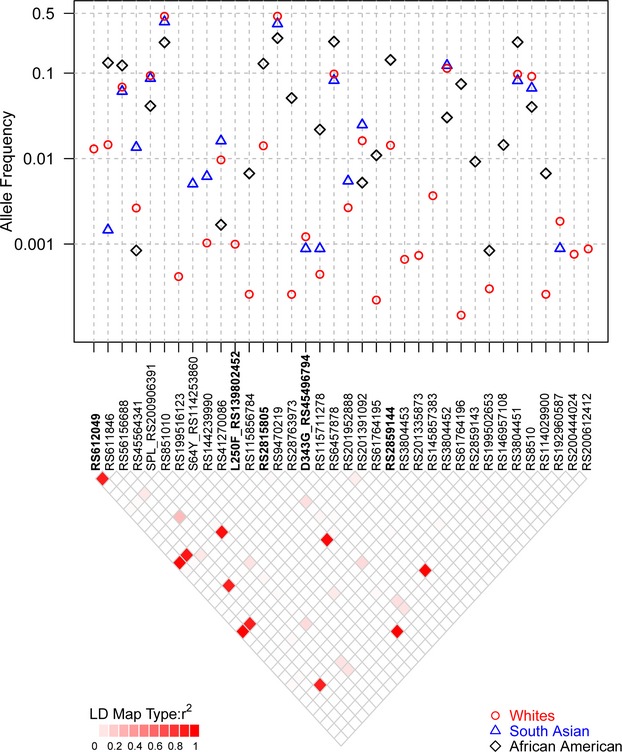
Sequence variants identified in European (whites), South Asian, and African American samples for MAPK14 gene. The figure summarizes the MAF for all variants with more than 10 copies observed in subjects sequenced in Nelson et al4 along with selected variants present in 1000 genomes and used in our analyses. Frequencies are shown by ethnic group. The 2 rare coding variants (D343G and L250F), 2 key SNPs (RS2859144 and RS2815805), and RS612049 from SLC6A8 in high LD are shown in bold. The LD for all variants in European subjects is shown as well. LD indicates linkage disequilibrium; MAF, minor allele frequency; SNPs, single nucleotide polymorphisms.
Figure 3.
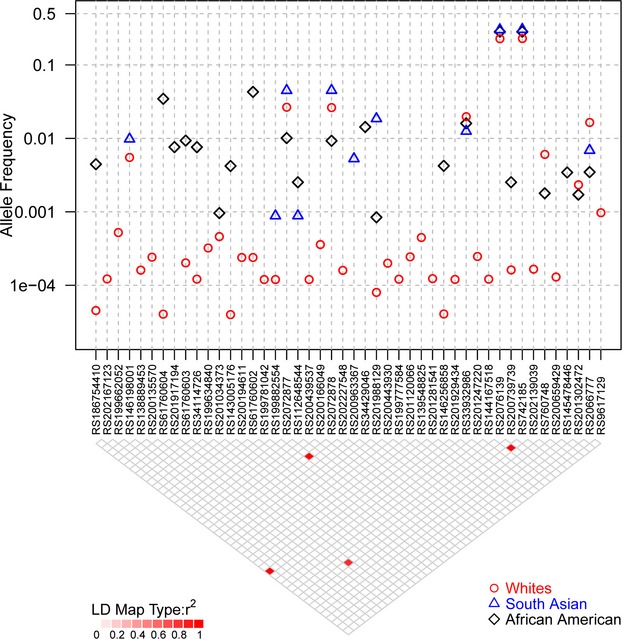
Sequence variants identified in European (whites), South Asian, and African American samples for the MAPK11 gene. The figure summarizes the minor allele frequency for all variants with more than 2 copies observed in subjects sequenced in Nelson et al4 Frequencies are shown by ethnic group. The LD for all variants in European subjects is shown as well. LD indicates linkage disequilibrium.
Common and rare variation in the MAPK14 and MAPK11 genes and flanking regions was analyzed for association with cardiovascular, metabolic, and inflammatory biomarker traits available in GEMS (n=1579) and the sequenced subset of CoLaus (n=2086). In GEMS, analyses for the MAPK14 and MAPK11 genes included 15 and 7 common (MAF>0.5%) variants individually, while aggregated analyses included 12 and 6 rare variants, respectively. In CoLaus, single‐variant analyses for the MAPK14 and MAPK11 genes included 13 and 6 common SNPs, while aggregated analyses included 10 and 5 rare variants, respectively. Analyses were conducted in the GEMS collection on all samples (38 traits) and within the dyslipidemic subjects (35 traits) and in the CoLaus collection on all samples (40 traits). From this initial round of analysis, a significant association was noted for a low‐frequency SNP, RS2859144 (1.5% MAF) in MAPK14 with MPO within dyslipidemic subjects (P=2.3×10−6) in GEMS. This association is illustrated in Figure 4. This SNP was in tight linkage disequilibrium (r2=1) with a synonymous SNP RS2815805 (H253H) and both had the same minor allele frequency. See Figure 5 for a gene schematic. Interestingly, RS2859144 was not associated with MPO in GEMS overall and on closer inspection, the allelic effect was observed to be opposite in normolipidemic controls as compared to the dyslipidemic cases (interaction P=1.7×10−6). Figure 6 summarizes the results for all traits within the GEMS (Figure 6A—dyslipidemic and normolipidemic combined) and CoLaus collections (B) for MAPK14, as well as MAPK11 (Figure 6C through 6E), including the minimum P‐value for common SNPs and the P‐value for aggregated analyses for rare variants. The thresholds for the analyses are highlighted in the graphs and specified in the figure legends. No other statistically significant associations (common or aggregated SNPs) were noted within the MAPK14 and MAPK11 genes; however, a nominally significant association (P=0.016) was observed between rare variants in MAPK14 and IL‐6 levels (Figure 7), a known pharmacodynamic marker of losmapimod.
Figure 4.
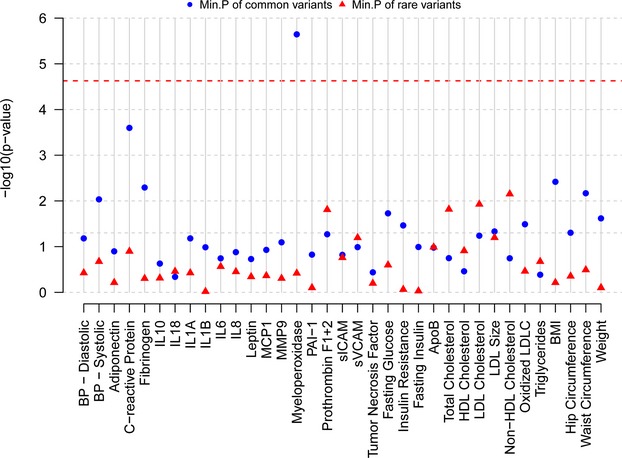
Analysis of cardiovascular, metabolic, and inflammatory biomarker phenotypes in GEMS collection for variants assayed via sequencing in the MAPK14 gene. The figure summarizes the P‐values for association tests performed for all available traits in the GEMS collection for the MAPK14 gene within dyslipidemic subjects. For each trait, the minimum of the P‐values for common variants and P‐values for the 3 rare variant aggregated tests are plotted in this graph. Common variants are defined as those with MAF >0.5%. P‐value thresholds are specified with a line and were calculated as 0.05/(no. of traits×no. of markers in both genes×no. of subgroups analyzed) P=0.05/(38×(22+6)×2)=2.35×10−5. ApoB, apolipoprotein B; BMI indicates body mass index; BP, blood pressure; GEMS, Genetic Epidemiology of Metabolic Syndrome Study; HDL, high‐density lipoprotein; IL, interleukin; LDL, low‐density lipoprotein; LDLC, low‐density lipoprotein cholesterol; MAF, minor allele frequency; PAI, plasminogen activator inhibitor; sICAM, soluble intercellular adhesion molecule; sVCAM, soluble vascular cell adhesion molecule.
Figure 5.
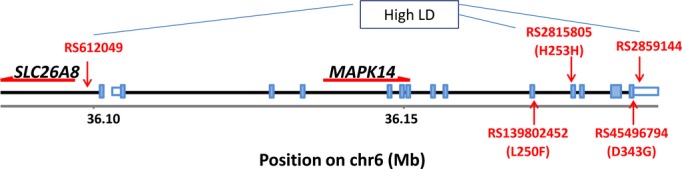
MAPK14 gene region. The location of the 2 rare coding variants is shown below and the 3 low‐frequency variants in high LD are shown above the gene schematic. LD indicates linkage disequilibrium.
Figure 6.
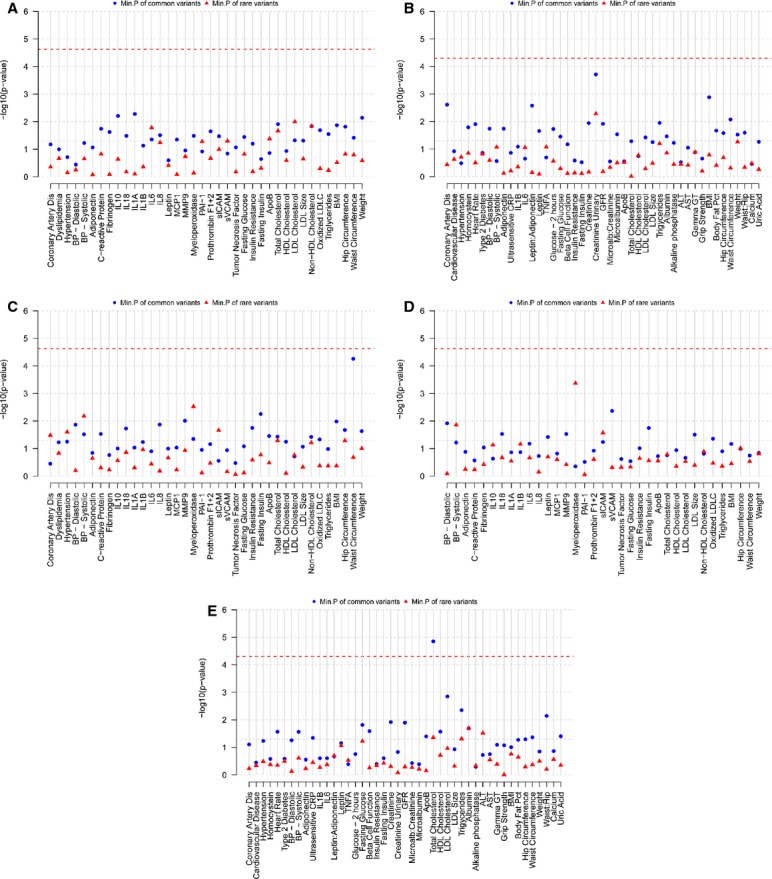
Analysis of cardiovascular, metabolic, and inflammatory biomarker phenotypes in GEMS and CoLaus collections for sequence variants in the MAPK14 and MAPK11 genes. The figure summarizes the P‐values for association tests performed for all available traits in the GEMS and CoLaus collections for MAPK14 and MAPK11. Analyses were performed in all GEMS subjects (A), and within 2086 CoLaus subjects (B) for MAPK14 and in all GEMS subjects (C), within dyslipidemic GEMS subjects (D), and within 2086 CoLaus subjects (E). For each trait, the minimum of the P‐values for common variants and P‐values for the 3 rare variant aggregated tests are plotted in this graph. Common variants are defined as those with MAF >0.5%. P‐value thresholds are specified with a line and were calculated as 0.05/(no. of traits×no. of markers in both genes×no. of subgroups analyzed) P=0.05/(38×(22+6)×2)=2.35×10−5 for the GEMS and P=0.05/(40×(19+6))=5.0×10−5 for CoLaus studies, respectively. 0.05/(no. of traits× no. of markers in both genes×no. of subgroups analyzed) P=0.05/(38×(22+6)×2)=2.35×10−5 for the GEMS and P=0.05/(40×(19+6))=5.0×10−5 for CoLaus studies, respectively. ALT, alanine aminotransferase; ApoB indicates apolipoprotein B; AST, aspartate aminotransferase; BMI, body mass index; BP, blood pressure; CoLaus, Cohorte Lausannoise; GEMS, Genetic Epidemiology of Metabolic Syndrome Study; GFR, glomerular filtration rate; gamma GT, gamma‐glutamyl transferase; HDL, high‐density lipoprotein; IL, interleukin; LDL, low‐density lipoprotein; LDLC, low‐density lipoprotein cholesterol; MAF, minor allele frequency; PAI–1, plasminogen activator inhibitor–1; sICAM, soluble intercellular adhesion molecule; sVCAM, soluble vascular cell adhesion molecule.
Figure 7.
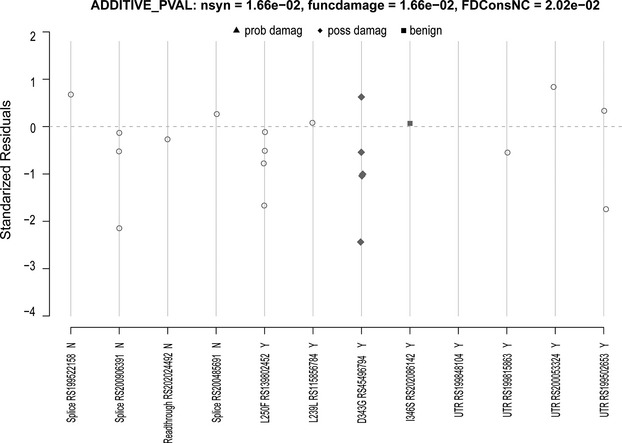
Rare variants in MAPK14 and interleukin‐6. Functional predictions are from SIFT.
As such interactions are often the result of chance, we sought replication in additional cohorts where MPO and dyslipidemia phenotypes were available. Neither RS2859144 nor RS2815805 were present on common genotyping platforms for CHS and FHS studies, but another marker in high linkage disequilibrium (r2=1) was available (RS612049). Despite their low frequency, these variants are well‐tagged by genome wide association study SNPs. We obtained genotype SNP data from CHS and FHS where RS612049, fasting lipids, and MPO were available in a population sample. Dyslipidemic and normolipidemic criteria from GEMS were applied to the 2 population‐based cohorts, and analysis was performed in those subgroups. RS612049 was also genotyped in an EPIC‐Norfolk sample where MPO levels were measured in nonfasting patients within a cardiovascular nested case–control study (cases n=1138 and controls n=2237).21 As the incident cases were enriched for high MPO levels, given the relationship between coronary artery disease and MPO observed in this sample,21 we included only the controls in the meta‐analysis. As shown in Figure 8A, the CHS and FHS studies, but not the EPIC controls, provided additional evidence demonstrating an association with higher MPO levels in the dyslipidemic group (meta‐P=7.56×10−6) and no association within the normolipidemic subgroup (meta‐P=0.62) in Figure 8B. The average effect size in the dyslipidemic group was quite substantial, with each additional copy of the minor allele corresponding to an average increase of 0.47 SD in MPO levels. The replication P value calculated by meta‐analyzing FHS, CHS, and EPIC controls was significant, P=0.009 (0.31 SD average increase) for the dyslipidemic groups.
Figure 8.
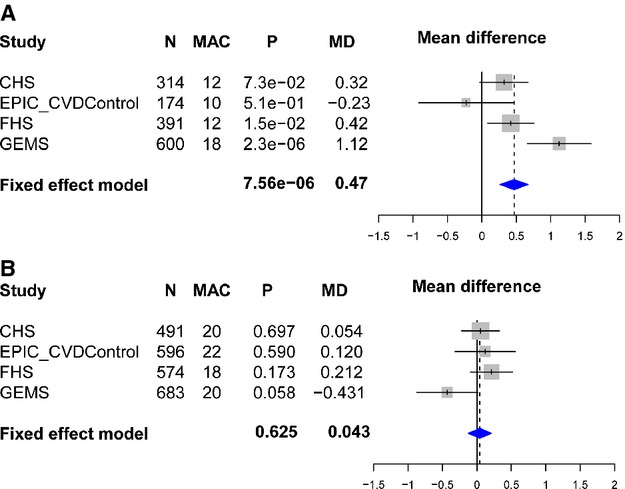
Association analyses for RS2859144/RS612049 and myeloperoxidase (MPO) levels by dyslipidemia status. The associations between MAPK14 single nucleotide polymorphisms and MPO levels are shown by dyslipidemia (A) and normolipidemia (B) status in separate Forest plots (mean difference and 95% CIs). CHS indicates Cardiovascular Health Study; EPIC‐CVD, European Prospective Investigation of Cancer–Cardiovascular Disease; FHS, Framingham Heart Study; GEMS, Genetic Epidemiology of Metabolic Syndrome Study; MAC, minor allele count; MD, mean difference; P, p value.
The variant associated with increased MPO levels was then analyzed for association with up to 40 cardiovascular and metabolic traits within the CoLaus, LOLIPOP, EPIC Norfolk, Ely, Fenland, and GenOA studies to provide further insight into the observed association. RS612049 was typed in Ely and Fenland, and RS2859144 was imputed in the GenOA case–control study and typed in LOLIPOP and remaining CoLaus samples to provide additional data. Although not significant after adjusting for the analysis of 40 traits (P=0.00125), there were some nominal associations that were directionally consistent (Figures 9 and 10). In the overall populations (Figure 10A), the same allele associated with higher MPO in dyslipidemic subjects appeared to confer a marginally lower GFR (Figure 9A, β −0.086, P=0.002) as well as an increased risk for obesity (Figure 9B, odds ratio 1.25, P=0.005). Nominal associations were also observed for higher uric acid (Figure 9C, P=0.01), though this was only available in a few studies. Within the dyslipidemic subgroups (Figures 9 and 10B), nominal associations were observed with lower systolic (Figure 9D, P=0.002) and diastolic blood pressures (Figure 9D, P=0.01) as well as lower HOMA‐B (Figure 9E, P=0.03) and higher fasting glucose (Figure 9F, P=0.04). In addition, there were many more inflammation‐linked markers available in GEMS than were available in the other studies and of those, RS2859144 was also nominally associated with oxidized‐LDL in the dyslipidemic group (β −0.4, P=0.03).
Figure 9.

Association analyses for RS2859144/RS612049 with metabolic variables in overall (A) glomerular filtration rate, (B) obesity status, (C) uric acid and dyslipidemic populations, (D) systolic blood pressure, (E) diastolic blood pressure, (F) HOMA‐B, and (G) glucose. CHS indicates Cardiovascular Health Study; CoLaus, Cohorte Lausannoise; EPIC, European Prospective Investigation of Cancer; FHS, Framingham Heart Study; GEMS, Genetic Epidemiology of Metabolic Syndrome Study; LOLIPOPW, London Life Sciences Prospective Population Study Whites; MAC, minor allele count; MD, mean difference; P, p value.
Figure 10.
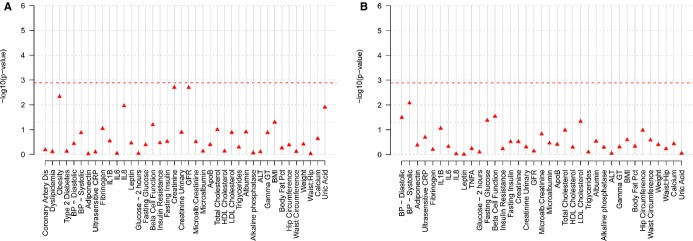
Analysis of cardiovascular and metabolic phenotypes in CoLaus, LOLIPOP, EPIC‐Norfolk, ELY, Fenland, and GenOA studies. The figure summarizes the P‐values for association tests performed for rs2859144 within the MAPK14 gene for 40 cardiovascular and metabolic traits combining results in a meta‐analysis from the CoLaus, LOLIPOP, EPIC‐Norfolk, ELY, Fenland, and GenOA studies. Analyses were performed in all subjects (A—40 traits) and within dyslipidemic subjects (B—37 traits). For each trait, the P‐values are shown. P‐value thresholds are specified with a line and were calculated as 0.05/(no. of traits) ≈0.00125. ALT indicates alanine aminotransferase; ApoB, apolipoprotein B; BMI, body mass index; BP, blood pressure; CoLaus, Cohorte Lausannoise; CRP, C‐reactive protein; EPIC, European Prospective Investigation of Cancer; GFR, glomerular filtration rate; Gamma GT, gamma‐glutamyl transferase; HDL, high‐density lipoprotein; IL, interleukin; LDL, low‐density lipoprotein; LOLIPOP, Life Sciences Prospective Population Study.
Discussion
We sought to characterize gene variation in MAPK11/14 genes in well‐phenotyped individuals to identify genetic variants that can be used to predict drug effects and generate hypotheses around alternative indications for MAPK‐α/β inhibitors including Losmapimod, a p38 map kinase inhibitor in development for acute coronary syndrome. Comprehensive sequencing meant that 94% of the variant alleles with minor allele frequency of 0.01% in Europeans were sampled at least once. The variants present in the GEMS and CoLaus collections were characterized using cardiovascular, metabolic, and biomarker phenotypes. We identified an association of RS2859144 in MAPK14 with MPO in a dyslipidemic population (GEMS, P=2.3×10−6). This variant RS2859144 (or proxy) was consistently associated with MPO in the FHS and CHS studies (replication meta‐P=0.003), leading to a meta‐P value of 9.96×10−7 in the 3 dyslipidemic groups. The variant or its proxy was then profiled in additional population‐based cohorts (up to a total of 58 930 subjects) including CoLaus, Ely, Fenland, EPIC, LOLIPOP, and the GenOA study obesity case–control for up to 40 cardiovascular and metabolic traits highlighting the same SNP, which was also nominally associated consistently with GFR (P=0.002) and risk of obesity (BMI≥30 kg/m2, P=0.004) in an overall analysis. BMI as a continuous trait was not nominally significant (P=0.052) and was not significant within the GIANT consortium,22 suggesting either this is a false positive or specific to a more extreme form of obesity.
Deep sequencing identified only 1 low‐frequency coding variant within MAPK11 (rs33932986, R275H, MAF=0.02) and no others with more than a few copies. We observed only 1 low‐frequency coding splice variant within MAPK14 (rs200906391, European MAF=0.0067), and only 2 other variants were present with more than a few copies within Europeans (L250F and D343G). The function of the 2 rare variants observed in this study (L250F and D343G) has not been studied, though a PHYLOP score of >2 indicates that they are evolutionarily conserved. No robust associations were observed with these 2 variants, though a nominally significant rare variant association was observed with IL‐6 levels in GEMS (Figure 11). Treatment with SB681323 (a back‐up P38 MAPK inhibitor) demonstrated a trend for reduced IL‐6 production 2 days after a percutaneous intervention1 and also significantly reduced IL‐6 in rheumatoid arthritis patients after a single dose.23
Figure 11.
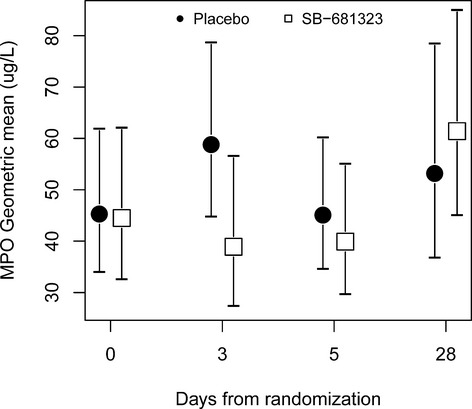
MPO plot from P38 MAPK inhibitor study. This figure is reproduced from data included in Sarov‐Blat et al1 showing a trend in post‐stent dampening at day 3 in MPO levels by SB681323. MPO indicates myeloperoxidase.
The 3 low‐frequency variants in high linkage disequilibrium that were found to be associated with MPO as well as obesity and GFR have not been functionally characterized. Two of these variants are within MAPK14, RS2859144 within the 3′untranslated region and RS2815805 within exon 9, RS612049 is 5′ to MAPK14 and also falls within the first exon of SLC26A8, which is transcribed in the opposite direction (Figure 4). SLC26A8 is an anion transporter expressed solely in the testes24 and therefore is not likely to explain the effects observed here. We searched ENCODE data through the RegulomeDB portal25 to see if there was any evidence for regulatory function of these SNPs. RS281505 showed only minimal binding evidence based on transcription factor binding and the presence of a DNase peak. Thus, the functionality underlying these associations remains to be determined. Lastly, according to genome wide association study central, RS612049 and other SNPs in linkage disequilibrium are close to genome‐wide significant (minimum P=3×10−8) for height only.26
The association of MPO levels and the MAPK14 variant provides valuable insight into the action of MAPK‐α/β inhibitors. High MPO levels are an important prognostic marker in predicting coronary events in healthy individuals21 and coronary artery disease patients16,27 and thus aligned well with results obtained in the SOLSTICE study where a reduction in post‐percutaneous intervention coronary events was observed in patients on Losmapimod.3 Moreover, there is some precedent for the dependence on HDL cholesterol, as MPO has been observed to predict carotid stenosis only on a background of low HDL cholesterol.28 MPO is a leukocyte‐derived enzyme that catalyzes the formation of a number of reactive oxygen species, which contribute to tissue damage during inflammation. It selectively modifies apolipoprotein A‐I, generating dysfunctional HDL cholesterol with impaired cholesterol efflux activity as well as generating pro‐atherogenic forms of modified LDL cholesterol.29 A major determinant of coronary plaque progression or regression rate is the balance between cholesterol uptake versus efflux pathways, and excess MPO would be likely to impair both processes. The relationship between MPO and p38 map kinase was further supported by a trend in reduction of MPO post‐percutaneous intervention with SB681323‐treated patients compared to an increase in placebo patients (Figure 11).1 Taken together, these results suggest a mechanistic link between P38 map kinase and MPO, thus providing evidence to support the current indication of acute coronary syndrome for Losmapimod. We could also hypothesize that patients with low HDL and/or high MPO levels might benefit more from Losmapimod treatment than those without such levels.
The same set of genetic markers that were associated with MPO also showed some suggestive associations with other traits in the overall sample. Lower GFR and increased risk of obesity as a binary trait exhibited directionally consistent associations across 6 datasets, although this was not statistically significant. The link between inflammation and obesity is well established; therefore, it is not surprising to observe an increased risk for obesity associated with the same allele linked with higher MPO, though the lack of a BMI association in the GIANT consortium might suggest that this is a false positive or related to more extreme obesity parameters. GFR is a commonly used clinical measure of kidney function, and evidence exists for an important pathogenic role for p38 MAPK activation in human glomerulonephritis.30 Furthermore, protection of kidney function conferred by a p38 map kinase inhibitor has been observed in hypertensive preclinical models.31 Thus, a p38 map kinase inhibitor would be predicted not only to have cardioprotective effects, but also a protective effect on the kidney and perhaps even generate some weight loss, though the latter would be unlikely with short‐term use. Lastly, elevated uric acid levels could be interpreted as a nonspecific indicator of increased cardiovascular risk, which is not thought to be in itself causal.32–33
The variables nominally associated with the MAPK14 variants within the dyslipidemia subgroup need to be interpreted with caution due to the modest significance levels and small number of carriers, but are provided for descriptive purposes. If they were to be confirmed in larger sample sizes, they would suggest a slightly worse metabolic syndrome phenotype in carriers as compared to noncarriers. The more general limitations of this report include the following: relatively small numbers of carriers due to the low frequency of the variant, limited power to detect associations with rare variants unless effects are large, data are restricted to populations of European origin, and the fact that the actual causal variant is not known.
In conclusion, by sequencing we have determined the full range of exonic variation, present in individuals of European ancestry, within MAPK14 and MAPK11 and tested for associations with a wide range of traits and conditions. We then followed up the most promising findings within up to 58 930 subjects. The most consistent associations observed were with a variant in MAPK14 and MPO on a low HDL‐C background and also more marginal associations with risk of obesity and kidney function in an overall analysis. These results also illustrate how mining of low‐frequency variation may reveal useful tools for associating genes with phenotypes of interest, when common variation has not been informative. This study also illustrates the value of having good intermediate phenotypes and how extreme sampling, as in the case of GEMS, can reveal context‐specific associations not observed in all‐comers. However, the low‐frequency variation does require access to substantial follow‐up samples to evaluate the consistency of the data, especially in the present case where genetic effects were partly context dependent. Lastly, sequencing across multiple ethnic groups indicates that subjects of African origin would be of particular interest for further follow‐up, as the RS2859144 and related markers are much more common in African‐Americans (MAF 14%) as compared to subjects of European origin.
Sources of Funding
The genotyping and analysis at the University of Cambridge was supported in part by funding from GlaxoSmithKline under a collaboration agreement. The GEMS study was sponsored in part by GlaxoSmithKline. The CoLaus study was supported by grants from GlaxoSmithKline, the Swiss National Science Foundation (Grant 33CSCO‐122661), and the Faculty of Biology and Medicine of Lausanne. Framingham Heart Study (FHS) research was conducted in part using data and resources of the National Heart, Lung, and BloodInstitute (NHLBI) and Boston University School of Medicine. The analyses reflect intellectual input and resource development from the FHS investigators participating in the SNP Health Association Resource (SHARe) project. This work was partially supported by NHLBI (contract no. N01‐HC‐25195) and its contract with Affymetrix for genotyping services (contract no. N02‐HL‐6‐4278). A portion of this research utilized the Linux Cluster for Genetic Analysis (LinGA‐II) funded by the Robert Dawson Evans Endowment of the Department of Medicine at Boston University School of Medicine and Boston Medical Center.
Disclosures
Waterworth, Li, Warren, Aponte, Sarov‐Blat, Sprecher, Chissoe, and Ehm are all full‐time employees of GlaxoSmithKline.
Acknowledgments
We thank the GSK sample preparation team, and D. Fraser for data preparation, and K. Johansson for consent review. We thank the GEMS Study Investigators: Philip Barter, PhD; Y. Antero Kesäniemi, PhD; Robert W. Mahley, PhD; Ruth McPherson, FRCP; and Scott M. Grundy, PhD. We also thank Dr Waeber MD, the CoLaus PI's Peter Vollenweider, MD and Gerard Waeber, MD, the LOLIPOP PI's, Jaspal Kooner, MD and John Chambers, MD, as well as the participants in all the studies. I would like to thank Vincent Mooser for his support for this work and valuable scientific discussions.
References
- 1.Sarov‐Blat L, Morgan JM, Fernandez P, James R, Fang Z, Hurle MR, Baidoo C, Willette RN, Lepore JJ, Jensen SE, Sprecher DL. Inhibition of p38 mitogen‐activated protein kinase reduces inflammation after coronary vascular injury in humans. Arterioscler Thromb Vasc Biol. 2010; 30:2256-2263. [DOI] [PubMed] [Google Scholar]
- 2.Melloni C, Sprecher DL, Sarov‐Blat L, Patel MR, Heitner JF, Hamm CW, Aylward P, Tanguay JF, DeWinter RJ, Marber MS, Lerman A, Hasselblad V, Granger CB, Newby LK. The study of LoSmapimod treatment on inflammation and InfarCtSizE (SOLSTICE): design and rationale. Am Heart J. 2012; 164:646-653. [DOI] [PubMed] [Google Scholar]
- 3.Newby LK, Marber MS, Melloni C, Sarov‐Blat L, Aberle LH, Aylward PE, Cai G, de Winter RJ, Hamm CW, Heitner JF, Kim R, Lerman A, Patel MR, Tanguay JF, Lepore JJ, Al‐Khalidi HR, Sprecher DL, Granger CBon behalf of the SOLSTICE Investigators. Losmapimod, a novel p38 mitogen‐activated protein kinase inhibitor, in non‐ST‐segment elevation myocardial infarction: a randomised phase 2 trial. Lancet. 2014; epub10.1016/S0140‐6736(14)60417‐7 [DOI] [PubMed] [Google Scholar]
- 4.Nelson MR, Wegmann D, Ehm MG, Kessner D, St JP, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D, Warren L, Aponte J, Zawistowski M, Liu X, Zhang H, Zhang Y, Li J, Li Y, Li L, Woollard P, Topp S, Hall MD, Nangle K, Wang J, Abecasis G, Cardon LR, Zollner S, Whittaker JC, Chissoe SL, Novembre J, Mooser V. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012; 337:100-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009; 461:747-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wyszynski DF, Waterworth DM, Barter PJ, Cohen J, Kesaniemi YA, Mahley RW, McPherson R, Waeber G, Bersot TP, Sharma SS, Nolan V, Middleton LT, Sundseth SS, Farrer LA, Mooser V, Grundy SM. Relation between atherogenic dyslipidemia and the Adult Treatment Program‐III definition of metabolic syndrome (Genetic Epidemiology of Metabolic Syndrome Project). Am J Cardiol. 2005; 15:194-198. [DOI] [PubMed] [Google Scholar]
- 7.Firmann M, Mayor V, Vidal PM, Bochud M, Pecoud A, Hayoz D, Paccaud F, Preisig M, Song KS, Yuan X, Danoff TM, Stirnadel HA, Waterworth D, Mooser V, Waeber G, Vollenweider P. The CoLaus study: a population‐based study to investigate the epidemiology and genetic determinants of cardiovascular risk factors and metabolic syndrome. BMC Cardiovasc Disord. 2008; 8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991; 1:263-276. [DOI] [PubMed] [Google Scholar]
- 9.Dawber TR, Meadors GF, Moore FE., Jr Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health. 1951; 41:279-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham Offspring Study: design and preliminary data. Prev Med. 1975; 4:518-525. [DOI] [PubMed] [Google Scholar]
- 11.Kooner JS, Chambers JC, Aguilar‐Salinas CA, Hinds DA, Hyde CL, Warnes GR, Gomez Perez FJ, Frazer KA, Elliott P, Scott J, Milos PM, Cox DR, Thompson JF. Genome‐wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat Genet. 2008; 40:149-151. [DOI] [PubMed] [Google Scholar]
- 12.Day N, Oakes S, Luben R, Khaw KT, Bingham S, Welch A, Wareham N. EPIC‐Norfolk: study design and characteristics of the cohort. European Prospective Investigation of Cancer. Br J Cancer. 1999; 80suppl 1:95-103. [PubMed] [Google Scholar]
- 13.Godino JG, van Sluijs EM, Marteau TM, Sutton S, Sharp SJ, Griffin SJ. Effect of communicating genetic and phenotypic risk for type 2 diabetes in combination with lifestyle advice on objectively measured physical activity: protocol of a randomised controlled trial. BMC Public Health. 2012; 12:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forouhi NG, Luan J, Hennings S, Wareham NJ. Incidence of type 2 diabetes in England and its association with baseline impaired fasting glucose: the Ely study 1990–2000. Diabet Med. 2007; 24:200-207. [DOI] [PubMed] [Google Scholar]
- 15.Swarbrick MM, Waldenmaier B, Pennacchio LA, Lind DL, Cavazos MM, Geller F, Merriman R, Ustaszewska A, Malloy M, Scherag A, Hsueh WC, Rief W, Mauvais‐Jarvis F, Pullinger CR, Kane JP, Dent R, McPherson R, Kwok PY, Hinney A, Hebebrand J, Vaisse C. Lack of support for the association between GAD2 polymorphisms and severe human obesity. PLoS Biol. 2005; 3:e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang WH, Wu Y, Nicholls SJ, Hazen SL. Plasma myeloperoxidase predicts incident cardiovascular risks in stable patients undergoing medical management for coronary artery disease. Clin Chem. 2011; 57:33-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keating BJ, Tischfield S, Murray SS, Bhangale T, Price TS, Glessner JT, Galver L, Barrett JC, Grant SF, Farlow DN, Chandrupatla HR, Hansen M, Ajmal S, Papanicolaou GJ, Guo Y, Li M, Derohannessian S, de Bakker PI, Bailey SD, Montpetit A, Edmondson AC, Taylor K, Gai X, Wang SS, Fornage M, Shaikh T, Groop L, Boehnke M, Hall AS, Hattersley AT, Frackelton E, Patterson N, Chiang CW, Kim CE, Fabsitz RR, Ouwehand W, Price AL, Munroe P, Caulfield M, Drake T, Boerwinkle E, Reich D, Whitehead AS, Cappola TP, Samani NJ, Lusis AJ, Schadt E, Wilson JG, Koenig W, McCarthy MI, Kathiresan S, Gabriel SB, Hakonarson H, Anand SS, Reilly M, Engert JC, Nickerson DA, Rader DJ, Hirschhorn JN, Fitzgerald GA. Concept, design and implementation of a cardiovascular gene‐centric 50 k SNP array for large‐scale genomic association studies. PLoS One. 2008; 3:e3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype‐phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009; 84:210-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008; 83:311-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petitti DB. Meta‐analysis, decision analysis, and cost‐effectiveness analysis. Statistical Methods in Meta‐Analysis. 2000New York, NY: Oxford University Press; 94-118. [Google Scholar]
- 21.Meuwese MC, Stroes ES, Hazen SL, van Miert JN, Kuivenhoven JA, Schaub RG, Wareham NJ, Luben R, Kastelein JJ, Khaw KT, Boekholdt SM, Shao B, Oda MN, Vaisar T, Oram JF, Heinecke JW, Exner M, Minar E, Mlekusch W, Sabeti S, Amighi J, Lalouschek W, Maurer G, Bieglmayer C, Kieweg H, Wagner O, Schillinger M. Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC‐Norfolk Prospective Population Study. J Am Coll Cardiol. 2007; 50:159-165. [DOI] [PubMed] [Google Scholar]
- 22.Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, Berndt SI, Elliott AL, Jackson AU, Lamina C, Lettre G, Lim N, Lyon HN, McCarroll SA, Papadakis K, Qi L, Randall JC, Roccasecca RM, Sanna S, Scheet P, Weedon MN, Wheeler E, Zhao JH, Jacobs LC, Prokopenko I, Soranzo N, Tanaka T, Timpson NJ, Almgren P, Bennett A, Bergman RN, Bingham SA, Bonnycastle LL, Brown M, Burtt NP, Chines P, Coin L, Collins FS, Connell JM, Cooper C, Smith GD, Dennison EM, Deodhar P, Elliott P, Erdos MR, Estrada K, Evans DM, Gianniny L, Gieger C, Gillson CJ, Guiducci C, Hackett R, Hadley D, Hall AS, Havulinna AS, Hebebrand J, Hofman A, Isomaa B, Jacobs KB, Johnson T, Jousilahti P, Jovanovic Z, Khaw KT, Kraft P, Kuokkanen M, Kuusisto J, Laitinen J, Lakatta EG, Luan J, Luben RN, Mangino M, McArdle WL, Meitinger T, Mulas A, Munroe PB, Narisu N, Ness AR, Northstone K, O'Rahilly S, Purmann C, Rees MG, Ridderstrale M, Ring SM, Rivadeneira F, Ruokonen A, Sandhu MS, Saramies J, Scott LJ, Scuteri A, Silander K, Sims MA, Song K, Stephens J, Stevens S, Stringham HM, Tung YC, Valle TT, van Duijn CM, Vimaleswaran KS, Vollenweider P, Waeber G, Wallace C, Watanabe RM, Waterworth DM, Watkins N, Witteman JC, Zeggini E, Zhai G, Zillikens MC, Altshuler D, Caulfield MJ, Chanock SJ, Farooqi IS, Ferrucci L, Guralnik JM, Hattersley AT, Hu FB, Jarvelin MR, Laakso M, Mooser V, Ong KK, Ouwehand WH, Salomaa V, Samani NJ, Spector TD, Tuomi T, Tuomilehto J, Uda M, Uitterlinden AG, Wareham NJ, Deloukas P, Frayling TM, Groop LC, Hayes RB, Hunter DJ, Mohlke KL, Peltonen L, Schlessinger D, Strachan DP, Wichmann HE, McCarthy MI, Boehnke M, Barroso I, Abecasis GR, Hirschhorn JN. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009; 41:25-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lukey PT, Perry HC, Yang S, Parry S, Dickson MC, Norris VH, Russell PG, Watissee M, Rioja I, Ray KP, Crowe S, Binks M. Single doses of p38 MAP kinase inhibitors or prednisolone affect CRP and IL‐6 in patients with active rheumatoid arthritis (RA). Open J Immunol; 2:85-97. [Google Scholar]
- 24.Lohi H, Kujala M, Makela S, Lehtonen E, Kestila M, Saarialho‐Kere U, Markovich D, Kere J. Functional characterization of three novel tissue‐specific anion exchangers SLC26A7, ‐A8, and ‐A9. J Biol Chem. 2002; 277:14246-14254. [DOI] [PubMed] [Google Scholar]
- 25.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012; 22:1790-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lango AH, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, Jackson AU, Vedantam S, Raychaudhuri S, Ferreira T, Wood AR, Weyant RJ, Segre AV, Speliotes EK, Wheeler E, Soranzo N, Park JH, Yang J, Gudbjartsson D, Heard‐Costa NL, Randall JC, Qi L, Vernon SA, Magi R, Pastinen T, Liang L, Heid IM, Luan J, Thorleifsson G, Winkler TW, Goddard ME, Sin LK, Palmer C, Workalemahu T, Aulchenko YS, Johansson A, Zillikens MC, Feitosa MF, Esko T, Johnson T, Ketkar S, Kraft P, Mangino M, Prokopenko I, Absher D, Albrecht E, Ernst F, Glazer NL, Hayward C, Hottenga JJ, Jacobs KB, Knowles JW, Kutalik Z, Monda KL, Polasek O, Preuss M, Rayner NW, Robertson NR, Steinthorsdottir V, Tyrer JP, Voight BF, Wiklund F, Xu J, Zhao JH, Nyholt DR, Pellikka N, Perola M, Perry JR, Surakka I, Tammesoo ML, Altmaier EL, Amin N, Aspelund T, Bhangale T, Boucher G, Chasman DI, Chen C, Coin L, Cooper MN, Dixon AL, Gibson Q, Grundberg E, Hao K, Juhani JM, Kaplan LM, Kettunen J, Konig IR, Kwan T, Lawrence RW, Levinson DF, Lorentzon M, McKnight B, Morris AP, Muller M, Suh NJ, Purcell S, Rafelt S, Salem RM, Salvi E, Sanna S, Shi J, Sovio U, Thompson JR, Turchin MC, Vandenput L, Verlaan DJ, Vitart V, White CC, Ziegler A, Almgren P, Balmforth AJ, Campbell H, Citterio L, De GA, Dominiczak A, Duan J, Elliott P, Elosua R, Eriksson JG, Freimer NB, Geus EJ, Glorioso N, Haiqing S, Hartikainen AL, Havulinna AS, Hicks AA, Hui J, Igl W, Illig T, Jula A, Kajantie E, Kilpelainen TO, Koiranen M, Kolcic I, Koskinen S, Kovacs P, Laitinen J, Liu J, Lokki ML, Marusic A, Maschio A, Meitinger T, Mulas A, Pare G, Parker AN, Peden JF, Petersmann A, Pichler I, Pietilainen KH, Pouta A, Ridderstrale M, Rotter JI, Sambrook JG, Sanders AR, Schmidt CO, Sinisalo J, Smit JH, Stringham HM, Bragi WG, Widen E, Wild SH, Willemsen G, Zagato L, Zgaga L, Zitting P, Alavere H, Farrall M, McArdle WL, Nelis M, Peters MJ, Ripatti S, van Meurs JB, Aben KK, Ardlie KG, Beckmann JS, Beilby JP, Bergman RN, Bergmann S, Collins FS, Cusi D, den HM, Eiriksdottir G, Gejman PV, Hall AS, Hamsten A, Huikuri HV, Iribarren C, Kahonen M, Kaprio J, Kathiresan S, Kiemeney L, Kocher T, Launer LJ, Lehtimaki T, Melander O, Mosley TH, Jr, Musk AW, Nieminen MS, O'Donnell CJ, Ohlsson C, Oostra B, Palmer LJ, Raitakari O, Ridker PM, Rioux JD, Rissanen A, Rivolta C, Schunkert H, Shuldiner AR, Siscovick DS, Stumvoll M, Tonjes A, Tuomilehto J, van Ommen GJ, Viikari J, Heath AC, Martin NG, Montgomery GW, Province MA, Kayser M, Arnold AM, Atwood LD, Boerwinkle E, Chanock SJ, Deloukas P, Gieger C, Gronberg H, Hall P, Hattersley AT, Hengstenberg C, Hoffman W, Lathrop GM, Salomaa V, Schreiber S, Uda M, Waterworth D, Wright AF, Assimes TL, Barroso I, Hofman A, Mohlke KL, Boomsma DI, Caulfield MJ, Cupples LA, Erdmann J, Fox CS, Gudnason V, Gyllensten U, Harris TB, Hayes RB, Jarvelin MR, Mooser V, Munroe PB, Ouwehand WH. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010; 467:832-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrow DA, Sabatine MS, Brennan ML, de Lemos JA, Murphy SA, Ruff CT, Rifai N, Cannon CP, Hazen SL. Concurrent evaluation of novel cardiac biomarkers in acute coronary syndrome: myeloperoxidase and soluble CD40 ligand and the risk of recurrent ischaemic events in TACTICS‐TIMI 18. Eur Heart J. 2008; 29:1096-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Exner M, Minar E, Mlekusch W, Sabeti S, Amighi J, Lalouschek W, Maurer G, Bieglmayer C, Kieweg H, Wagner O, Schillinger M. Myeloperoxidase predicts progression of carotid stenosis in states of low high‐density lipoprotein cholesterol. J Am Coll Cardiol. 2006; 47:2212-2218. [DOI] [PubMed] [Google Scholar]
- 29.Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005; 25:1102-1111. [DOI] [PubMed] [Google Scholar]
- 30.Stambe C, Nikolic‐Paterson DJ, Hill PA, Dowling J, Atkins RC. p38 Mitogen‐activated protein kinase activation and cell localization in human glomerulonephritis: correlation with renal injury. J Am Soc Nephrol. 2004; 15:326-336. [DOI] [PubMed] [Google Scholar]
- 31.Olzinski AR, McCafferty TA, Zhao SQ, Behm DJ, Eybye ME, Maniscalco K, Bentley R, Frazier KS, Milliner CM, Mirabile RC, Coatney RW, Willette RN. Hypertensive target organ damage is attenuated by a p38 MAPK inhibitor: role of systemic blood pressure and endothelial protection. Cardiovasc Res. 2005; 66:170-178. [DOI] [PubMed] [Google Scholar]
- 32.Culleton BF, Larson MG, Kannel WB, Levy D. Serum uric acid and risk for cardiovascular disease and death: the Framingham Heart Study. Ann Intern Med. 1999; 131:7-13. [DOI] [PubMed] [Google Scholar]
- 33.Moriarity JT, Folsom AR, Iribarren C, Nieto FJ, Rosamond WD. Serum uric acid and risk of coronary heart disease: Atherosclerosis Risk in Communities (ARIC) Study. Ann Epidemiol. 2000; 10:136-143. [DOI] [PubMed] [Google Scholar]