

Abstract
Background
C‐reactive protein (CRP) binds to damaged cells, activates the classical complement pathway, is elevated in multiple inflammatory conditions, and provides prognostic information on risk of future atherosclerotic events. It is controversial, however, as to whether inhibiting CRP synthesis would have any direct anti‐inflammatory effects in humans.
Methods and Results
A placebo‐controlled study was used to evaluate the effects of ISIS 329993 (ISIS‐CRPRx) on the acute‐phase response after endotoxin challenge in 30 evaluable subjects. Healthy adult males were randomly allocated to receive 6 injections over a 22‐day period of placebo or active therapy with ISIS 329993 at 400‐ or 600‐mg doses. Eligible subjects were subsequently challenged with a bolus of endotoxin (2 ng/kg). Inflammatory and hematological biomarkers were measured before and serially after the challenge. ISIS‐CRPRx was well tolerated with no serious adverse events. Median CRP levels increased more than 50‐fold from baseline 24 hours after endotoxin challenge in the placebo group. In contrast, the median increase in CRP levels was attenuated by 37% (400 mg) and 69% (600 mg) in subjects pretreated with ISIS‐CRPRx (P<0.05 vs. placebo). All other aspects of the acute inflammatory response were similar between treatment groups.
Conclusion
Pretreatment of subjects with ISIS‐CRPRx selectively reduced the endotoxin‐induced increase in CRP levels in a dose‐dependent manner, without affecting other components of the acute‐phase response. These data demonstrate the specificity of antisense oligonucleotides and provide an investigative tool to further define the role of CRP in human pathological conditions.
Keywords: acute phase response, antisense inhibitor, C‐reactive protein, endotoxin, healthy volunteers
Introduction
Creactive protein (CRP) is an acute‐phase reactant synthesized primarily by hepatocytes.1–2 Plasma CRP levels are normally <3 mg/L, but can increase by over 1000‐fold in response to inflammatory stimuli. Induction of CRP expression occurs at the transcriptional level through cytokine signaling, predominantly interleukin‐6 (IL‐6).2–3 Human CRP binds to damaged cells and can activate the classical complement pathway, augmenting inflammation and furthering tissue damage.4 Moreover, CRP is elevated in many chronic inflammatory disorders and has consistently been shown to provide prognostic information on the risk of future vascular events that is comparable in magnitude to that provided by elevations of cholesterol and blood pressure.5–7 It is controversial, however, as to whether CRP in healthy subjects has direct proinflammatory effects, because findings from infusion studies using bacterial recombinant CRP8–10 have not been replicated in studies using pharmaceutical‐grade human CRP.11
ISIS 329993 (ISIS‐CRPRx) is an antisense oligonucleotide (ASO) drug, complementary to the coding region of the human CRP messenger RNA (mRNA). ISIS‐CRPRx is a second‐generation ASO drug 20 nucleotides in length comprised of a phosphorothioate backbone, a central 2′‐deoxyribose region to support the recruitment of the ubiquitous ribonucleolytic enzyme, RNase H, and 2′‐O‐methoxyethyl modifications in the 3′ and 5′ flanking regions to enhance binding affinity for the target mRNA and drug half‐life.12–15 Watson‐Crick base‐pair association, and subsequent elicitation of RNase H activity by ISIS‐CRPRx, leads to degradation of the human CRP mRNA transcript to effectively block its translation.16
Initial preclinical studies of CRP antisense inhibitors demonstrated pharmacological activity at both the mRNA and protein levels in transgenic (Tg) mouse models, as well as in monkeys challenged with IL‐6.17 Subsequent investigations found that antisense reduction of CRP with ISIS‐CRPRx in CRP Tg mice with established collagen‐induced arthritis significantly improved clinical signs of disease.18 In humans, single and multiple doses of ISIS‐CRPRx from 50 to 600 mg were safe and well‐tolerated in the initial phase I study involving healthy volunteers. In addition to these findings, serum CRP levels were reduced from 54% to 83% from baseline in a small cohort of subjects with elevated baseline CRP levels who received 5 doses intravenously of 600 mg of ISIS‐CRPRx over a 3‐week treatment period.18
Purified endotoxin, a cell wall component of Gram‐negative bacteria, has been extensively used as a model to study acute inflammatory response in humans. After an intravenous bolus of endotoxin, there is a well‐characterized response that consists of mild flu‐like symptoms, hematological changes, marked increases in cytokines and acute‐phase proteins, as well as activation of coagulation and complement pathways.19–22 We thus sought to determine the effect of the novel antisense CRP inhibitor, ISIS‐CRPRx, on the acute‐phase response induced by an endotoxin challenge.
Methods
Study Design
We conducted a phase I, double‐blind, placebo‐controlled, adaptive, dose‐response study to evaluate the effects of ISIS‐CRPRx on the acute‐phase response to endotoxin challenge in healthy volunteers. The study consisted of a 28‐day screening period, a 22‐day treatment period, a 5‐day endotoxin challenge period, and a 63‐day safety follow‐up period (Figure 1). Study subjects were enrolled into 2 sequential dose cohorts, each randomized at a 2:1 ratio, active to placebo, in order to achieve 10 evaluable subjects per treatment arm. The first dose cohort was 400 mg of ISIS‐CRPRx, or placebo. The dose for the second cohort (600 mg) was decided by a data monitoring committee based on the results from the treatment period and endotoxin challenge of the first cohort. All subjects, monitors, study center personnel, and the sponsor were blinded during the course of the study, except the pharmacist, who prepared the study drug. The protocol was approved by an independent institutional review board (Copernicus Group, Durham, NC), and the study was performed in compliance with the standards of good clinical practice and the Declaration of Helsinki in its revised edition (Washington, DC, 2002).
Figure 1.
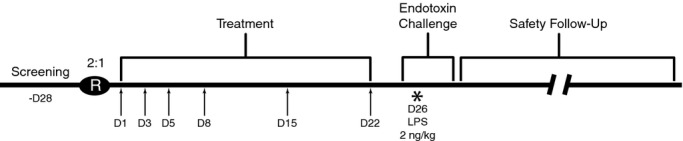
Study schedule. *Subjects who participated in the endotoxin challenge were admitted to the study center on day 25 for 3 overnight stays. On day 26, subjects received an intravenous bolus of Escherichia coli lipopolysaccharide (LPS) at 2 ng/kg body weight. Arrow symbols indicate study drug dosing days; D, day; IV, intravenous; R, randomization.
Study Participants
Eligible subjects were male volunteers (18 to 40 years in age and 55 to 95 kg in body weight) who were in good health and without clinically significant abnormalities in their medical history, physical examination or laboratory evaluations. The study excluded subjects who had a CRP level >10 mg/L, were current smokers, had a history of any clinically important allergy or a known allergy to lactose or polyethylene glycol (PEG; excipients in the endotoxin formulation), were currently using any medication or health supplement, received a vaccination within 6 months of screening, participated in a clinical trial of an immunosuppressive drug or one using endotoxin within 6 months of screening, had received another investigational drug, medical device, or medical procedure within 90 days or 5 half‐lives of screening, or participated in an investigational study involving systemic administration of an oligonucleotide within 9 months of screening. Subjects were enrolled at a single site in the United States (Duke University Medical Center, Durham, NC) from April 23, 2012 to August 9, 2012. All participants gave written informed consent before enrollment. The last subject completed the safety follow‐up period on December 6, 2012.
Study Drug
ISIS 329993 was supplied in 2‐mL stoppered glass vials as a 1‐mL solution (200 mg/mL) for single use only by Isis Pharmaceuticals, Inc. (Carlsbad, CA). Placebo was 0.9% sterile saline. Study drug was administered by a 2‐hour intravenous infusion.
Treatment Period
Eligible subjects were admitted to the study center on day −1 for 5 consecutive overnight stays. Study drug was administered on days 1, 3, and 5. Subjects were discharged on day 5 after all study procedures were completed. Subjects returned to the study center for an overnight stay on days 7, 14, and 21. Study drug was administered on days 8, 15, and 22. Subjects were discharged after drug administration and completion of all study procedures. Safety and clinical laboratory evaluations were performed periodically throughout the treatment period, and adverse events (AEs) were recorded.
Endotoxin
Endotoxin was supplied as a sterile, white, lyophilized powder in a clear glass, aluminum‐sealed, stoppered 5‐mL vial. Each vial contained 10 000 EU (≈1 μg) of lyophilized reference endotoxin, 10 mg of lactose, and 1 mg of PEG 6000, which was reconstituted with 5 mL of sterile, preservative‐free water to a final concentration of 200 ng/mL.
Reference Endotoxin is a purified lipopolysaccharide prepared from Escherichia coli O:113 (U.S. Reference Standard Endotoxin; Clinical Center reference endotoxin, Lot 3) under good manufacturing practice guidelines by the Pharmacy Development Service, Clinical Center, National Institutes of Health (NIH; Bethesda, MD). This material has been approved by the U.S. Food and Drug Administration for investigational use only.
Endotoxin Challenge
Subjects were admitted to the study center on day 25 and examined for evidence of infection or any other condition that precluded participation in the endotoxin challenge. Assessments included vital signs, 12‐lead electrocardiogram (ECG), clinical laboratory tests, urinalysis, and a drug/alcohol screen.
On day 26, eligible subjects received an intravenous bolus of reconstituted reference endotoxin in the amount of 2 ng/kg body weight. Vital signs were recorded with subjects at rest in an approximate 45‐degree supine position before and at various time points after administration of the endotoxin bolus. Blood samples were also collected before and after endotoxin challenge for assessment of known markers of the acute‐phase response. Markers evaluated included those for inflammation (tumor necrosis factor alpha [TNF‐α], IL‐1β, IL‐6, monocyte chemoattractant protein 1 [MCP‐1], CRP, serum amyloid A [SAA], fibrinogen, soluble E‐selectin, and lipopolysaccharide [LPS]‐binding protein), complement activation (Bb, C5a, and C4), coagulation (prothrombin [PT] fragment [F1+2], thrombin‐antithrombin complex, and endogenous thrombin potential), and fibrinolysis (D‐dimer). On day 27, after collecting the 24‐hour postendotoxin samples, subjects underwent safety assessments (vital signs, safety labs, ECG, AE check, and a brief physical examination). Subjects were discharged from the facility on day 28 after the 48‐hour postendotoxin samples were drawn. On day 29, subjects returned to the study center for collection of 72‐hour postendotoxin samples, vital signs, and AEs.
Safety Follow‐Up Period
Subjects were followed until day 92. During this time, subjects returned to the study center on days 43 (±7 days) and 92 (±7 days) for safety and clinical laboratory evaluations. Any AEs were recorded.
Lifestyle Restrictions
Alcohol consumption was not permitted from days −3 through 29. Subjects also refrained from alcohol consumption for 48 hours before their follow‐up visits to the study center on study days 43 and 92. Subjects were required to fast for at least 6 hours before all safety laboratory samples were collected.
For the endotoxin challenge, subjects were required to refrain from consuming food or drinks containing caffeine, including tea, coffee, chocolate, and carbonated drinks, from 48 hours before check‐in on days 25 through 29. Subjects were not permitted sunbathing or any physical exercise, sports, or exertion other than normal walking within 72 hours before study days 25 through 29.
Concomitant Medications
The use of prescription and over‐the‐counter medications (with the exception of occasional acetaminophen) was prohibited during this study, unless the occurrence of an AE required a drug therapy.
Any antibiotic use within 30 days before the endotoxin challenge on day 26 was prohibited. Subjects were not permitted any prescribed medication or any other over‐the‐counter medication, health/herbal supplement, or vitamin by any route of administration within 7 days of study day 26, or 5 half‐lives of the drug, whichever was longer. Subjects requiring any medication concurrent with the study were not eligible for participation.
Medications used to manage the expected clinical effects from endotoxin, including headache and fever, may blunt the cytokine response. To evaluate the potential of cytokine responses, prescribed medication was used only if nonpharmacological approaches were insufficient to manage emergent symptoms.
Safety Monitoring
Safety and tolerability were assessed by determining the incidence, severity, and dose relationship of AEs and changes in laboratory parameters.
Laboratory Analysis
Standard laboratory tests were performed by LabCorp (Research Triangle Park, NC). CRP was measured with a high‐sensitivity assay by MedPace Reference Labs (Cincinnati, OH), complement factors by National Jewish Health (Denver, CO), inflammation markers by Aushon (Billerica, MA), and coagulation markers by the Hemostasis and Thrombosis Center at Duke University Medical Center (Durham, NC).
Data Analysis
The safety population consisted of all subjects who received at least 1 dose of study drug. Analysis of the response to endotoxin challenge was performed on the per‐protocol population. This population consisted of all subjects who completed dosing, received the endotoxin challenge, and had at least 1 postendotoxin challenge CRP value.
Subjects assigned to placebo were pooled and analyzed as a group. Baseline was defined as the last evaluation preceding the first dose of study drug, unless specified otherwise. Statistical tests were 2‐sided with a type I error rate controlled at an alpha level of 0.05. Statistical significance was determined using Wilcoxon's rank‐sum test based on sample size and data distribution. Analysis of data was performed using SAS software (version 9.2; SAS Institute Inc., Cary, NC).
Results
Subjects
Forty‐two eligible adult males were enrolled into the study in order to achieve 10 evaluable subjects per treatment arm. Subjects were assigned sequentially to 1 of 2 dose cohorts at a 2:1 randomization ratio of active to placebo. The first cohort assigned was for treatment with a 400‐mg dose and the second was for treatment with a 600‐mg dose. A total of 35 of 42 subjects were dosed with study drug or placebo before the endotoxin challenge (23 active and 12 placebo). Baseline characteristics of subjects who received at least 1 dose of study drug are summarized by treatment group in Table 1.
Table 1.
Subject Demographics and Baseline Characteristics
| Placebo (N=12) | 400 mg (N=12) | 600 mg (N=11) | |
|---|---|---|---|
| Race, n (%) | |||
| White | 7 (58) | 6 (50) | 4 (36) |
| Black | 4 (33) | 6 (50) | 6 (55) |
| Other | 1 (8) | 0 (0) | 1 (9) |
| Age, y | 22.7±3.7 | 28.7±6.5 | 27.7±5.8 |
| Weight, kg | 78.8±10.5 | 77.2±9.8 | 80.4±11.4 |
| BMI, kg/m2 | 25.5±2.6 | 24.8±2.9 | 25.2±3.1 |
| CRP, mg/L | 0.85 (0.30, 1.15) | 0.60 (0.55, 1.40) | 0.50 (0.20, 1.10) |
Values represent the mean±SD for age, weight, and BMI; and the median (Q1, Q3) for CRP. BMI indicates body mass index; CRP, C‐reactive protein.
Thirty‐three of 35 (94%) treated subjects received each of the 6 scheduled doses of study drug. One subject assigned to placebo discontinued after receiving 4 doses because of a low white blood cell (WBC) count, and 1 subject withdrew consent after receiving 5 doses of 600 mg of ISIS‐CRPRx. Two subjects withdrew consent after receiving the 6 doses of study drug (n=1, placebo; n=1, 400 mg of ISIS‐CRPRx) and consequently did not participate in the endotoxin challenge. Thirty of 31 subjects who participated in the endotoxin challenge (10 subjects per treatment assignment) were evaluable for analysis of the effects of treatment on the response to endotoxin challenge. A complete representation of the flow of study participants is shown in Figure 2.
Figure 2.
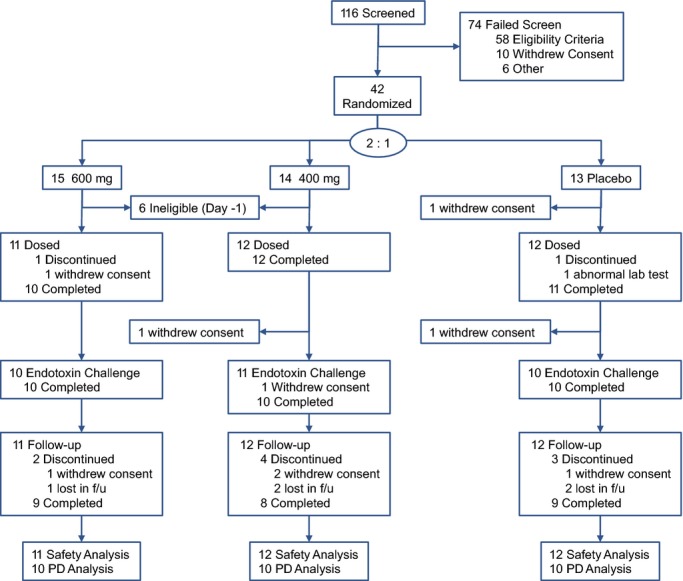
Flow of study participants. PD indicates pharmacodynamic.
Safety and AEs in the Treatment Period Preceding Endotoxin Challenge
ISIS‐CRPRx was well tolerated at both doses tested. Reported AEs were, if anything, more common in the pooled placebo group during the treatment period (12 events in 13 participants) than in the 2 ISIS‐CRPRx‐treated groups (10 events in 23 participants). AEs were predominantly associated with intravenous infusion of study drug. These AEs occurred in both placebo and ISIS‐CRPRx‐treated subjects (Table 2). In addition to these events, 3 of 23 (13%) subjects dosed with ISIS‐CRPRx reported headache. Otherwise, no unexpected safety concerns occurred in association with study drug during this period. There were no unexplained or clinically remarkable changes in routine laboratory measures, nor was there a significant change in CRP levels from baseline to the end of treatment between dose groups (data not shown).
Table 2.
Adverse Events Reported During Treatment Period (Days 1 Through 25/ET, Safety Population)
| Preferred MedDRA Term, n (%) | Placebo (N=12) | 400 mg (N=12) | 600 mg (N=11) |
|---|---|---|---|
| Headache | 0 (0) | 1 (8) | 2 (18) |
| Infusion site hematoma | 1 (8) | 0 (0) | 2 (18) |
| Infusion site extravasation | 1 (8) | 0 (0) | 1 (9) |
| Infusion site pain | 2 (17) | 0 (0) | 0 (0) |
| Pain in extremity | 0 (0) | 1 (8) | 0 (0) |
| Dermatitis contact | 0 (0) | 1 (8) | 0 (0) |
| Gastroenteritis | 0 (0) | 1 (8) | 0 (0) |
| Abdominal pain | 0 (0) | 0 (0) | 1 (9) |
| Infusion‐related reaction | 1 (8) | 0 (0) | 0 (0) |
| WBC count decreased | 1 (8) | 0 (0) | 0 (0) |
ET indicates endotoxin; WBC, white blood cell.
Endotoxin Challenge
Pretreatment of subjects with ISIS‐CRPRx produced a dose‐dependent, statistically significant reduction in endotoxin‐induced CRP levels (Figures 3 and 4), as well as the area under the curve over time. Specifically, among those allocated to placebo, median CRP levels increased more than 50‐fold from baseline 24 hours after endotoxin challenge. By contrast, the median increase in CRP levels was attenuated by 37% (400 mg) and 69% (600 mg) in subjects pretreated with ISIS‐CRPRx (P<0.05 vs. placebo; Figure 3B). Maximum CRP levels remained below 10 mg/L in 8 of 10 subjects in the 600‐mg dose group. There was no effect, however, from CRP antisense treatment on induction of SAA expression (Figure 3C). Similarly, there was no effect on upstream induction of proinflammatory cytokines and chemokines, including TNF‐α, IL‐6, and MCP‐1 (Figure 5). Endotoxin‐induced activation of the coagulation cascade, fibrinolytic pathway, and endothelium was also unaffected by pretreatment with ISIS‐CRPRx, relative to placebo, as measured by changes in D‐dimer, PT fragment (F1+2), and soluble E‐selectin levels (Figure 6). Similarly, there was no differential effect between treatment groups on complement factors or split products, C4, Bb, and C5a (Figure 7).
Figure 3.
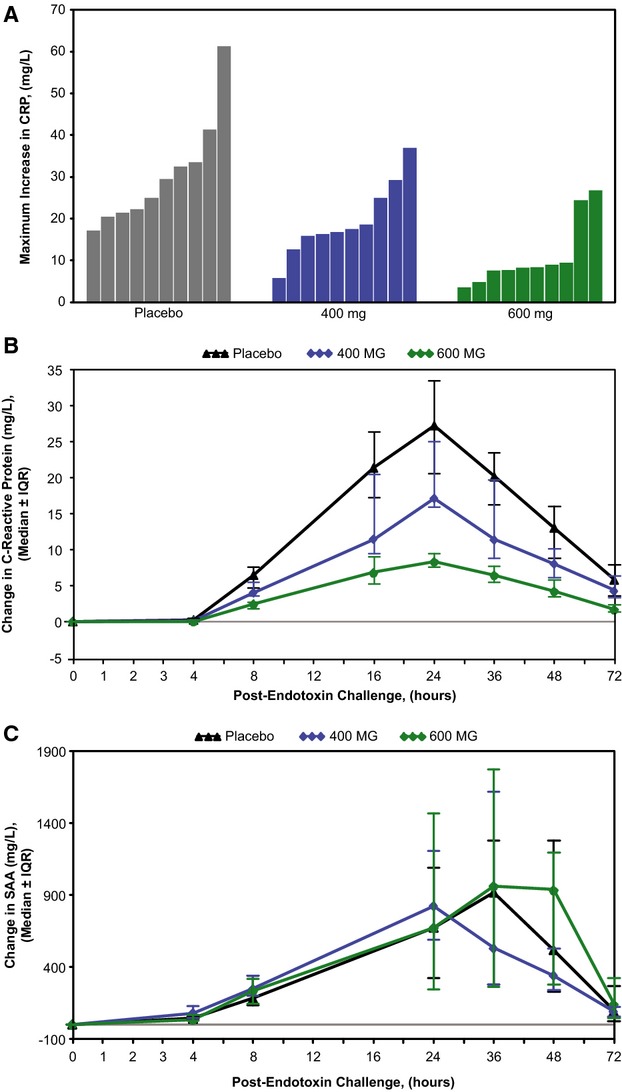
Pretreatment with ISIS‐CRPRx attenuated endotoxin‐induced increase in CRP levels in a dose‐dependent and selective manner. (A) Maximum change in CRP levels by subject, (B) median change in CRP levels over time by treatment group, and (C) median change in SAA over time by treatment group, relative to preendotoxin baseline levels. CRP indicates C‐reactive protein; IQR, interquartile range; SAA, serum amyloid A.
Figure 4.
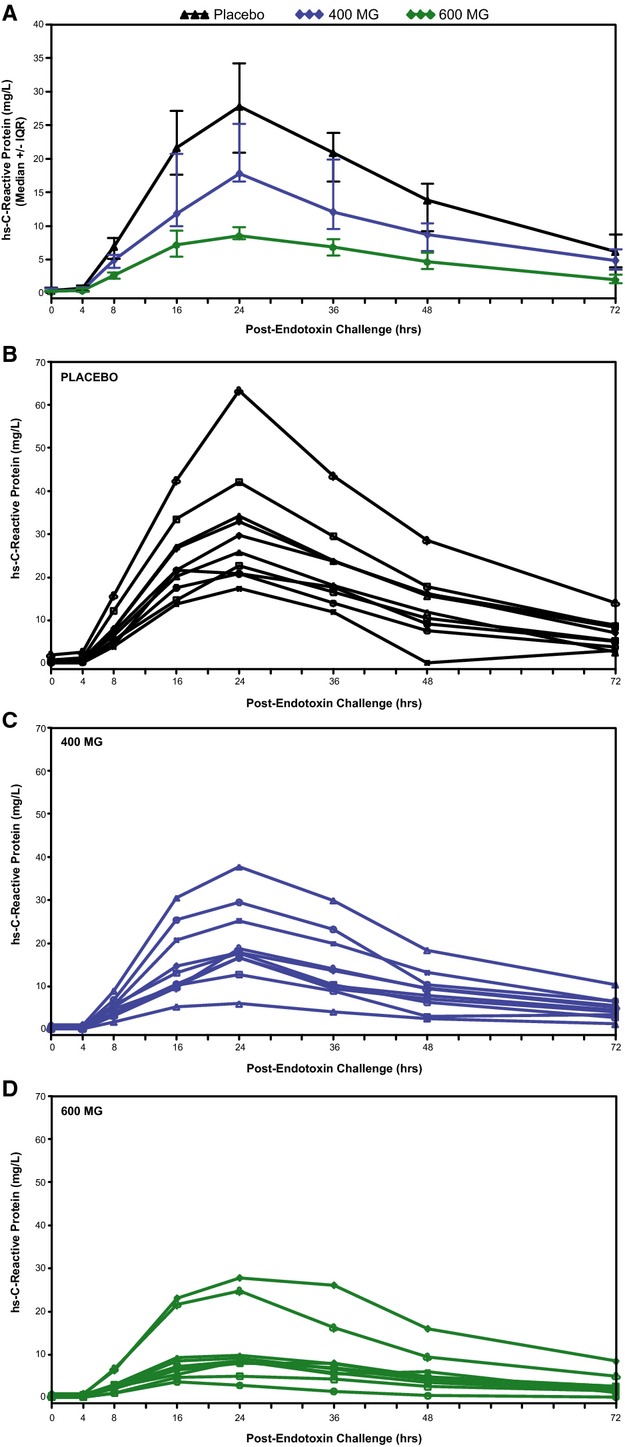
Absolute CRP concentrations pre– and post–endotoxin challenge in (A) treatment groups, (B) placebo‐treated subjects, (C) 400‐mg ISIS‐CRPRx‐treated subjects, and (D) 600‐mg ISIS‐CRPRx‐treated subjects. CRP indicates C‐reactive protein; IQR, interquartile range.
Figure 5.
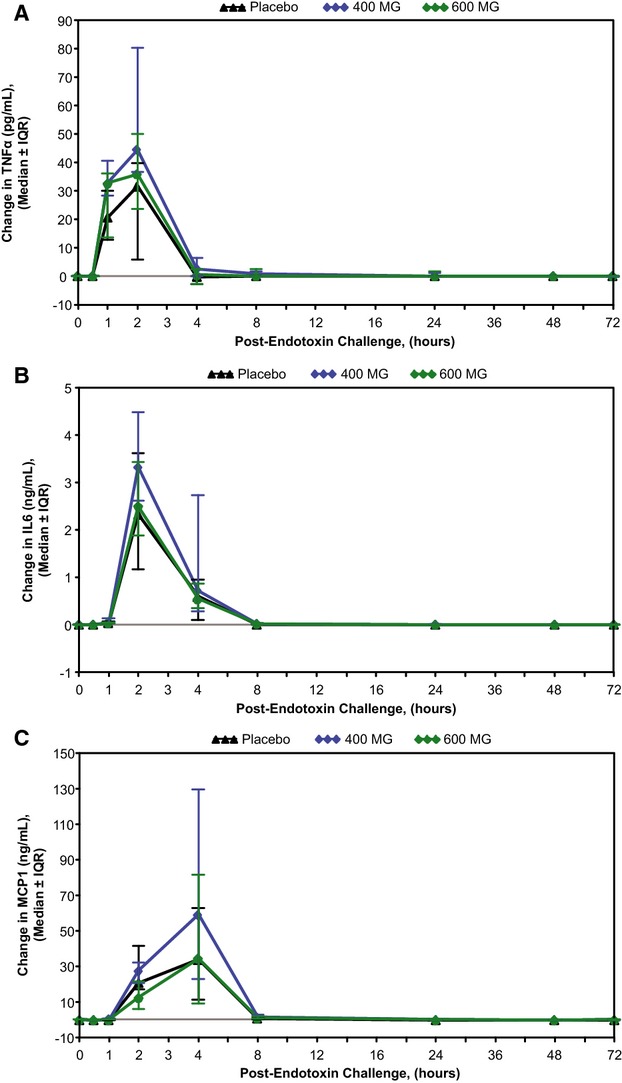
ISIS‐CRPRx pretreatment had no effect on endotoxin‐induced increases in cytokine and chemokine levels. Median changes in (A) TNF‐α, (B) IL‐6, and (C) MCP‐1, over time by treatment group. IL indicates interleukin; IQR, interquartile range; MCP‐1, monocyte chemoattractant protein 1; TNF‐α, tumor necrosis factor alpha.
Figure 6.
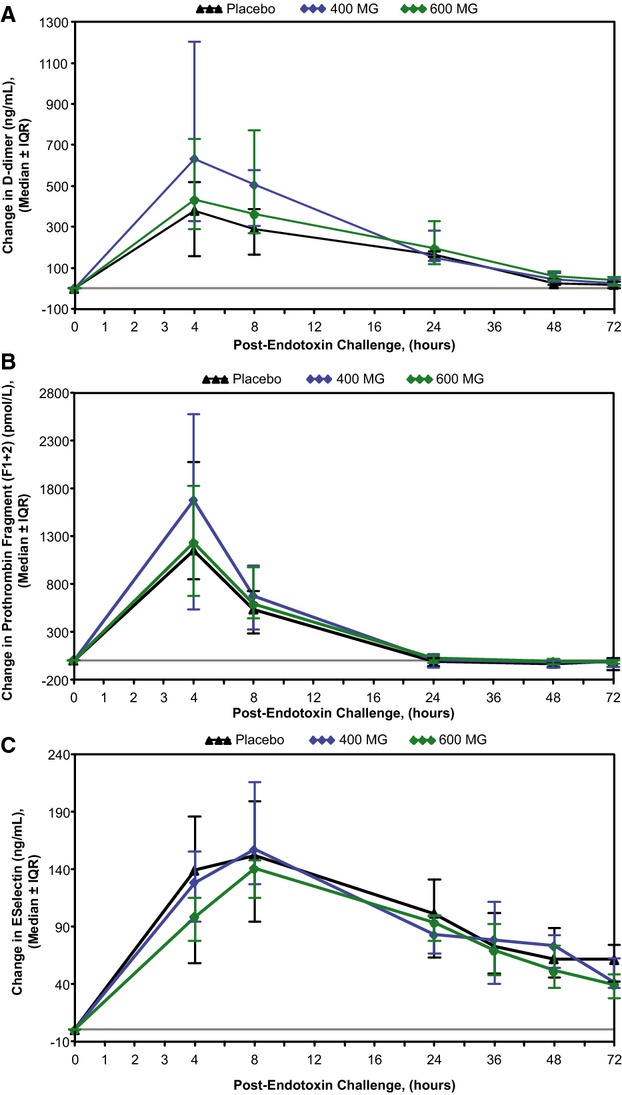
ISIS‐CRPRx pretreatment had no effect on endotoxin‐induced changes in coagulation parameters. Median changes in (A) D‐dimer, (B) prothrombin fragment [F1+2], and (C) E‐selectin, over time by treatment group. IQR indicates interquartile range.
Figure 7.
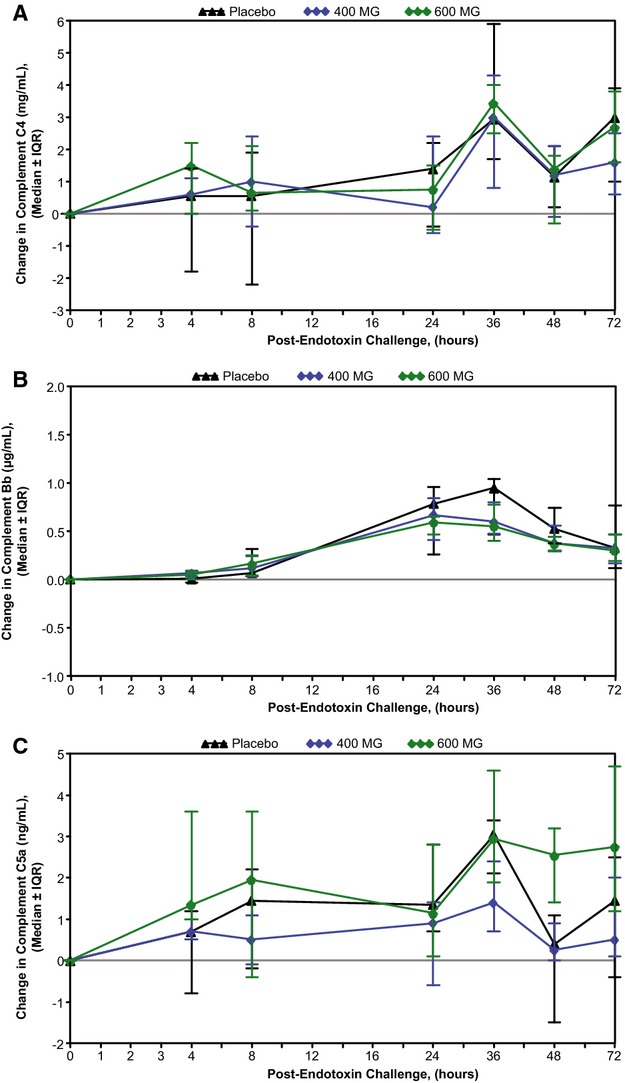
ISIS‐CRPRx pretreatment had no effect on endotoxin‐induced changes in complement factors. Median change in (A) complement C4, (B) complement split product Bb, and (C) complement split product C5a, over time by treatment group. IQR indicates interquartile range.
Expected endotoxin‐induced changes in blood cell counts were similar in subjects pretreated with ISIS‐CRPRx, compared to placebo‐pretreated subjects. As expected, total WBC counts initially decreased postchallenge, reaching a nadir after 1 hour, and then increased above baseline to counts, ranging from 7100 to 17 500 cells/mm3 at 8 hours. The associated subtype profiles, marked by concordant lympho‐ and monocytopenia, were also unaltered by pretreatment with ISIS‐CRPRx, relative to placebo, based on both the absolute and differential counts. Affected parameters returned toward baseline values by 24 hours after the challenge.
The physiological response to endotoxin challenge was similar across treatment groups, as reflected by changes from baseline in body temperature, heart rate, and blood pressure during the 72‐hour postchallenge period (Figure 8). Increases in median body temperature peaked 3.5 hours post–endotoxin challenge. Maximum increases in individual subjects ranged from 0.5 to 2.1°C. Heart rates increased sharply 1 hour after endotoxin exposure, peaking 3 to 4 hours postchallenge, with maximum increases from baseline in individual subjects of 18 to 69 beats per minute. Subjects' heart rates gradually returned toward baseline by 12 to 24 hours after the challenge. A transient increase in systolic and diastolic blood pressure also occurred in all treatment groups, with median values peaking at the 1.5 hour time point. Maximum increases from baseline in systolic and diastolic pressure by subject ranged from 2 to 32 and 1 to 31 mm Hg, respectively. For all physiological parameters, no remarkable differences were observed after endotoxin challenge for those pretreated with ISIS‐CRPRx, as compared to placebo.
Figure 8.
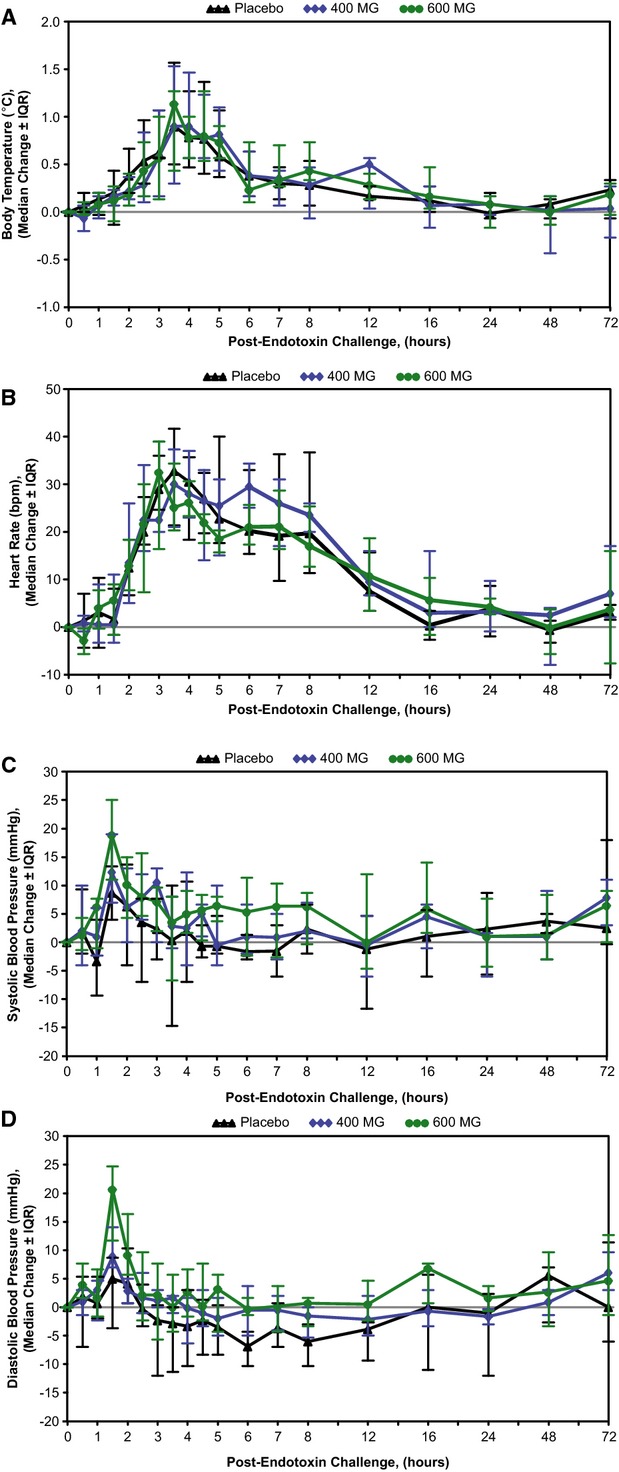
ISIS‐CRPRx pretreatment had no effect on endotoxin‐induced changes in vital signs. Median changes in (A) body temperature, (B) heart rate, (C) systolic blood pressure, and (D) diastolic blood pressure, over time by treatment group. IQR indicates interquartile range.
Classical endotoxin‐induced flu‐like symptoms (eg, headache, fever, and chills) were reported by 30 of 30 (100%) should be subject's, that is, irrespective of treatment group (Table 3). All symptoms resolved spontaneously without sequelae. Other routine laboratory measures, such as liver transaminases, were unremarkable.
Table 3.
Adverse Events Reported During Endotoxin Challenge Period (Days 26 to 29)
| Preferred MedDRA Term, n (%) | Placebo (N=10) | 400 mg (N=11) | 600 mg (N=10) |
|---|---|---|---|
| Headache | 8 (80) | 10 (91) | 10 (100) |
| Pyrexia | 8 (80) | 9 (82) | 9 (90) |
| Chills | 8 (80) | 9 (82) | 8 (80) |
| Myalgia | 6 (60) | 7 (64) | 5 (50) |
| Nausea | 3 (30) | 5 (45) | 4 (40) |
| Tachycardia | 5 (50) | 3 (27) | 2 (20) |
| Vomiting | 2 (20) | 0 (0) | 1 (10) |
| Dizziness | 2 (20) | 1 (9) | 0 (0.0) |
| Palpitations | 0 (0) | 1 (9) | 1 (10) |
| Fatigue | 0 (0) | 1 (9) | 1 (10) |
| Catheter site pain | 0 (0) | 1 (9) | 0 (0) |
| Flushing | 0 (0) | 1 (9) | 0 (0) |
| Hyperhidrosis | 0 (0) | 1 (9) | 0 (0) |
| Photophobia | 0 (0) | 0 (0) | 1 (10) |
| Hypotension | 1 (10) | 0 (0) | 0 (0) |
| Lethargy | 1 (10) | 0 (0) | 0 (0) |
| Musculoskeletal pain | 1 (10) | 0 (0) | 0 (0) |
| Cyanosis | 1 (10) | 0 (0) | 0 (0) |
| Livedo reticularis | 1 (10) | 0 (0) | 0 (0) |
Follow‐up Period
Subjects were monitored for 63 days after the endotoxin challenge or otherwise after the last dose of study drug. Twenty‐six of 35 (74%) subjects completed the follow‐up period (Figure 2). Three AEs were reported during this period, 2 in the placebo group and 1 in the 600‐mg dose group (Table 4). The latter event was characterized as a mild increase in creatinine, as measured on the last visit, day 92 (1.29 mg/dL, upper limit of normal=1.27), and then confirmed 7 days later (1.35 mg/dL). The subjects' blood urea nitrogen (BUN) concentration, BUN/creatinine ratio, and serum potassium concentration were within the range of normal. Serum creatinine levels returned to normal within 3 weeks.
Table 4.
Adverse Events Reported During Follow‐up Period (Day 30/ET to End of Study, Safety Population)
| Preferred MedDRA Term, n (%) | Placebo (N=12) | 400 mg (N=12) | 600 mg (N=11) |
|---|---|---|---|
| Blood creatinine increased | 0 (0) | 0 (0) | 1 (9) |
| Headache | 1 (8) | 0 (0) | 0 (0) |
| Sinus headache | 1 (8) | 0 (0) | 0 (0) |
Discussion
In this phase I, double‐blind, placebo‐controlled study conducted in healthy volunteers, we observed that the ASO, ISIS‐CRPRx, selectively attenuated the endotoxin‐induced increase in CRP levels in a dose‐dependent manner. Other signals and processes of the acute response to endotoxin challenge remained intact, however, including the transient increases in cytokines and chemokines levels, activation of both the coagulation cascade and fibrinolytic process, fluctuations in WBC counts and differentials, increased body temperature and heart rate, as well as anticipated clinical signs and symptoms. Overall, ISIS‐CRPRx was well tolerated and with no evident safety issues during the treatment period at both the 400‐ and 600‐mg doses tested.
We believe our data to be of clinical as well as pathophysiological interest for several reasons. First, these data provide a robust demonstration of the high degree of selectivity and specificity that can be achieved by antisense technology. In this regard, the observed dose‐dependent effects of ISIS‐CRPRx on CRP synthesis without alteration of other acute‐phase parameters provides in vivo confirmation of ISIS‐CRPRx as an ASO designed specifically to accelerate degradation of human CRP mRNA without inducing other secondary effects.
Second, our data lend further support to the concept that pentameric CRP is predominantly a biomarker of disease, rather than a causal factor, in the pathophysiology of inflammation. Endotoxin infusion led to the anticipated increases in cytokine and chemokine expression, and leukocytosis, whereas the secondary increase in CRP had no effect on these parameters. Thus, in spite of a 3‐fold decrease in CRP peak at 24 hours in the antisense‐treated group, compared to placebo, none of the parameters reflecting inflammation, coagulation, or complement activation revealed any difference in the time frame between 24 and 48 hours. These data do not support the concept that pentameric CRP itself has a direct, active effect on these pathways in otherwise healthy individuals. In fact, these results complement recent work from Lane et al.,11 showing that infusion of pharmaceutical‐grade CRP had little effect on the induction of inflammation itself, again in healthy individuals. The current data are also consistent with Mendelian randomization studies that have found CRP to be a predictor of vascular events, but unlikely in itself to be in a critical causal pathway. However, these data are not informative about the potential role of monomeric CRP in these processes.23
Neither the current results nor those of Lane et al.11 reduce the clinical utility of CRP as a biomarker of inflammation or as a predictor of future vascular risk. However, the neutral results of both studies do emphasize the importance of targeting upstream mediators of inflammation as potential treatments for vascular disease, rather than a downstream biomarker, such as CRP itself.24 In this regard, 2 large‐scale trials directly testing the inflammation hypothesis of atherothrombosis are underway, including the 10 000 participant Canakinumab Anti‐Inflammatory Thrombosis Outcomes Study (CANTOS) targeting upstream IL‐1β production25 and the 7000 patient NIH‐sponsored Cardiovascular Inflammation Reduction Trial (CIRT) testing low‐dose methotrexate.26–27 Whereas both canakinumab and low‐dose methotrexate reduce downstream CRP, they do so by also inhibiting the function and/or expression of upstream IL‐6. This is likely to be biologically relevant because Mendelian randomization studies similar to those that have been neutral for CRP are strongly positive for IL‐6 signaling. Specifically, 2 studies have now been presented indicating that polymorphism in the IL‐6 receptor pathway associates with lifelong reductions in CRP and lifelong reductions in rates of vascular events.28–29 Data from the CANTOS and CIRT hard outcome vascular prevention trials are anticipated within the next 3 to 4 years.
Finally, the unique specificity of ISIS‐CRPRx provides a new investigative tool for those interested in CRP function in a variety of settings. For example, the ability to dose‐titrate inhibition of CRP synthesis may provide a method to reduce the AEs of complement‐induced CRP damage known to occur in the setting of tissue damage. To our knowledge, the only other inhibitor of CRP uses a small‐molecule approach to bind and interfere with the acute‐phase protein's function in vivo.30 However, the respective small molecule evaluated in the reported animal model studies is no longer being considered for development in the clinic (http://pentraxin.wordpress.com/rd-programs/).
Limitations of our study merit consideration. First, we studied only men, and thus our data cannot be generalized to women. Issues such as hormone use are known to increase CRP production. Second, endotoxin challenge models are a robust acute induction of the innate immune response and may mask, or not effectively model, any potential aberrant functions and processes associated with abnormally elevated CRP levels. Third, the effects of CRP inhibition in healthy volunteers challenged with endotoxin may not reflect effects that occur in subjects burdened with disease, a chronic pathological condition, or acute cell damage. Finally, it is theoretically possible that ISIS‐CRPrx did not affect secondary cytokine and chemokine expression because the net 69% reduction in the 600‐mg dose might have been insufficient. We think this unlikely because many individuals had CRP reductions of 85% to 90%, and these individuals also did not demonstrate secondary cytokine or chemokine changes. Nonetheless, whether complete inhibition of CRP synthesis might have this effect cannot be fully addressed here. Alternative approaches, such as RNAi therapeutics, might be able to provide such levels of inhibition, as has recently been shown for proprotein convertase subtilisin/kexin type 9.31 However, most RNAi therapeutics are formulated in lipid nanoparticle (LNP) delivery systems to overcome stability and delivery limitations. Because LNPs themselves may elicit significant proinflammatory cytokine and gene expression effects and can produce infusion‐related reactions,32 patients receiving RNAi therapeutics typically require premedication with dexamethasone, nonsteroidal anti‐inflammatory drugs, and antihistamines, which have the potential to confound effects on the inflammatory pathways being interrogated here.31,33 Second‐generation ASO approaches that do not require liposomal encapsulation might provide a direction for future research, particularly because the predominant site of CRP synthesis are hepatocytes.
In conclusion, in this placebo‐controlled study, we demonstrate that pretreatment of healthy young men with ISIS‐CRPRx selectively reduces the endotoxin‐induced increase in CRP levels in a dose‐dependent manner, but does not inhibit other components of the classical acute‐phase response. These data demonstrate the specificity of ASOs and provide an investigative tool to further define the role of CRP in human pathological conditions. For example, it will be of interest to know if the ASO approach outlined here alters the dissociation between pentameric CRP to monomeric CRP, particularly in settings with ongoing inflammation.
Sources of Funding
Isis Pharmaceuticals, Inc.
Disclosures
Ridker is a consultant for Isis Pharmaceuticals, Inc., and is listed as a coinventor on patents held by the Brigham and Women's Hospital that relate to the use of inflammatory biomarkers in cardiovascular disease and diabetes that have been licensed to Siemens and AstraZeneca; Noveck was the principle investigator for this study; Stroes consulted for Isis Pharmaceuticals, Inc., during the design and conduct of the study; Flaim, Baker, Hughes, Graham, and Crooke are employees of Isis Pharmaceuticals, Inc.
Acknowledgments
The authors thank Shuting Xia for statistical support and review of the manuscript and Tracy Reigle for graphics support, both of Isis Pharmaceuticals, Inc.
References
- 1.Pepys MB, Hirschfield GM. C‐reactive protein: a critical update. J Clin Invest. 2003; 111:1805-1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black S, Kushner I, Samols D. C‐reactive protein. J Biol Chem. 2004; 279:48487-48490. [DOI] [PubMed] [Google Scholar]
- 3.Castell JV, Gómez‐Lechón MJ, David M, Fabra R, Trullenque R, Heinrich PC. Acute‐phase response of human hepatocytes: regulation of acute‐phase protein synthesis by interleukin‐6. Hepatology. 1990; 12:1179-1186. [DOI] [PubMed] [Google Scholar]
- 4.Griselli M, Herbert J, Hutchinson WL, Taylor KM, Sohail M, Krausz T, Pepys MB. C‐reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J Exp Med. 1999; 190:1733-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997; 336:973-979. [DOI] [PubMed] [Google Scholar]
- 6.Ridker PM, Hennekens CH, Buring JE, Rifai N. C‐reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000; 342:836-843. [DOI] [PubMed] [Google Scholar]
- 7.Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J. C‐reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta‐analysis. Lancet. 2010; 375:132-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bisoendial RJ, Kastelein JJ, Levels JH, Zwaginga JJ, van den Bogaard B, Reitsma PH, Meijers JC, Hartman D, Levi M, Stroes ES. Activation of inflammation and coagulation after infusion of C‐reactive protein in humans. Circ Res. 2005; 96:714-716. [DOI] [PubMed] [Google Scholar]
- 9.Bisoendial RJ, Kastelein JJ, Peters SL, Levels JH, Birjmohun R, Rotmans JI, Hartman D, Meijers JC, Levi M, Stroes ES. Effects of CRP infusion on endothelial function and coagulation in normocholesterolemic and hypercholesterolemic subjects. J Lipid Res. 2007; 48:952-960. [DOI] [PubMed] [Google Scholar]
- 10.Birjmohun RS, Bisoendial RJ, van Leuven SI, Ackermans M, Zwinderman A, Kastelein JJ, Stroes ES, Sauerwein HP. A single bolus infusion of C‐reactive protein increases gluconeogenesis and plasma glucose concentration in humans. Metabolism. 2007; 56:1576-1582. [DOI] [PubMed] [Google Scholar]
- 11.Lane T, Wassef NL, Poole S, Mistry Y, Lachmann H, Gillmore J, Hawkins PN, Pepys MB. Infusion of pharmaceutical grade natural human C reactive protein is not pro inflammatory in healthy adult human volunteers. Circ Res. 2014; 114:672-676. [DOI] [PubMed] [Google Scholar]
- 12.Crooke ST, Vickers T, Lima WH. In: Crooke ST. (ed.). Mechanisms of antisense drug action, an introduction. Antisense Drug Technology: Principles, Strategies, and Applications. 20072nd edBoca Raton, FL: CRC Press/Taylor & Francis Group; 3-46. [Google Scholar]
- 13.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. 2010; 50:259-293. [DOI] [PubMed] [Google Scholar]
- 14.Yu RZ, Grundy JS, Geary RS. Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin Drug Metab Toxicol. 2013; 9:169-182. [DOI] [PubMed] [Google Scholar]
- 15.Bennett CF. In: Crooke ST. (ed.). Pharmacological properties of 2′‐O‐methoxyethyl‐modified oligonucleotides. Antisense Drug Technology: Principles, Strategies, and Applications. 20072nd edBoca Raton, FL: CRC Press/Taylor & Francis Group; 273-303. [Google Scholar]
- 16.Wu H, Lima WF, Zhang H, Fan A, Sun H, Crooke ST. Determination of the role of the human RNase H1 in the pharmacology of DNA‐like antisense drugs. J Biol Chem. 2004; 279:17181-17189. [DOI] [PubMed] [Google Scholar]
- 17.Crooke R, Baker B, Wedel M. In: Crooke ST. (ed.). Cardiovascular therapeutic applications. Antisense Drug Technology: Principles, Strategies, and Applications. 20072nd edBoca Raton, FL: CRC Press/Taylor & Francis Group; 626-628. [Google Scholar]
- 18.Jones NR, Pegues MA, McCrory MA, Singleton W, Bethune C, Baker BF, Norris DA, Crooke RM, Graham MJ, Szalai AJ. A selective inhibitor of human C‐reactive protein translation is efficacious in vitro and in C‐reactive protein transgenic mice and humans. Mol Ther Nucleic Acids. 2012; 1:e5210.1038/mtna.2012.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suffredini AF, Hochstein HD, McMahon FG. Dose‐related inflammatory effects of intravenous endotoxin in humans: evaluation of a new clinical lot of Escherichia coli O:113 endotoxin. J Infect Dis. 1999; 179:1278-1282. [DOI] [PubMed] [Google Scholar]
- 20.van Deventer SJH, Büller HR, ten Cate JW, Aarden LA, Hack CE, Sturk A. Experimental endotoxemia in humans: analysis of cytokine release and coagulation, fibrinolytic and complement pathways. Blood. 1990; 76:2520-2526. [PubMed] [Google Scholar]
- 21.Andreasen AS, Krabbe KS, Krogh‐Madsen R, Taudorf S, Pedersen BK, Møller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem. 2008; 15:1697-1705. [DOI] [PubMed] [Google Scholar]
- 22.Hudgins LC, Parker TS, Levine DM, Gordon BR, Saal SD, Jiang XC, Seidman CE, Tremaroli JD, Lai J, Rubin AL. A single intravenous dose of endotoxin rapidly alters serum lipoproteins and lipid transfer proteins in normal volunteers. J Lipid Res. 2003; 44:1489-1498. [DOI] [PubMed] [Google Scholar]
- 23.C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC). Wensley F, Gao P, Burgess S, Kaptoge S, Di Angelantonio E, Shah T, Engert JC, Clarke R, Davey‐Smith G, Nordestgaard BG, Saleheen D, Samani NJ, Sandhu M, Anand S, Pepys MB, Smeeth L, Whittaker J, Casas JP, Thompson SG, Hingorani AD, Danesh J. Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ. 2011; 342:d548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ridker PM. Targeting inflammatory pathways for the treatment of cardiovascular disease. Eur Heart J. 2014; 35:540-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin‐1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti‐inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J. 2011; 162:597-605. [DOI] [PubMed] [Google Scholar]
- 26.Everett BM, Pradhan AD, Solomon DH, Paynter N, Macfadyen J, Zaharris E, Gupta M, Clearfield M, Libby P, Hasan AA, Glynn RJ, Ridker PM. Rationale and design of the Cardiovascular Inflammation Reduction Trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013; 166:199-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ridker PM. Testing the inflammatory hypothesis of atherothrombosis: scientific rationale for the cardiovascular inflammation reduction trial (CIRT). J Thromb Haemost. 2009; 7suppl 1:332-339. [DOI] [PubMed] [Google Scholar]
- 28.IL6R Genetics Consortium Emerging Risk Factors Collaboration. Sarwar N, Butterworth AS, Freitag DF, Gregson J, Willeit P, Gorman DN, Gao P, Saleheen D, Rendon A, Nelson CP, Braund PS, Hall AS, Chasman DI, Tybjærg‐Hansen A, Chambers JC, Benjamin EJ, Franks PW, Clarke R, Wilde AA, Trip MD, Steri M, Witteman JC, Qi L, van der Schoot CE, de Faire U, Erdmann J, Stringham HM, Koenig W, Rader DJ, Melzer D, Reich D, Psaty BM, Kleber ME, Panagiotakos DB, Willeit J, Wennberg P, Woodward M, Adamovic S, Rimm EB, Meade TW, Gillum RF, Shaffer JA, Hofman A, Onat A, Sundström J, Wassertheil‐Smoller S, Mellström D, Gallacher J, Cushman M, Tracy RP, Kauhanen J, Karlsson M, Salonen JT, Wilhelmsen L, Amouyel P, Cantin B, Best LG, Ben‐Shlomo Y, Manson JE, Davey‐Smith G, de Bakker PI, O'Donnell CJ, Wilson JF, Wilson AG, Assimes TL, Jansson JO, Ohlsson C, Tivesten Å, Ljunggren Ö, Reilly MP, Hamsten A, Ingelsson E, Cambien F, Hung J, Thomas GN, Boehnke M, Schunkert H, Asselbergs FW, Kastelein JJ, Gudnason V, Salomaa V, Harris TB, Kooner JS, Allin KH, Nordestgaard BG, Hopewell JC, Goodall AH, Ridker PM, Hólm H, Watkins H, Ouwehand WH, Samani NJ, Kaptoge S, Di Angelantonio E, Harari O, Danesh J. Interleukin‐6 receptor pathways in coronary heart disease: a collaborative meta‐analysis of 82 studies. Lancet. 2012; 379:1205-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hingorani AD, Casas JP. The interleukin‐6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. 2012; 379:1214-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pepys Nature 2006 Pepys MB. Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, Hawkins PN, Myers RM, Smith MD, Polara A, Cobb AJ, Ley SV, Aquilina JA, Robinson CV, Sharif I, Gray GA, Sabin CA, Jenvey MC, Kolstoe SE, Thompson D, Wood SP. Targeting C‐reactive protein for the treatment of cardiovascular disease. Nature. 2006; 440:1217-1221. [DOI] [PubMed] [Google Scholar]
- 31.Fitzgerald K, Frank‐Kamenetsky M, Shulga‐Morskaya S, Liebow A, Bettencourt BR, Sutherland JE, Hutabarat RM, Clausen VA, Karsten V, Cehelsky J, Nochur SV, Kotelianski V, Horton J, Mant T, Chiesa J, Ritter J, Munisamy M, Vaishnaw AK, Gollob JA, Simon A. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single‐blind, placebo‐controlled, phase 1 trial. Lancet. 2014; 383:60-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abrams MT, Koser ML, Seitzer J, Williams SC, DiPietro MA, Wang W, Shaw AW, Mao X, Jadhav V, Davide JP, Burke PA, Sachs AB, Stirdivant SM, Sepp‐Lorenzino L. Evaluation of efficacy, biodistribution, and inflammation for a potent siRNA nanoparticle: effect of dexamethasone co‐treatment. Mol Ther. 2010; 18:171-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, Perez J, Chiesa J, Warrington S, Tranter E, Munisamy M, Falzone R, Harrop J, Cehelsky J, Bettencourt BR, Geissler M, Butler JS, Sehgal A, Meyers RE, Chen Q, Borland T, Hutabarat RM, Clausen VA, Alvarez R, Fitzgerald K, Gamba‐Vitalo C, Nochur SV, Vaishnaw AK, Sah DW, Gollob JA, Suhr OB. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013; 369:819-829. [DOI] [PubMed] [Google Scholar]