

SUMMARY
Colitis results from breakdown of homeostasis between intestinal microbiota and the mucosal immune system, with both environmental and genetic influencing factors. Flagellin receptor TLR5-deficient mice (T5KO) display elevated intestinal pro-inflammatory gene expression and colitis with incomplete penetrance, providing a genetically sensitized system to study the contribution of microbiota to driving colitis. Both colitic and non-colitic T5KO exhibited transiently unstable microbiotas, with lasting differences in colitic T5KO while their non-colitic siblings stabilized their microbiotas to resemble wild-type mice. Transient high levels of Proteobacteria, especially Enterobacteria species including E. coli, observed in close proximity to the gut epithelium was a striking feature of colitic microbiota. A Crohn’s disease-associated E. coli strain induced chronic colitis in T5KO, which persisted well after the exogenously introduced bacterial species had been eliminated. Thus, an innate immune deficiency can result in unstable gut microbiota associated with low-grade inflammation and harboring Proteobacteria can drive and/or instigate chronic colitis.
Keywords: TLR5, colitis, gut microbial diversity, 16S rRNA gene pyrosequencing, adherent-invasive Escherichia coli
INTRODUCTION
Inflammatory bowel disease (IBD) is thought to result from a breakdown in the relationship between the intestinal mucosal immune system and the large microbial biomass, including about 100 trillion bacteria, that inhabit this organ (Kaser et al., 2010; Xavier and Podolsky, 2007). The underlying cause(s) of the disturbance is not well defined but clearly involves both environmental and genetic factors. Prominent among environmental factors is the gut microbiota in that IBD has been associated with broad changes in composition of the microbiota in general and with select bacterial strains that have been suggested to be opportunistic pathogens (Dupont and Dupont, 2011). For example, a number of studies of IBD in humans have described a functional class of Escherichia coli referred as Adherent-Invasive E. coli (AIEC) (Darfeuille-Michaud, 2002). While E. coli, in general, is a fairly common component of the human microbiota, AIEC were identified as E. coli that had seemingly reached inappropriate locations either adherent to, within, or beyond the gut epithelium whereas the vast majority of gut microbiota are not in direct contact with cells but, rather, are clearly physically separated by the mucus layer; i.e. glycocalyx (Derrien et al., 2010). The AIEC reference strain bacteria LF82 displays relatively high invasiveness in vitro and has been shown to exacerbate murine colitis in a manner dependent upon its expression of flagella (Carvalho et al., 2008). While flagellin is a microbe-associated molecular pattern that is present at the bacterial surface of both pathogenic and nonpathogenic E. coli strains, differential regulation of flagellin expression has been suggested between commensal bacteria and Crohn’s Disease (CD)-associated AIEC, and differential regulation under the two-component EnvZ/OmpR regulatory system (Rolhion et al., 2007).
Several genetic polymorphisms in innate immunity have been described to increase risk of developing IBD (Hugot et al., 2001; Ogura et al., 2001; Rioux et al., 2007). These associations support the possibility that IBD can result from a primary innate immune deficiency. For example, persons homozygous for a frame-shift mutation in the Nod2 gene that ablates its ability to recognize bacterial-derived glycol-peptides have a 17-fold increased risk of developing IBD (Cho and Brant, 2011). Mechanistic investigations into how such an innate immune deficiency might eventuate in chronic intestinal inflammation have been stymied by the experimental result that mice engineered to lack Nod2 function do not develop intestinal inflammation (Kobayashi et al., 2005) despite exhibiting altered gut microbiota composition (Mondot et al., 2011; Rehman et al., 2011). However, some other mice strains with engineered innate immune deficiencies are prone to developing gut inflammation and may ultimately prove to shed light on the pathogenesis of IBD. For example, mice broadly lacking epithelial innate immune function, due to deletion of IKK in intestinal epithelial cells, develop uniform severe colitis (Nenci et al., 2007). Such colitis is not due to microbial onslaught per se but, rather appears to be driven by a secondary, TNF-α-mediated, immune response presumably driven by altered gut bacteria.
A paradigmatically similar, but more subtle, example of an innate immune deficiency promoting gut inflammation are mice engineered to lack the flagellin receptor, Toll-like receptor 5 (TLR5). TLR5-deficient mice (T5KO) are prone to developing elevated pro-inflammatory gene expression in the intestine (Carvalho et al., 2012; Vijay-Kumar et al., 2010; Vijay-Kumar et al., 2007). The extent to which such pro-inflammatory gene expression results in overt colitis or “low-grade” inflammation (i.e. the intestine appears histologically normal but exhibits modest elevation in pro-inflammatory gene expression) that correlates with development of metabolic syndrome characterized by mild obesity and insulin-resistance appears to be modulated by the microbiota. Specifically, antibiotics ameliorate robust colitis (Vijay-Kumar et al., 2007) and both low-grade inflammation and metabolic syndrome could be transferred to germ-free mice by transferring the T5KO microbiota (Vijay-Kumar et al., 2010). The notion that microbiota composition is a key determinant of colitis in T5KO mice is further supported by the observation that embryo transplant of T5KO into mice purchased from Jackson Labs, so as to “Jacksonize” their microbiota, eliminated development of spontaneous overt colitis (Vijay-Kumar et al., 2010). That analogous observations have been made with murine models of colitis (e.g. IL-10 KO (Matharu et al., 2009; Sellon et al., 1998)) provides broad support for the notion that the composition of the gut microbiota is an important environmental determinant of colitis, but such approaches of complete microbiota exchange or ablation have not been especially informative in defining which changes are closely correlated with colitis development nor shed much light on underlying mechanism. In the absence of such Jacksonization, continued inter-breeding/back-crossing of T5KO mice and their WT control relatives eventuates in spontaneous colitis in 10-25% of mice. Such incomplete penetrance poses logistical challenges in experimental design when using this model. However, it is a higher penetrance rate than that seen for persons homozygous for the Nod2 frame-shift mutation (in which a 17-fold increase results in a disease risk of perhaps 5%), and well beyond the 4% IBD concordance seen amongst dizygotic twins (Cho and Brant, 2011). Thus, we reasoned that examination of the microbiota in colitic T5KO mice, their non-colitic littermates, and closely related WT mice might shed light upon the mechanisms that dictate the extent to which an innate immune deficiency will eventuate in intestinal inflammation. In the present study, we observed that such an innate immune deficiency resulted in inability to stably manage the gut microbiota in general and select flagellate Enterobacteria in particular, which had the ability to drive colitis in T5KO mice.
RESULTS
TLR5-deficient mice are prone to develop post-weaning gut inflammation
To better understand the role of gut microbiota in the development in T5KO colitis, we temporally analyzed the composition of the gut microbiota in colitic T5KO mice, littermate T5KO mice that did not develop overt colitis, and closely related WT mice.We prospectively collected fecal samples weekly starting at weaning from a series of litters of WT (n=18) and T5KO mice (n=110). The WT and T5KO mice studied were offspring of mice that were littermates (i.e. born of a heterozygous parent) thus providing a high likelihood those differences between WT and T5KO microbiota would be driven by host genotype. When examined at 12 weeks of age for evidence of colitis, the majority of T5KO mice lacked evidence of colitis while a subset exhibited both colomegaly and splenomegaly (Figure 1A-B), features not present in any of the WT mice and which we have previously observed reliably correlate with robust histopathologic evidence of colitis in T5KO mice (Vijay-Kumar et al., 2007). The colitic T5KO mice did not cluster within individual cages but rather had a seemingly random distribution among the T5KO mice. We next retrospectively analyzed fecal levels of lipocalin-2 (Lcn-2), which we have recently shown is a sensitive and broadly dynamic marker of intestinal inflammation able to mark mice with both low-grade inflammation and robust colitis (our manuscript under review - attached). Relative to WT mice, non-colitic T5KO mice exhibited moderately elevated levels of fecal Lcn-2 in accord with our report of low-grade inflammation in these mice (Carvalho et al., 2011; Carvalho et al., 2012). Moreover, that fecal Lcn-2 levels were further elevated about 10-fold in colitic T5KO mice supported that stratifying based on gross measures reliably differentiated mice with robust and low-grade inflammation (Figure 1C). While the difference in fecal Lcn-2 between colitic and non-colitic T5KO mice was already evident upon weaning, it decreased in the post-weaning period in non-colitic T5KO while increasing during this period in mice that developed colitis suggesting that non-colitic mice had better managed a pro-inflammatory challenge. Retrospective analysis of post-weaning growth curves echoed our previous findings (Vijay-Kumar et al., 2010) that T5KO mice developing colitis exhibited modest reductions in weight gain while non-colitic T5KO mice displayed increased post-weaning weight gain relative to WT mice (Figure 1D). Together, these results demonstrate that T5KO mice exhibit intestinal inflammation with most mice maintaining low-grade inflammation but some mice developing robust colitis in the post-weaning period.
Figure 1. Development of spontaneous inflammation and microbial dysbiosis in T5KO mice.
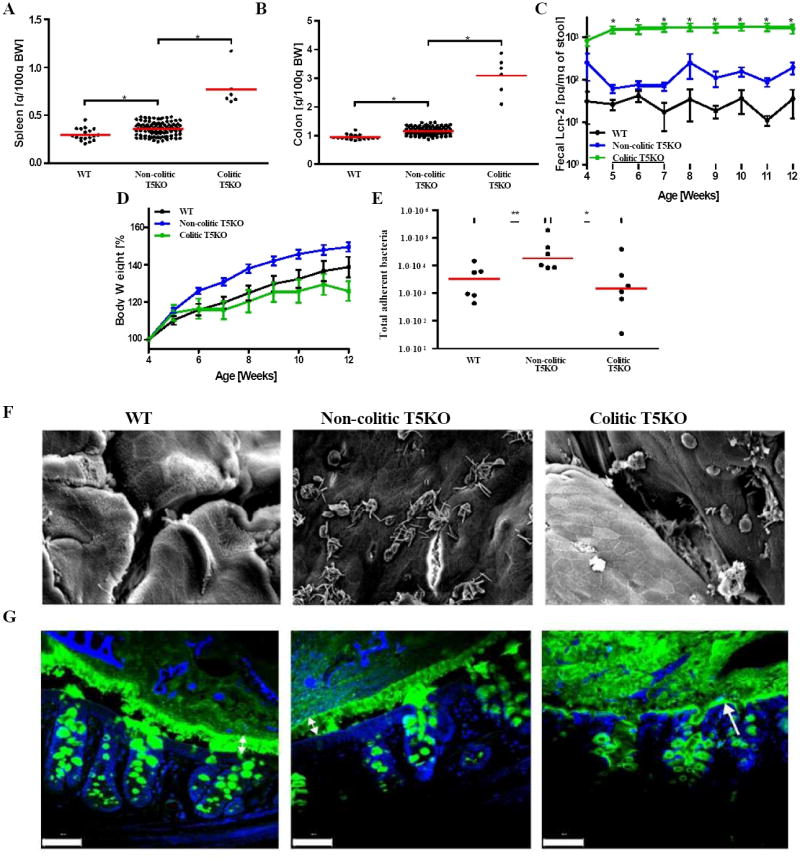
Wild-type (n=18) and T5KO mice (n=110) were housed for 12 weeks in the animal facility to track spontaneous colitic mice (defined as described in Methods). (A) Following euthanasia, spleen was isolated and mass measured. (B) Colon mass. (C) Stool was collected weekly after weaning and diluted in 500 μL of PBS. Then, supernatant was assayed for lipocalin-2 (Lcn-2) expression by ELISA. (D) Body mass was monitored weekly from week 4 to week 12. (E) Colon was washed carefully with PBS to remove any stool and bacterial DNA was isolated. Total adherent bacteria was measured by quantitative PCR analysis using universal 16S rRNA primers. (F) Representative electron microscopy observation of colon (magnification: 1500X). (G) Muc 2 mucin immunostaining - green. Nuclei were stained using DAPI - blue (Scale bar = 20 μM). The data on panels E-G is representative of 2 independent experiments.* p<0.05.
Increased Proteobacteria in colitic T5KO mice
Changes in the microbiota associated with colitis were analyzed in WT, non-colitic T5KO, and colitic T5KO mice by quantitation of total levels of bacteria, using 16S rRNA gene quantitative PCR (qPCR).This approach revealed the presence of increased numbers of intestinal mucosa-associated bacteria in non-colitic compared to colitic T5KO mice (Figure 1E). In accordance, examination of the intestinal mucosal surface by electron microscopy revealed frequent presence of clusters of bacteria in non-colitic T5KO mice (Figure 1F). Such clusters of bacteria were rarely found in WT mice likely reflecting that the fixatives/solvents utilized in this method disassociated most bacterial-intestinal interactions. Clusters of bacteria were also rarely seen in colitic T5KO mice. Rather, colitic T5KO mice displayed occasional patches of bacteria and in proximity to cells appearing to be phagocytes. To better visualize intestinal bacteria in their native location relative to the gut epithelium and the mucus layer, we utilized fixatives that preserve the juxtaposition of these structures. WT mice displayed a well-organized mucus layer similar to that reported previously (Johansson et al., 2008) (Figure 1G). The mucus layer in non-colitic T5KO appeared relatively normal although it was modestly thinner than that of WT mice. In contrast, the mucus structure of colitic T5KO appeared disorganized and, moreover, lacked a well-defined firm inner layer with bacteria close to or in contact with the gut epithelium.
The composition of the microbiota was assessed by pyrosequencing the 16S rRNA genes of cecal bacteria (12 week old mice). When viewed at the phylum level, such end point analysis indicated that the microbiota of WT mice and non-colitic T5KO mice were quite similar, while the most notable feature of colitic T5KO mice was the increase in Proteobacteria species (Figure 2A). Principal coordinates analysis (PCoA) plots of unweighted UniFrac (Lozupone and Knight, 2005) demonstrated dramatic differences in the species level composition of colitic T5KO mice and comparatively modest differences in non-colitic T5KO relative to WT mice (Figure 2B). Together, these results suggest that loss of TLR5 resulted in alterations the gut microbiota especially in mice that developed colitis.
Figure 2. End point microbiota composition in WT, non-colitic and colitic T5KO mice.
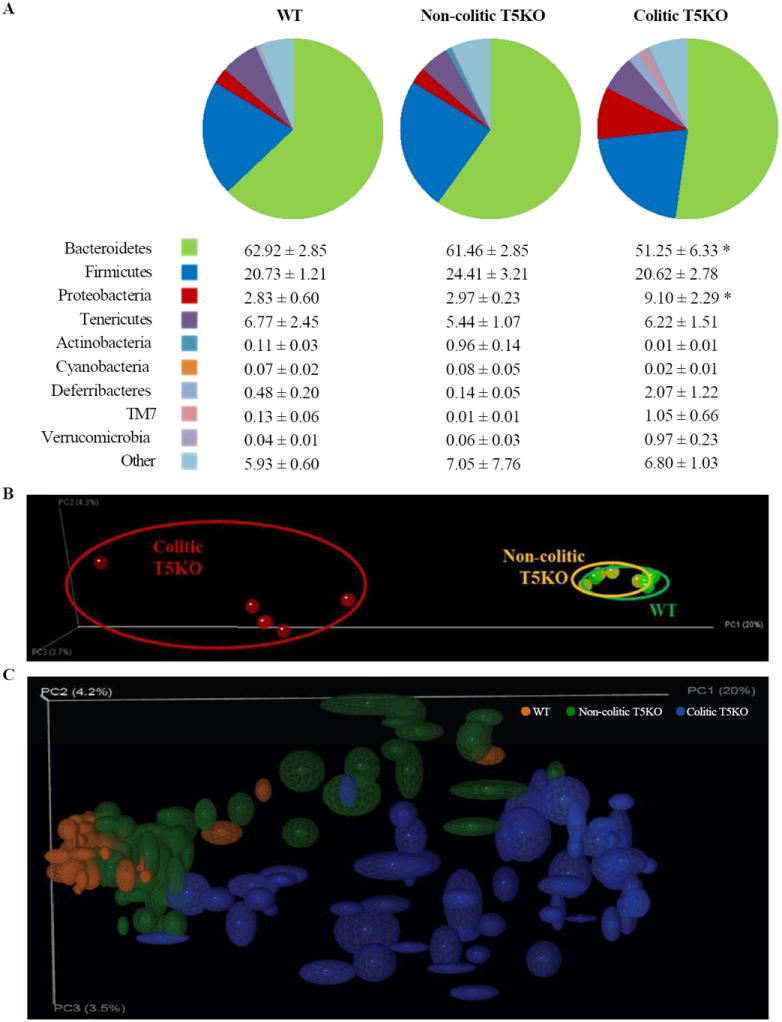
Wild-type, non-colitic and colitic T5KO mice (n=6-8 mice per group) were euthanized at 12-week old. Cecal contents were collected and microbiota composition was analyzed via 16S rRNA analysis. (A) Relative abundance of phyla in cecal bacteria. Table provides mean for each phyla as the percentage of total sequences analyzed. (B) Mouse cecal bacterial communities were clustered using principal coordinates analysis (PCoA) of the UniFrac unweighted distance matrix. PC1, PC2 and PC3 are plotted. The percentage of the variation explained by the plotted principal coordinates is indicated in the axis labels. Results are from an analysis of 5-6 mice per group. (C) Jackknifing PCoA plot of cecal and fecal samples from all time points. Point locations are the average location of 10 jackknife replicates using 1000 random sequences per sample and ellipses show the confidence based on these randomizations. Analysis was done by ANOVA and statistical significance (P<0.01) is denoted by asterisk (*).
Increased volatility in TLR5-deficient mouse gut microbiota
To better understand the extent to which the changes we observed in the cecal microbiota might drive and/or be a consequence of colitis, we examined the composition of the gut microbiota in the fecal samples that were serially collected from each mouse 1-9 weeks post-weaning. The composition of the 9-week samples were similar to that of cecal contents collected shortly thereafter indicating that analysis of these materials reflects similar parameters. Collective analysis of all temporal fecal samples collected showed relatively tight clustering of microbiotas from WT mice while a broader and dramatically different distribution was seen in colitic T5KO mice (Figure 2C). An intermediate pattern was seen in non-colitic T5KO mice with many samples clustering near those of WT mice and others distributed more broadly. These results suggested that, although WT and non-colitic mice eventuate fairly similar microbiotas, they might have dissimilar temporal patterns of development. To further investigate this notion, we next compared relative composition over time from weaning to 11 weeks of age. Such analysis, namely plot of PC1 vs. time, showed clear clustering at initial time points that remained stable over time amongst WT mice whereas T5KO lacked a clear cluster upon weaning but began clustering based upon eventual development of colitis at 4-5 weeks post-weaning (Figure 3A). In accordance with such delayed stabilization of their microbiotas, greater variability in microbiotas was observed among all T5KO mice, that did, and did not, develop colitis. Such microbial volatility was evident when examining week to week changes in individual mice or looking at the average weekly changes in UniFrac distances in our groups of mice (Figure 3B). Increased volatility was also evident by looking at the phylum level. Specifically, the average composition was relatively constant over the 8-week period of study in WT mice but appeared to change considerably in colitic and non-colitic T5KO mice (Figure 3C). Moreover, plotting temporal phylum composition in individual mice with most T5KO (non-colitic and colitic) mice showed markedly greater week to week changes in T5KO mice (both non-colitic and colitic) relative to WT mice (Figure 3D). The greater variability correlated with greater community dissimilarity which increased over time in colitic T5KO as reflected by the greater slope in Figure 3E.
Figure 3. Increased volatility in microbiota of T5KO mice.
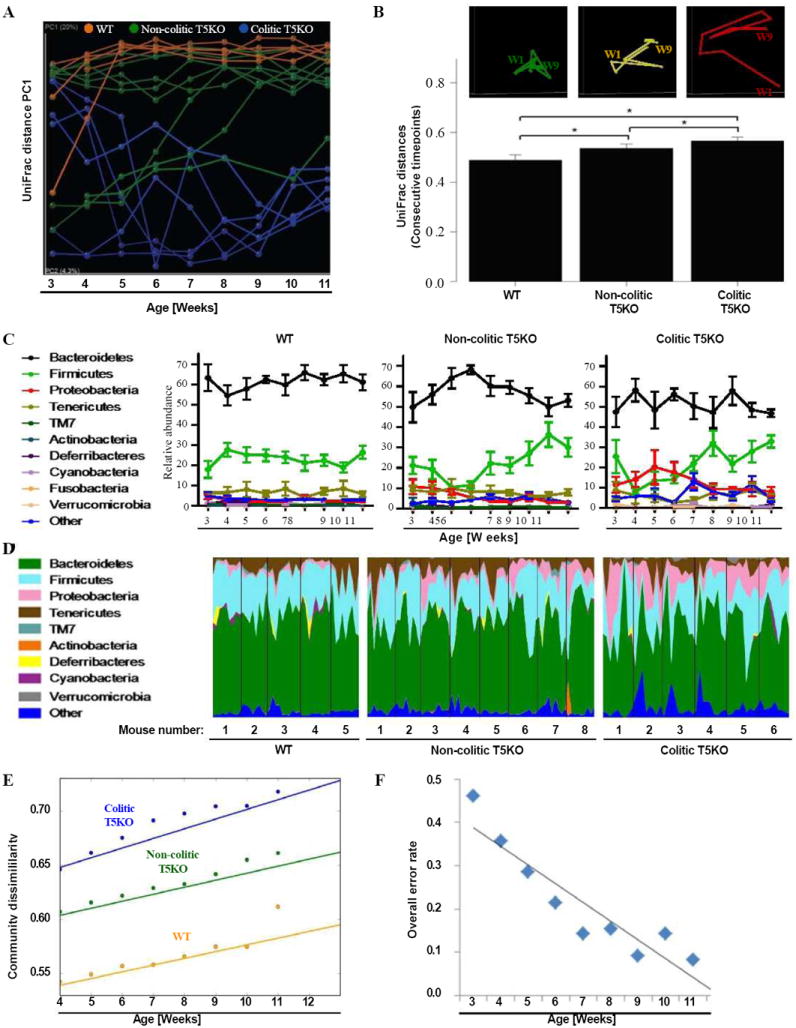
Stool from wild-type, non-colitic and colitic T5KO mice (n=5-8 mice per group) were collected weekly for 9 weeks after weaning (from 3-week to 11-week old). Stool microbiota composition was analyzed via 16S rRNA analysis. (A) Mouse cecal bacterial communities were clustered using principal coordinates analysis (PCoA) of the UniFrac unweighted distance matrix. PC1 is plotted for each time point (from 3-week to 11-week old). The time is expressed on the x-axis and the percentage of the variation explained by the plotted principal coordinates is indicated in the y-axis labels. Results are from an analysis of 5-8 mice per group (samples generating less than 1000 sequences were removed from analysis). (B) After clustering of mouse cecal bacterial communities using principal coordinates analysis (PCoA) of the UniFrac unweighted distance matrix, a representative mouse has been used to illustrate the time point evolution of the microbiota (top panel). The average of the UniFrac unweighted distance for each category (WT, non-colitic and colitic T5KO) between consecutive time points has been calculated (bottom panel). (C) Relative abundance of phyla in stool bacteria from WT (left panel), non-colitic T5KO (middle panel) and colitic T5KO (right panel) mouse group. (D) Relative abundance of phyla over time (from 3-week to 11-week old) in stool bacteria from individual WT (left panel), non-colitic T5KO (middle panel) and colitic T5KO (right panel) mice. (E) Semivariogram plot of community dissimilarity (UniFrac, y axis) vs. days dissimilarity (Euclidean, x axis). (F) Overall misclassification error rates using 9 families at each week. Analysis was done by ANOVA and statistical significance (P<0.01) is denoted by asterisk (*). Related materials can be found in supplementary figure 1.
When analyzed at the phylum level, much of the increased microbial volatility appeared to be driven by differences in Proteobacteria, which further analysis showed was mostly due to Enterobacteria. Yet, removal of all of the Enterobacteria operational taxonomic units (OTUs) from the analysis did not significantly impact the UniFrac-based volatility analysis (Supplemental figure 1A) arguing that Enterobacteria are but one family whose abundance changes in T5KO mice. In accordance with this notion, use of the “nearest shrunken centroid” method (PAM package for R) to define patterns that could identify colitic mice found high predictability based on relative abundance of 7 bacterial families (Supplemental figure 1B). Use of these criteria to identify colitic mice based on fecal microbiota composition had little predictive power upon weaning (error rate close to 50%), but greatly increased in the following weeks (Figure 3F). Using this method to predict based on class or order showed only modest predictive power (Supplemental figures 1C-D). These results suggest that broad changes across multiple microbial families are associated with, and could perhaps be predictive of, development of colitis.
TLR5-deficient mice exhibit increased mucosal-associated Enterobacteria
In light of our above results, and reports of others, that elevations in Proteobacteria species correlate with colitis (Lupp et al., 2007; Nagalingam et al., 2011), we further mined our sequencing data to examine subgroups of this family. Most notably, we observed markedly higher levels of Enterobacteria throughout the interval studied but particularly prominent in the first few weeks post weaning (Figure 4A). The dominant OTU of this group, comprising up to 6% of fecal bacteria, was a 16S rRNA sequence consistent with E. coli (99% match). Consistent with the fact that the colitic and non-colitic T5KO mice were littermates and close relatives of the WT mice, this OTU was detectable in most non-colitic mice studied but was present in much lower abundance. The presence/localization of Enterobacteria was examined by FISH using a specific probe and performed on intestinal samples fixed in a manner that preserves the mucus layer. Such analysis confirmed the large increase in abundance of Enterobacteria in colitic mice and, moreover, indicated that some bacteria had penetrated the mucus layer and were very close to, or in direct contact with, the gut epithelium (Figure 4B). Thus overt colitis in colitic T5KO correlated with the presence of Enterobacteria, whose abundance was greatest in the early post-weaning period.
Figure 4. Colitic T5KO mice harbored abnormal amount of Enterobacteria.
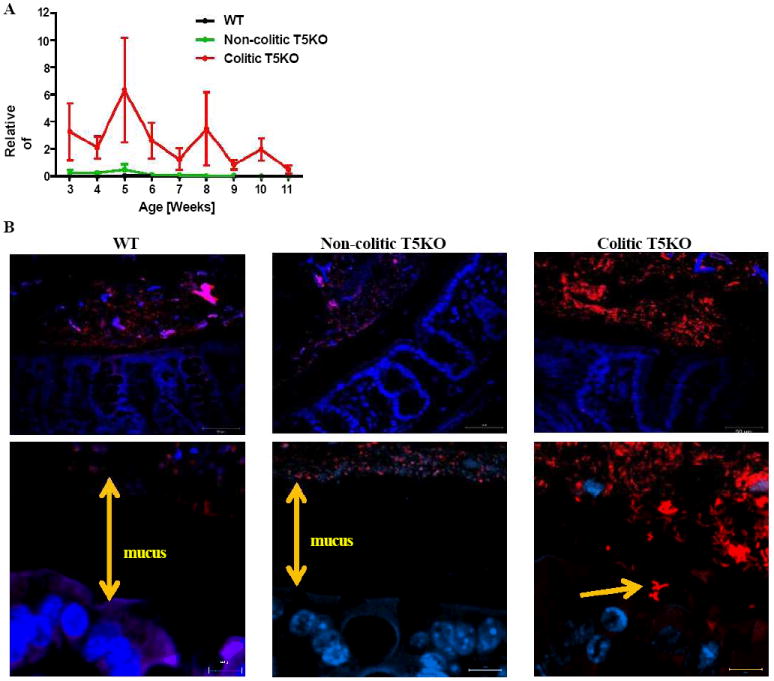
(A) Stool from wild-type, non-colitic and colitic T5KO mice (n=6-8 mice per group) were collected weekly for 9 weeks after weaning (from 3-week to 11-week old). Stool microbiota composition was analyzed via 16S rRNA analysis to determine the relative abundance of Enterobacteria in stool from WT, non-colitic T5KO and colitic T5KO mouse group (B) Microscopic pictures of colon after FISH using an Alexa 555 conjugated Enterobacteria specific probe (red) and nuclei in blue after DAPI staining (magnification: top-40X, bottom-100X). Related materials can be found in supplementary figure 2.
Increased susceptibility of TLR5-deficient mice to E. coli (AIEC) LF82 infection
The association of Enterobacteria with colitis could be a consequence of colitis and/or reflect a role for such bacteria in driving disease. While it might seem reasonable to investigate the latter possibility by simply exposing WT and non-colitic T5KO mice to select Enterobacteria species, interpreting such experiments is often not straightforward in that basal differences in gut microbiota, and consequently, basal state of immune activation, can have marked effects on the ability of exogenously-administered bacteria to colonize the gut. As an initial attempt to obviate this concern, we treated WT and non-colitic T5KO mice with streptomycin (to “level the field”) and then challenged them with an Enterobacteria species associated with Crohn’s disease in humans, namely Adherent-Invasive E. coli (AIEC) reference strain LF82. Levels of AIEC LF82 in feces, known to reflect colonization levels, were about 10-fold greater in T5KO mice from days 3-6 suggesting loss of TLR5 impaired the host’s ability to manage these flagellate bacteria (Supplemental figure 2A). Such delayed bacterial clearance by T5KO, relative to WT, mice was not seen upon exposure to a flagellin-deficient AIEC LF82 mutant indicating that the increased susceptibility of T5KO to AIEC LF82 colonization reflects a direct role for TLR5 in recognition of AIEC LF82 flagella. In fact, we observed that T5KO mice cleared aflagellate AIEC LF82 more efficiently than WT mice (Supplemental figure 2B) perhaps reflecting that the previously-defined T5KO basal phenotype of non-specific resistance to some bacteria (Vijay-Kumar et al., 2008) was not fully corrected by reducing total bacterial loads with streptomycin treatment. In any event, the importance of the TLR5-flagellin interaction in host defense against AIEC LF82 is highlighted by the observation that loss of flagellin markedly enhances colony forming unit (CFU) levels in WT mice but has no effect in T5KO mice. To investigate the functional consequences of the inability to manage AIEC LF82, we examined extent to which these bacteria could induce gut inflammation in these mice, focusing on ceca where colonization of AIEC LF82 is highest in mice. Cecal inflammation is known to correlate with changes in gross morphology, particularly contraction or shrinkage, of this organ. This phenotype was readily apparent in T5KO mice colonized with flagellate, but not in T5KO mice infected with aflagellate AIEC LF82 nor WT mice colonized with either bacterial strain (Supplemental figure 2C). In accordance with this observation, use of myeloperoxidase activity as a marker of inflammation and histopathologic examination of the ceca confirmed that AIEC LF82, particularly when flagellate, was a significant trigger of gut inflammation in T5KO, but not WT mice (Supplemental figure 2D-F).
AIEC LF82 infection induces early gut inflammation in TLR5-deficient mice
A major limitation to the use of antibiotics to study role of the gut microbiota is that antibiotics alter but do not eliminate this complex microbial community (Antunes and Finlay, 2011). Thus, to better investigate the role of TLR5 upon exposure to AIEC strain LF82, we next utilized the approach of using germ-free (GF) T5KO and WT mice. In contrast to our colony of non-colitic T5KO mice, which have elevated pro-inflammatory/anti-microbial gene expression and metabolic syndrome, germ-free T5KO mice were metabolically indistinguishable from WT GF mice and lacked elevation in the pro-inflammatory marker Lcn-2 that sensitively reflects gut inflammation in T5KO mice (Supplemental figure 3A-F). The germ-free status of T5KO and WT mice was confirmed by verifying a greater than 6 log reduction in 16S fecal DNA and a reduction by 10-fold of fecal Lcn-2 expression (Supplemental figure 3 F-G). Subsequently, GF WT and T5KO mice were colonized (mono-associated) by oral gavage with 107 AIEC LF82 bacteria and assayed for development of colitis 2 weeks later. Mono-association with AIEC LF82 did not induce inflammation in WT mice but caused moderate colitis in T5KO mice (as assessed by gross, biochemical, histopathological, and the inflammatory marker Lcn-2 measurements (Figure 5 A-G)). Microscopic examination of localization of E. coli in the mono-associated mice failed to detect closely adherent bacteria at the 2-week time point suggesting the bacteria may only colonize rare breeches in the mucosa in accordance with earlier studies on the interaction of this microbes with the murine intestine (Carvalho et al., 2009). That AIEC LF82 failed to induce colitis in WT mice and induced only moderate colitis in T5KO mice is in accord with a recent report that microbial virulence factor expression requires commensal microbiota (Kamada et al., 2012) and suggests the importance of examining how mono-associated mice might fare when not maintained in isolators. Thus, we next administered AIEC strain LF82 to GF mice that were subsequently removed from GF isolators and placed in specific pathogen free housing. Under these conditions, AIEC LF82 persistence would require outcompeting other microbes that quickly colonize GF mice placed in SPF housing regardless of whether they are, or are not, deliberately “conventionalized” (Supplemental figure 4A). WT GF mice administered AIEC LF82 did not exhibit any indication of illness whereas T5KO mice exhibited mild weight loss and diarrhea from day 3 to 7 post-infection (Figure 6A). Such clinical-type symptoms paralleled reduced clearance of AIEC LF82, which became apparent 5d post-infection (Figure 6B). Mice were euthanized 7d post-infection and subsequently examined for various inflammatory indices and immunological parameters previously used to characterize spontaneous T5KO colitis. T5KO mice exhibited all examined typical features of spontaneous T5KO colitis including splenomegaly, shortened but thickened colon (i.e. colomegaly), elevated levels of colonic MPO, histopathological scoring, bacterial translocation, and colon Lcn-2 expression (Figure 6C-K). Such gut inflammation was not readily apparent in GF T5KO mice colonized with the commensal flagellate E. coli strain F-18, which is commonly used as a commensal E. coli isolate (Supplemental figure 4B-J). Colon cytokine levels of AIEC LF82 colonized mice also reflected what we previously observed in T5KO mice with low-grade inflammation (Vijay-Kumar et al., 2007), namely reduced levels of the cytokines CXCL1 and IL-6, which are secreted by epithelial cells in response to flagellin and elevated levels of “master pro-inflammatory cytokines” IL-1β and TNF-α, which are predominantly secreted by immune cells (Figure 6L-O). A similar pattern of cytokine levels was seen in sera (Supplemental figure 5). Together, these results suggest that colitis in T5KO mice can be triggered by selected E. coli pathobionts such as AIEC that might also be present, but not promote colitis, in WT mice.
Figure 5. Mono-association of Crohn’s disease associated Adherent-Invasive E. coli (AIEC) strain LF82 infection increased intestinal inflammation in T5KO mice.
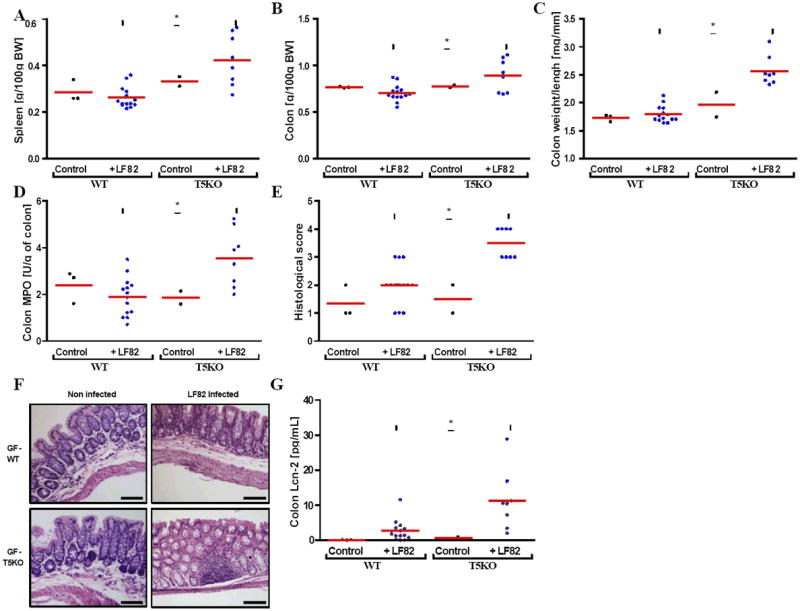
(A) Following euthanasia, spleen was isolated and mass measured. (B) Colon mass. (C) Ratio between colon weight and colon length. (D) Colon MPO activity. (E-F) Histological score and representative H&E stained colon (magnification, 100×). (G) Colon was cultured for 24h, at which time supernatant was assayed for Lcn-2 by ELISA. * p<0.05. Related materials can be found in supplementary figure 3.
Figure 6. Germ-free T5KO mice are highly susceptibility to early AIEC LF82 infection.
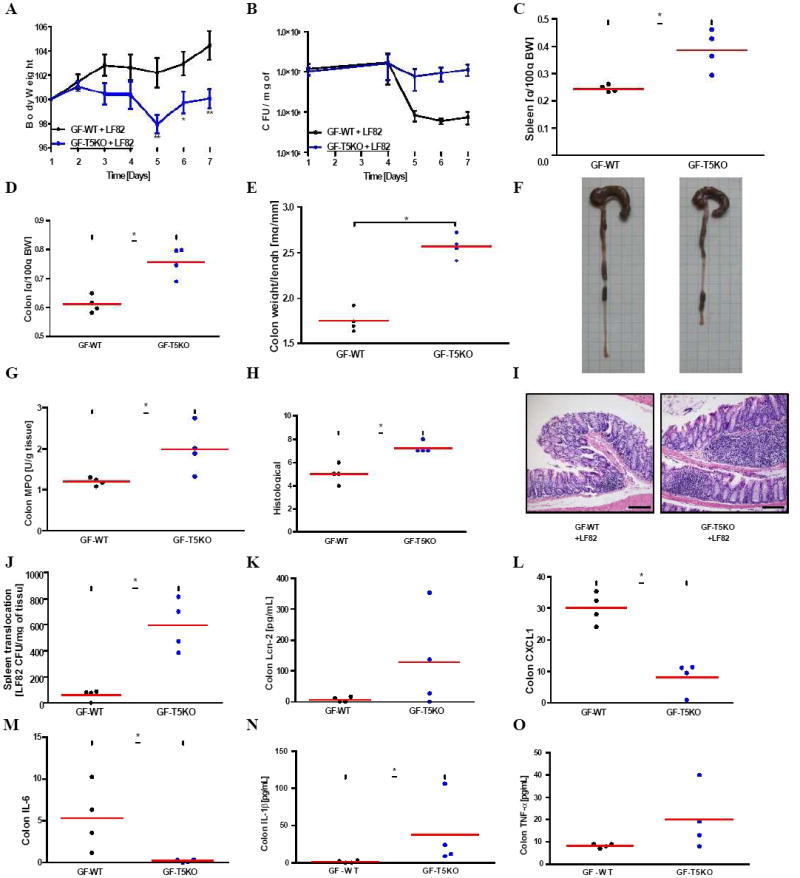
Germ-free wild-type and T5KO mice (n=4 mice per group) were orally infected with 107 flagellate AIEC LF82 bacteria. (A) Body mass was monitored daily during the treatment. (B) Numeration of AIEC LF82 present in the WT or T5KO mouse stool from day 1 to day 7 post infection. (C) Following euthanasia, spleen was isolated and mass measured. (D) Colon mass. (E) Ratio between colon weight and colon length. (F) Gross picture of colon. (G) Colon MPO activity. (H-I) Histological score and representative H&E stained colon (magnification, 100×). (J) Bacterial translocation by numbering AIEC LF82 CFU present in the spleen at day 7 post infection. (K-O) Colon was cultured for 24h, at which time supernatant was assayed for Lcn-2 (K) and several pro-inflammatory cytokines, namely CXCL1 (L), IL-6 (M), IL-1β (N) and TNF-α (O), by ELISA. * p<0.05. Related materials can be found in supplementary figure 4.
Delayed clearance of AIEC LF82 promotes chronic intestinal inflammation
While use of antibiotics or germ-free mice can overcome the inability of some bacteria to colonize the mouse intestine, stopping use of the antibiotics or permitting acquisition of a microbiota by removal from GF housing, often results in quick depletion of such administered bacteria. Thus, any bacterium able to play a significant role in the spontaneous chronic colitis exhibited by some T5KO mice would either require an ability to persist beyond acquisition of a microbiota and/or trigger events that would result in chronic inflammation after the bacteria had been cleared from the GI tract. To investigate the extent to which AIEC might display such characteristics in T5KO mice, we colonized GF WT and T5KO mice with AIEC, or as controls treated with sterile PBS, placed in SPF housing, and monitored for 119 days. Analogous to our shorter term experiment, T5KO mice lacked the rapid reduction in AIEC LF82 levels exhibited by WT mice although, like WT mice, AIEC levels were very low by 5 weeks post-inoculation and did not differ significantly from WT mice from that time through the end of the experiment (Figure 7A). Such delayed clearance correlated with transient diarrhea 5-15 days post-inoculation that was accompanied with transient weight loss during this time. Despite the lack of difference in AIEC LF82 levels beyond 42d post-inoculation, the reduced weight gain of T5KO mice relative to WT persisted over the following 90d (Figure 7B). Analogously, higher levels of the fecal inflammatory marker Lcn-2 persisted throughout the 119 day experiment despite the levels of AIEC LF82 being similar in WT and T5KO mice by 42 days (Figure 7C). Examination of the murine intestine at the 120 day time point by the aforementioned parameters revealed that T5KO mice that were exposed to AIEC LF82 exhibited typical features of chronic colitis including splenomegaly, colomegaly, histopathological scoring, and elevated inflammatory markers such as MPO and Lcn-2 (Figure 7D-N). In accordance with our previous work, chronic colitis in T5KO mice associated with elevated levels of CXCL1 and IL-6 (Vijay-Kumar et al., 2007a). Importantly, neither relatively delayed bacterial clearance nor chronic intestinal inflammation was observed in GF T5KO mice colonized with aflagellate AIEC LF82 indicating a direct role for TLR5 in recognizing AIEC flagellin to protect against this pathobiont. Together, these results indicate that loss of TLR5 renders mice susceptible to colonization by select flagellate bacteria that can drive gut inflammation capable of persisting even when the bacteria is present at only very low levels and support the general notion that chronic intestinal inflammation can result from an innate immune deficiency and be triggered by transient colonization by selected bacteria.
Figure 7. Transient AIEC LF82 infection resulted in a chronic basal inflammation in germ-free T5KO mice.
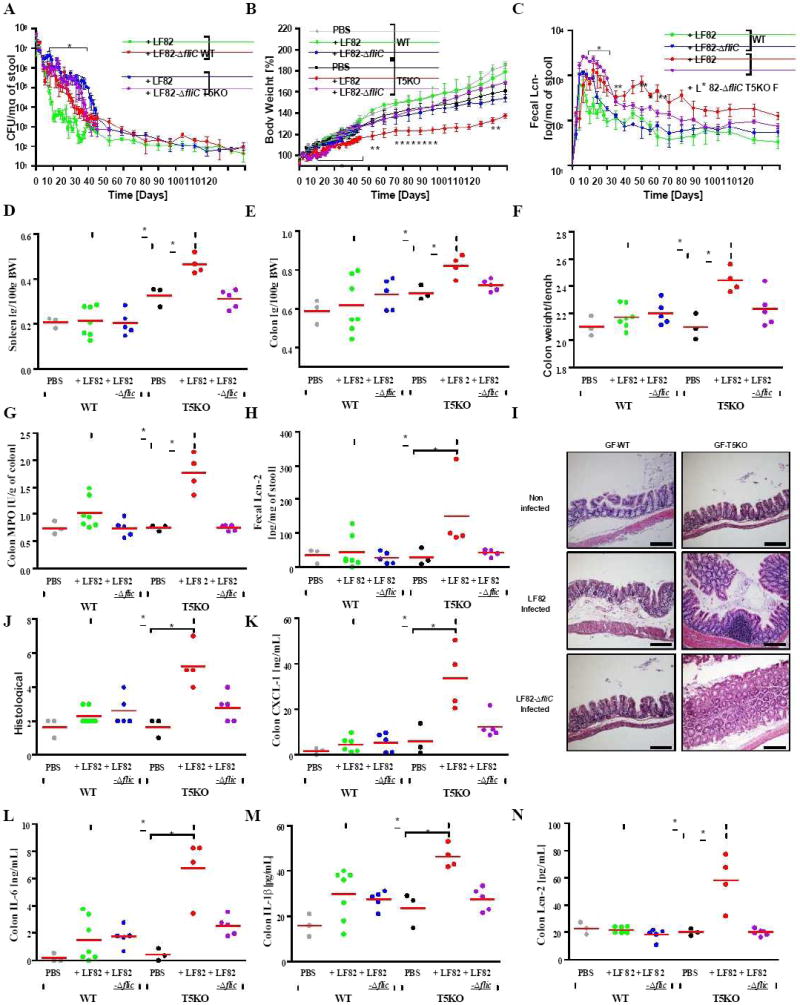
Germ-free wild-type and T5KO mice (n=3-7 mice per group) were orally infected with 107 flagellate AIEC LF82 bacteria. As control, uninfected germ-free WT or T5KO mice were transferred to a sterile cage without treatment. (A) Numeration of AIEC LF82 present in the WT or T5KO mouse stool. (B) Body mass was monitored daily from day 0 to day 35 post infection and then weekly from day 42 to day 119 post infection. (C) Stool was collected daily (D0 to D35) or weekly (D42 to D119) after AIEC LF82 infection and diluted in 500 μL of PBS. Then, supernatant was assayed for Lcn-2 expression by ELISA. (D) Following euthanasia, spleen was isolated and mass measured. (E) Colon mass. (F) Ratio between colon weight and colon length. (G) Colon MPO activity. (H) Fecal Lcn-2 was assayed by ELISA at the end of the experiment. (I-J) Histological score and representative H&E stained colon (magnification, 100×). (K-N) Colon was cultured for 24h, at which time supernatant was assayed for several pro-inflammatory cytokines, namely CXCL1 (K), IL-6 (L), IL-1β (M), and Lcn-2 (N) by ELISA. * p<0.05. Related materials can be found in supplementary figure 5.
DISCUSSION
Alterations in the gut microbiota have long been suspected to play an important role in the pathogenesis of IBD. Consequently, there has been considerable interest in applying recent advances in DNA sequencing technology to interrogate the gut microbiota in this disorder (Walker et al., 2011a; Walker et al., 2011b; Willing et al., 2010). While inter-person variance, combined with the rapid and dramatic effects of diet, pose a considerable obstacle to this endeavor, perhaps the greatest obscurant to understanding the role of the microbiota in IBD, and other diseases, is that it has generally only been possible to examine the microbiota in patients after they have been diagnosed, which typically takes place well after initial symptoms occurred. Thus, while a number of studies have reported alterations in the microbiota in IBD, the extent to which these occur late or early in the disease process, and whether they are a cause or consequence of disease, remain undefined. Herein, to address these challenges and better understand the role of the microbiota in colitis, we temporally examined, from weaning to colitis establishment, the microbiota in T5KO mice, which are prone to developing intestinal inflammation.
Consistent with the notion that gut microbiota is acquired from one’s early environment, overall microbiota composition was similar shortly-post-weaning in T5KO mice that developed colitis and their littermates that did not. Nor was there a stark difference between microbiota of T5KO and closely related WT mice upon this initial sampling. Rather, the most striking overall difference that we observed was the greater volatility (week to changes) that occurred in T5KO mice that did, or did not, develop colitis. We hypothesize that such volatility reflects that, consistent with its preferential expression on mucosal tissues, TLR5 is a “first responder” to occasional breaches that sporadically occur throughout the mucosa – a vision promoted by the notion of 100 trillion bacteria populating a surface the size of a tennis court. Accordingly, we interpret that such volatility is accompanied by elevated expression of Lcn-2, and likely numerous elevations in anti-bacterial/pro-inflammatory gene expression (Carvalho et al., 2012; Vijay-Kumar et al., 2007), reflects the complex redundant ability of the immune system to secondarily manage such disturbances. From an evolutionary perspective, we propose such machinations of the TLR5-deficient immune system are highly effective in that most mice did not develop overt colitis but rather have increases in weight/adipose tissue that often correlate with increased breeding in mice (Vijay-Kumar et al., 2010). Accordingly, non-colitic T5KO mice breed at least as well as WT mice whereas, like other colitic mice, colitic T5KO produce very few offspring.
The other major pattern evident in these results was the increase in Proteobacteria, specifically Enterobacteria in colitic T5KO. While it is known that increases in Proteobacteria can be a consequence of inflammation (Lupp et al., 2007; Nagalingam et al., 2011), the levels of Proteobacteria/Enterobacteria were greatest 3-weeks post weaning and were seemingly controlled well in advance of when the mice were euthanized and colitis realized, which suggests a potentially causative role. To investigate this notion, we colonized WT and T5KO with an Enterobacteria species known to associate with Crohn’s disease, namely AIEC reference strain LF82 (Darfeuille-Michaud et al., 1998). While neither WT nor T5KO mice are normally colonizable by this bacterium (Carvalho et al., 2009), the restriction could be overcome by use of antibiotics and/or use of germ-free mice. While both approaches afforded detectable colonization, the latter approach resulted in a more lasting, albeit still transient colonization by this bacterium in accord with our result that the bacterium does not tightly adhere to the mucosa. In either case, clearance of flagellate AIEC strain LF82, but not an aflagellate isogenic mutant strain was delayed in T5KO mice indicating a role for TLR5 in protecting against this, and perhaps other motile, bacteria. Failure to efficiently clear AIEC strain LF82 correlated with development of colitis. Such colitis was evident within a few days of colonization and persisted for at least 119 days even though levels of the bacteria were similar between WT and T5KO mice by 42 days. We envisage that such persistent inflammation could reflect a need for chronic elevation of the pathways that contain the bacteria in the absence of TLR5 or that priming of the adaptive immune system has taken place during the period in which the mucosal immune system-microbiota relationship was stabilizing that resulted in a chronic inflammatory state. Together, these results suggests that the elevated Enterobacteria observed in colitic T5KO mice are likely both a cause and consequence of colitis and, moreover, that classic concepts of microbial causation of disease such as Koch’s postulates may not be sufficient to describe the role of the microbiota in disease.
The observation that several polymorphisms in genes with innate immune function confer risk for IBD makes it tempting to speculate that the mechanistic paradigms underlying T5KO colitis may be similar to those underlying IBD. Accordingly, our results suggest that IBD might clinically manifest following a prolonged, ultimately unsuccessful, attempt by the mucosal immune system to maintain/contain a stable microbiota while avoiding the excessive immune responses that can be detrimental to the host. In this scenario, a more successful, albeit still sub-optimal, consequence of altered intestinal/microbiota relationship would be the maintenance of “low-grade” inflammation that correlates with diseases such as obesity, type 2 diabetes, and cancer whose effects generally do not manifest during one’s prime reproductive years.
Another possibility suggested by our results is that some IBD-associated bacteria such as AIEC strain LF82 may not merely colonize the gut as a consequence of disease and/or be an aggravating factor of established disease but rather might play an early role in disease pathogenesis particularly as instigators of inflammation. We anticipate that ongoing development of fecal sample banks that catalog specimens throughout life on persons who develop IBD and control subjects will make it possible to better address if such instigator bacteria exist in humans who eventually develop chronic gut inflammation. We would predict that elevated levels of select bacteria might be predictors, and causative factors of IBD and perhaps other chronic inflammatory disorders. Should either general developmental microbiota volatility or select bacterial taxa correlate with future disease development, it would suggest the possibility of therapeutic manipulations of the microbiota, particularly early in life might be a reasonable means of preventing diseases associated with active and low-grade intestinal inflammation.
EXPERIMENTAL PROCEDURES
Mice maintenance
WT and T5KO mice (back crossed to C57BL/6 mice for 10 generations) were bred and maintained as previously described. All experiments were approved by appropriate Institutional animal use committees. Experimental mice were weaned at 21d and maintained for 10 weeks thereafter. Body mass was measured and stools collected weekly. Mice were stratified as colitic exhibiting splenomegaly (>0.5g/100g of body weight) and colomegaly (>1.5g/100g of body weight).
Mucosal Analysis
Electron microscopy (Gilmor et al., 1996), mucous staining (Johansson et al., 2008), and localization of bacteria therein by fluorescent in situ hybridization was performed as previously described using probes described in Supplementary info. Myeloperoxidase activity (Castaneda et al., 2005), histologic scoring of inflammation (Onyeagocha et al., 2009), and assay of intestinal cytokines (Vijay-Kumar et al., 2007) was measured as previously described.
Microbiota Analysis
Bacterial 16s DNA on fecal and cecal samples was performed using the Quantitative Insights Into Microbial Ecology pipeline as detailed in Supplementary Information.
Bacterial infections
CD-associated AIEC strain LF82 (Darfeuille-Michaud, 2002), isogenic non flagellate mutant LF82-ΔfliC (Barnich et al., 2003), S. typhimurium SL3201, and E. coli F18 were grown over night at 37°C without agitation in Luria-Bertani (LB) medium. After centrifugation, bacteria were resuspended in PBS and used to inoculate mice. Acute (Barthel et al., 2003) or chronic colitis was assessed as described in Results/figure legends.
Statistical analysis
Significance was determined using the Mann Whitney t test or one-way ANOVA (GraphPad Prism software). Differences were noted as significant * p<0.05.
Supplementary Material
Highlights.
TLR5-deficient mice (T5KO) are prone to develop post-weaning gut inflammation
Colitic T5KO are associated with increased microbial volatility and Proteobacteria
T5KO show increased susceptibility to Crohn’s disease-associated E. coli (AIEC)
AIEC persists longer in T5KO and induces chronic intestinal inflammation
Acknowledgments
We thank Catherine Paul and Yueju Su for technical support and Gunnar Hannson for helpful discussions. We also gratefully acknowledge Maureen Bower, Dr. Balfour Sartor, and the National Gnotobiotic Rodent Resource Center (NGRRC) at UNC. This work was supported by NIH grants DK061417 and DK083890 to ATG. FAC is a recipient Research Fellowship award from the Crohn’s and Colitis Foundation of America (CCFA). GRRC is supported by grants from NIH (P40RR018603 and P30 DK 34987) and CCFA. RK is supported in part by the NIH, CCFA and Howard Hughes Medical Institute.
References
- Antunes LC, Finlay BB. A comparative analysis of the effect of antibiotic treatment and enteric infection on intestinal homeostasis. Gut Microbes. 2011;2:105–108. doi: 10.4161/gmic.2.2.15610. [DOI] [PubMed] [Google Scholar]
- Barnich N, Boudeau J, Claret L, Darfeuille-Michaud A. Regulatory and functional co-operation of flagella and type 1 pili in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn’s disease. Mol Microbiol. 2003;48:781–794. doi: 10.1046/j.1365-2958.2003.03468.x. [DOI] [PubMed] [Google Scholar]
- Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Russmann H, Hardt WD. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun. 2003;71:2839–2858. doi: 10.1128/IAI.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Aitken JD, Gewirtz AT, Vijay-Kumar M. TLR5 activation induces secretory interleukin-1 receptor antagonist (sIL-1Ra) and reduces inflammasome-associated tissue damage. Mucosal Immunol. 2011;4:102–111. doi: 10.1038/mi.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Barnich N, Sauvanet P, Darcha C, Gelot A, Darfeuille-Michaud A. Crohn’s disease-associated Escherichia coli LF82 aggravates colitis in injured mouse colon via signaling by flagellin. Inflamm Bowel Dis. 2008;14:1051–1060. doi: 10.1002/ibd.20423. [DOI] [PubMed] [Google Scholar]
- Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CH, Stanners CP, Darfeuille-Michaud A. Crohn’s disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med. 2009;206:2179–2189. doi: 10.1084/jem.20090741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Nalbantoglu I, Ortega-Fernandez S, Aitken JD, Su Y, Koren O, Walters WA, Knight R, Ley RE, Vijay-Kumar M, et al. Interleukin-1beta (IL-1beta) promotes susceptibility of Toll-like receptor 5 (TLR5) deficient mice to colitis. Gut. 2012;61:373–384. doi: 10.1136/gut.2011.240556. [DOI] [PubMed] [Google Scholar]
- Castaneda FE, Walia B, Vijay-Kumar M, Patel NR, Roser S, Kolachala VL, Rojas M, Wang L, Oprea G, Garg P, et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology. 2005;129:1991–2008. doi: 10.1053/j.gastro.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology. 2011;140:1704–1712. doi: 10.1053/j.gastro.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darfeuille-Michaud A. Adherent-invasive Escherichia coli: a putative new E. coli pathotype associated with Crohn’s disease. Int J Med Microbiol. 2002;292:185–193. doi: 10.1078/1438-4221-00201. [DOI] [PubMed] [Google Scholar]
- Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115:1405–1413. doi: 10.1016/s0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- Derrien M, van Passel MW, van de Bovenkamp JH, Schipper RG, de Vos WM, Dekker J. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes. 2010;1:254–268. doi: 10.4161/gmic.1.4.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont AW, Dupont HL. The intestinal microbiota and chronic disorders of the gut. Nat Rev Gastroenterol Hepatol. 2011;8:523–531. doi: 10.1038/nrgastro.2011.133. [DOI] [PubMed] [Google Scholar]
- Gilmor ML, Nash NR, Roghani A, Edwards RH, Yi H, Hersch SM, Levey AI. Expression of the putative vesicular acetylcholine transporter in rat brain and localization in cholinergic synaptic vesicles. J Neurosci. 1996;16:2179–2190. doi: 10.1523/JNEUROSCI.16-07-02179.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105:15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, Nunez G. Regulated Virulence Controls the Ability of a Pathogen to Compete with the Gut Microbiota. Science. 2012 doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:204. doi: 10.1016/j.chom.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Matharu KS, Mizoguchi E, Cotoner CA, Nguyen DD, Mingle B, Iweala OI, McBee ME, Stefka AT, Prioult G, Haigis KM, et al. Toll-like receptor 4-mediated regulation of spontaneous Helicobacter-dependent colitis in IL-10-deficient mice. Gastroenterology. 2009;137:1380–1390. e1381–1383. doi: 10.1053/j.gastro.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondot S, Barreau F, Al Nabhani Z, Dussaillant M, Le Roux K, Dore J, Leclerc M, Hugot JP, Lepage P. Altered gut microbiota composition in immune-impaired Nod2-/- mice. Gut. 2011 doi: 10.1136/gutjnl-2011-300478. [DOI] [PubMed] [Google Scholar]
- Nagalingam NA, Kao JY, Young VB. Microbial ecology of the murine gut associated with the development of dextran sodium sulfate-induced colitis. Inflamm Bowel Dis. 2011;17:917–926. doi: 10.1002/ibd.21462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, Huth M, Nikolaev A, Neufert C, Madison B, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- Onyeagocha C, Hossain MS, Kumar A, Jones RM, Roback J, Gewirtz AT. Latent cytomegalovirus infection exacerbates experimental colitis. Am J Pathol. 2009;175:2034–2042. doi: 10.2353/ajpath.2009.090471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman A, Sina C, Gavrilova O, Hasler R, Ott S, Baines JF, Schreiber S, Rosenstiel P. Nod2 is essential for temporal development of intestinal microbial communities. Gut. 2011;60:1354–1362. doi: 10.1136/gut.2010.216259. [DOI] [PubMed] [Google Scholar]
- Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolhion N, Carvalho FA, Darfeuille-Michaud A. OmpC and the sigma(E) regulatory pathway are involved in adhesion and invasion of the Crohn’s disease-associated Escherichia coli strain LF82. Mol Microbiol. 2007;63:1684–1700. doi: 10.1111/j.1365-2958.2007.05638.x. [DOI] [PubMed] [Google Scholar]
- Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Kumar A, Neish AS, Uematsu S, Akira S, Gewirtz AT. Toll-like receptor 5-deficient mice have dysregulated intestinal gene expression and nonspecific resistance to Salmonella-induced typhoid-like disease. Infect Immun. 2008;76:1276–1281. doi: 10.1128/IAI.01491-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, Neish AS, Uematsu S, Akira S, Williams IR, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117:3909–3921. doi: 10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011a;5:220–230. doi: 10.1038/ismej.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, Brostoff J, Parkhill J, Dougan G, Petrovska L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011b;11:7. doi: 10.1186/1471-2180-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Jarnerot G, Tysk C, Jansson JK, Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854. e1841. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.