

INTRODUCTION
PED, known also as PED/PEA-15, is a Death Effector Domain (DED)-family member of 15 kDa having a variety of effects on cell growth and metabolism [1,2]. PED has a broad anti-apoptotic action, being able to inhibit both the intrinsic and the extrinsic apoptotic pathways [3,4,5,6]. Inhibition of the extrinsic pathway is accomplished through its DED, which likely acts as a competitive inhibitor for pro-apoptotic molecules during the assembly of the death-inducing signaling complex (DISC) [1,7]. To data, although PED has been reported to be over-expressed in a number of different cancer types, such as gliomas, squamous carcinoma, breast, lung cancer, and B cells chronic lymphocytic leukemia, the mechanisms that regulate its expression have not been fully addressed [3,5,8,9]. PED interacts with different molecules, among these ERK 1/2, altering their nuclear localization by sequestering them into the cytosol [10] .
PED is present in the cells in either an unphosphorylated or a phosphorylated form. Phosphorylation occurs at Ser104 by protein kinase C (PKC), and on Ser116 by AKT or calcium calmodulin kinase II (CamK II) [11,12]. Interestingly, PED may interact with ERK1/2 only in its unphosphorylated form and it is associated to an increased ERK 1/2 phosphorylation [12]. Based on its phosphorylation status, PED might play a role as either a tumor-promoter factor (in the phosphorylated form) or tumor-suppressor (in the unphosphorylated form) factor [12].
To get further insights on the role of PED in cancer, we aimed to find new PED interactors. Using Tandem Affinity Purification (TAP), we previously identified and characterized, among others, Rac1, a member of mammalian Rho GTPase protein family, as a PED-interacting protein. PED-Rac1 interaction resulted in the modulation of cell migration/invasion processes in lung cancer cells through ERK1/2 pathway [13]. Another attractive PED-interacting protein was Hsc70 (Heat Shock cognate protein of 70 KDa), a chaperone protein, reported to be involved in a multitude of housekeeping chaperoning functions including folding of nascent polypeptides, protein translocation across membranes, prevention of protein aggregation under stress conditions, disassembly of clathrin coated vesicles and also in chaperone-mediated autophagy (CMA) [14,18]. CMA is a uniquely selective form of autophagy by which specific cytosolic proteins are transported one-by-one across the lysosomal membrane for degradation [14,15]-]. CMA is constitutively active in many cell types, but it is maximally activated under stress conditions (inducible CMA) such as nutritional stress, starvation or cellular stresses leading to protein damage [14]. CMA is selective for a subset of cytosolic soluble proteins. The selectivity is determined by the presence of a recognition-targeting motif in the amino acid sequence of the substrate proteins. All CMA substrates contain in their amino acid sequence a pentapeptide motif biochemically related to KFERQ, known as the CMA targeting motif [17]. The KFERQ sequence is recognized in the cytosol by Hsc70. Hsc70 not only targets the CMA substrate to the lysosomal membrane, where it can interact with the CMA receptor the lysosome-associated protein type 2A (LAMP-2A), but, along with its cochaperones it likely facilitates substrate unfolding, which is require for substrate translocation across the lysosomal membrane [14,18, 21].
In this manuscript we describe the interaction of PED with Hsc70 and demonstrate that Hsc70 targets the unphosphorylated PED for CMA.
RESULTS
PED interacts with Hsc70
Using Tandem Affinity Purification (TAP) (described in [13]) we identify Hsc70 and PED interaction. PED-Hsc70 interaction was confirmed by several approaches: co-immunoprecipitation (Figure 1A-B), GST pull-down (Figure 1C) and we also demonstrated their co-localization by confocal microscopy (Figure 1D). The immunoprecipitation experiment was performed in A549 cells transfected with Myc-PED cDNA, by incubating cell extracts with anti-Myc antibody. The interaction was revealed by western blot with Hsc70 antibody (Figure 1A). Alternatively, we immunoprecipitated with anti-Hsc70 antibody in the same cells and blotted for Myc (Figure 1B). As shown in Figure 1A-B, in both experiments, Hsc70 co-immunoprecipitates along with PED protein. In order to asses a direct binding within PED and Hsc70, A549 cell extracts were incubated with recombinant GST-PED fusion protein or, as a negative control, with Glutathione-Sepharose 4B beads that were not prebound to the recombinant protein. As shown in Figure 1C, Hsc70 was pulled down by GST-PED recombinant protein.
Figure 1. Validation of the interaction between PED and Hsc-70. In vitro immunoprecipitation.

HEK-293 were transfected with PED-Myc cDNA. After 48 hrs, 1 mg of protein extracts were immunoprecipitated with anti-PED (A) or anti-Hsc70 antibody (B) or with control IgG, as indicated. Samples were then resolved with SDS-PAGE and western blot with anti-Hsc70 or anti-Myc antibodies. 10 μg of total cellular extracts were loaded as control. (C)- GST pull down. A549 cellular extracts were incubated with Glutathione-Sepharose 4B beads pre-bound to GST-PED fusion protein. Negative control was obtained by incubating cellular extracts to Glutathione-Sepharose 4B beads not pre-bound to GST-PED. Positive control was obtained loading the pre-bound GST PED fusion beads (10 μg). 50 μg of total cellular extract was loaded. (D) Confocal experiments. PED and Hsc70 distribution in A549 cells was determined by immunofluorescence analysis using confocal microscopy. A549 cells were fixed with 4% PFA, permeabilized in blocking solution (0.1% Saponin, 10% FBS in PBS), incubated with PED or Hsc70 antibodies and then with secondary antibodies conjugated respectively with a green (anti-rabbit) or red (anti-mouse) fluorochrome. The intensity of the yellow signals represents the degree of co-localization of the two proteins.
Hsc70-PED interaction was further supported by the fact that both proteins co-localized in A549 cells when we used indirect immunofluorescence by confocal microscopy. As shown in Figure 1D, we observed a clear co-localization between PED and Hsc70 mostly in the cytosol but also in punctuate structures compatible with intracellular vesicles.
PED-Hsc70 interaction during CMA
We next asked if PED was associated to Hsc70 at least in part for its degradation by CMA. We evaluated by confocal microscopy the levels and intracellular distribution of PED and its association with Hsc70 and LAMP-2A-positive lysosomes under conditions of CMA up-regulation. For this purpose, A549 cells were grown in the presence of serum (low CMA activity) or after prolonged (24 h) serum removal (high CMA activity), and CMA activation was initially monitored by immunofluorescence for LAMP-2A and Hsc70 [19]. As shown in Supplementary Figure 1, under starving conditions, CMA is upregulated and the lysosomes involved in the CMA process (those enriched in LAMP-2A and Hsc-70), clearly relocate to the perinuclear region [26]. The increase in CMA activity was associated also with an increase in the amounts of CMA-active lysosomes, assessed as an increment of the co-localization of Hsc-70 and LAMP-2A in vesicular structures (Supplementary Figure 1). We then assessed the co-localization of PED with CMA-positive lysosomes, during CMA activation. To this end we performed two different indirect immunofluorescences, for PED and LAMP-2A and for PED and Hsc70 in conditions of low or high CMA activity. Interestingly, PED co-localized with both Hsc70 and LAMP-2A and this co-localization increased in high CMA activity conditions (Figure 2).
Figure 2. PED localizes to CMA-positive lysosomes.

(A) Cellular localization of PED and LAMP-2A protein in serum deprived A549 cells. Upon serum starvation, PED-LAMP-2A co-localization was evident (yellow signal) indicating the co-localization of PED with CMA-positive lysosomes. (B) Cellular localization of PED and Hsc70 protein in serum deprived A549 cells. After starvation, in A459 a yellow signalling was evident, indicating PED- Hsc70 co-localization.
Uptake of PED by isolated lysosomes
The most direct way to determine if a protein is a substrate for CMA, is to be able to reproduce its direct translocation into isolated lysosomes [26]. To that purpose, intact rat liver lysosomes were incubated with purified recombinant GST-PED protein and lysosomal uptake was determined by comparing the amount of PED associated to lysosomes previously treated or not with protease inhibitors. All the protein associated to non-treated lysosomes is located on the cytosolic side of the lysosomal membrane because the protein transported inside is rapidly degraded. However, the protein associated to lysosomes previously treated with a cocktail of protease inhibitors to block the three main groups of proteases present in lysosomes (serine, cystein and aspartic proteases), corresponds to the protein bound to the lysosomal membrane and the one translocated inside. Uptake can be calculated as the difference between the amount of protein detectable by immunoblot in both groups of lysosomes at the end of the incubation. We found that a fraction of PED binds to the lysosomal membrane but in addition, the increase in the amount of PED associated to lysosomes pre-treated with lysosomal inhibitors confirmed that PED can be transported inside the lysosomes (Figure 3A). Binding of PED to lysosomes was saturable, as it was its uptake into lysosomes (Figure 3B). This supports that PED uptake by lysosomes may take place by CMA, as this is known to be a unique characteristic of this pathway when compared to other types of autophagy [20]. Uptake is saturated at lower concentrations than biding probably because unfolding is not required for substrate binding to the lysosomal membrane but it is essential for its translocation into the lumen [21].
Figure 3. Lysosomal uptake of PED by chaperone-mediated autophagy.

(A) Intact rat liver lysosomes pretreated or not with protease inhibitors (PI) were incubated with purified PED (0.1mg) and at the end of the incubation collected by centrifugation and subjected to SDS-PAGE and immunoblot for PED. Left: representative immunoblot (input: 1ng). Right: binding, association and uptake of PED by lysosomes calculated as described under material and methods. Values are expressed as percentage of the protein added and are mean + S.E. n=4. (B) Lysosomes were incubated with increased concentrations of PED and processed as in A. Left: representative immunoblot. Right: Quantification of the percentage of PED bound to lysosomes. (C) Lysosomes were incubated with PED in the presence of ovalbumin or GAPDH, a non-CMA and a CMA substrate respectively and processed as in A. Left: representative immunoblot. Right: PED uptake in lysosomes incubated with PED alone or in the presence of the indicated proteins. Values are expressed as percentage of the protein added and are mean+ S.E. n=4. Percentage of inhibition is indicated. D-E. Lysosomes were incubated with a fix concentration of PED and increasing concentrations of Rnase A (D) or with RNase A and increasing concentrations of PED (E). Samples were subjected to immunoblot for both proteins.
To further confirm that lysosomal internalization of PED was taking place via CMA, we compared the effect that other proteins had on PED uptake. As shown in Figure 3C, ovalbumin, a protein lacking any KFERQ-like motif and previously shown to do not undergo CMA [20], did not have any effect on lysosomal uptake of PED. In contrast, glyceraldheyde-3-phosphate-dehydrogenase (GAPDH), a well-characterized CMA substrate [20], was able to partially inhibit PED uptake, supporting that both proteins competed for the components involved in CMA translocation (Figure 3C). In fact, using ribonuclease A (RNase A) another well characterized CMA substrate [20] we demonstrated that this competition was reciprocal, as increasing concentrations of RNase A were able to gradually decrease the amount of PED associated to lysosomes (Figure 3D) and similarly, increasing concentrations of PED also reduced the amount of lysosome-associated RNase A (Figure 3E).
These results support that PED is a bona fide CMA substrate as it can be internalized directly into isolated lysosomes using similar mechanisms to those described for other CMA substrates.
PED levels are regulated by lysosomal activity
We then tested whether the modulation of lysosome activity in intact cells, could affect PED intracellular levels. To this end, A549 cells were subjected to conditions described to activate or inhibit CMA, and intracellular PED levels were then tested. To activate CMA, cells were incubated in serum-deprived medium (Figure 4A), or transfected with LAMP-2A cDNA, the protein limiting for CMA (Figure 4B) [22]. In both experiments, we observed that under conditions of CMA activation PED intracellular levels decreased. We then incubated cells with chloroquine to inhibit lysosomal degradation by all types of autophagy (Figure 4C) and found that blockage of lysosomal proteolysis lead to an increase of PED intracellular levels. To determine if this lysosomal degradation occurred via CMA we transfected the cells with RNAi for LAMP2A (Figure 4D) or Hsc70 (Figure 4E). Directly targeting of either CMA components lead to similar increase in intracellular PED levels confirming that blockage of CMA prevent PED from degradation. Induction of CMA or blockage of lysosomal degradation (two different inhibitors shown here), were able to modulate levels of PED when expressed as exogenous protein (Figure 4F). Overall these findings support that part of cellular PED normally undergoes degradation in lysosomes via CMA.
Figure 4. PED expression is affected by CMA.

(A) CMA activation. PED protein level analyzed in A549 cells by western blot upon A549 serum deprivation for 24 and 48 hr or (B) in A549 cells transfected for 24 hr with LAMP-2A cDNA. (C) CMA inhibition. PED protein level analyzed by western blot upon A549 treatment with different doses of cloroquine or (D) in A549 transfected for 24 hr with ShRNA-LAMP-2A or (E) with siRNA Hsc70 for 48hr. (F) A549 transfected for 24 hr with PED-Myc cDNA and then serum deprived or treated with 10μM of chloroquine for 24 hrs or 15mM NH4CL for 4 hrs. PED Myc levels were analyzed by Western blot.
Role of PED phosphorylation in PED degradation by CMA
The analysis of PED protein sequence revealed the presence of two possible CMA-targeting motifs, NKLDK (amino acids 52-57), and DIIRQ (amino acids 110-115) (Figure 5A). Interestingly, the latter of this motifs resides close to two major PED phosphorylation sites, Ser114 and Ser116 [5,11,23]. We therefore tested whether PED phosphorylation may affect its interaction with Hsc70 and thus its degradation by CMA. For this purpose, we transfected HEK-293 cells with a phosphomimetic PED mutant, where Ser104/116 were substituted with aspartic acid (HA-PED-S104/116D) or with a death-phosphorylation mutant where Ser 104/116 were substituted with alanines (HA-PED-S104/116A) [12,24]. As shown in Figure 5B, PED-S104/116A interacted at a greater extent with Hsc70 compared to PED-S104/116D.
Figure 5. CMA regulates the unphosphorylated form of PED.

(A) Schematic representation of PED phosphorylation sites and putative KFEQR sites. (B) HEK-293 were transfected with PED-Myc cDNA, HA-PED-S104/116A and HA-PED-S104/116D. After 48 hrs, 1 mg of protein extracts were immunoprecipitated with anti-PED antibody or with control IgG. Samples were then resolved with SDS-PAGE and western blot with anti-Hsc70 antibodies. 10 μg of total cellular extract was loaded as control. (C) A549 cells transfected with PED-Myc, HA-PED-S104/116A and HA-PED-S104/116D were serum deprived for 24 hrs. Protein levels of PED wt and phosphorylation mutant were analyzed by Western blot (D) Western blot analysis of PED-Myc and HA-PED-S104/116A, HA-PED-S104/116D after A549 co-transfection with LAMP2A cDNA upon 24 hr. (E) Western blot analysis of PED-Myc and HA-PED-S104/116A, HA-PED-S104/116D upon A549 treatment with 15 mMol of NH4Cl for 4 hr. (F) Western blot analysis of PED-Myc and HA-PED-S104/116A, HA-PED-S104/116D upon A549 treatment with 10 μMol of chloroquine for 24 hr.
Changes in PED phosphorylation state modified its amenability for lysosomal degradation. Thus, blockage of lysosomal degradation by treatment with NH4Cl (Figure 5C) or chloroquine (Figure 5D), induced accumulation of the wild-type and S104/116A PED, whereas levels of S104/116D remained unchanged. Furthermore, to confirm that these differences in lysosomal degradation were related to CMA we induced CMA with either serum starvation (Fig. 5E) or LAMP-2A transfection (Figure 5F). We found that up-regulation of CMA by either of these approaches resulted in a more pronounced decrease in levels of PED-S104/116A than the one observed for wild-type PED (Figure 5C), in agreement with the increase in its interaction with Hsc70. In contrast, levels of PED-S104/116D remained unchanged upon CMA up-regulation (Figure 5D) supporting a failure to undergo degradation through this pathway.
Taken together those data demonstrate that CMA may preferentially degrade the non phosphorylated form of PED and that phosphorylation of this protein may be used in cells to increase its stability by preventing its degradation by CMA..
Physiological relevance of CMA on PED function
The main activity of PED, in particular in NSCLC, is the regulation of cell apoptosis/viability [1,2,3,23] .Therefore, we investigated whether, the different PED mutants may affect cell viability and TRAIL induced apoptosis, during CMA activation.
We activated CMA by serum starvation and then verified cell viability and response to TRAIL in A549 cells overexpressing PED-S104/116D or PED-S104/116A. As shown in Figure 6A-B, the phosphomimetic PED mutant conferred to the cells a growth advantage compared to the non phosphorylated mutant.
Figure 6. Role of PED phosphorylation on cell growth and apoptosis.
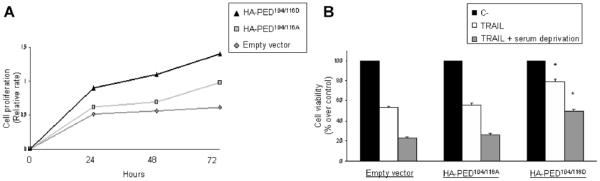
(A) A549 were transfected with different PED mutant as indicated. After 24 hrs, cells were serum starved for different times point and cell viability was assessed with an MTT assay. (B) A549 were transfected with different PED mutant as indicated. After 24 hrs, cells were treated with SuperKiller TRAIL (50ng/ml) alone o associated with serum deprivation for 24hrs. Cell viability was assessed with an MTT assay.
Materials and Methods
Cell lines and reagents
Human A549 cell line was purchased from American Type Culture Collection (ATCC, Milan Italy) and was maintained in RPMI containing 10% heat-inactivated FBS and with 2mM L-glutamine and 100 U/ml penicillin streptomycin. HEK 293 cell line was grown in DMEM medium containing 10% heat-inactivated FBS, with 2mM L-glutamine and 100 U/ml penicillin–streptomycin. Media, sera, and antibiotics for cell culture were from Sigma Aldricht (Milan, Italy). Protein electrophoresis reagents were from Bio-Rad (Richmond, VA), and Western blotting and ECL reagents were from GE Healthcare (Milan, Italy). Hsc70 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), human LAMP-2A antibody from Abcam (Abcam Inc, MA, USA), anti-β-Actin antibodies from Sigma, anti Myc from Cell signalling Technologies (Milan, Italy). PED antibody was generated as decribed before (Condorelli et al., 2002). For transfection Lipofectamine 2000 was purchased by Invitrogen (Milan, Italy) The plasmids used were PED-Myc (ref), the phosphorylation mutant HA-PED(S104/116)A and HA-PED(S104/116)D were a kindly gift of Dr Ginsberg, La Jolla, San Diego, USA. Plasmid encoding for shRNA LAMP-2A and LAMP-2A cDNA was a kindly gift of Ana Maria Cuervo [16]. SiRNA Hsc70 was from SantaCruz Biotechnologies (Santa Cruz, CA, USA). Cloroquine and NH4Cl were from Sigma Aldrich. All the chemicals used for in vitro lysosome experiments were as described previously [20].
Immunoprecipitation
HEK-293 cells were cultured at a final concentration of 90% in p100 plates. The cells were harvested with RIPA Buffer on a shaker for 30 min. One milligram of total extract was immunoprecipitated using 4mg/ml Anti-PED, for 16 h on shaker. Then, A/G beads (Santa Cruz Biotechnology Inc., Santa Cruz, CA) were added for two hrs. The beads were washed for three times with washing buffer (50mM Tris–HCl pH 7.5, 150mMNaCl, 0.1% Triton, 10% glycerol), and then 30μl of sample buffer was added; the samples were boiled at 100°C for 5 min and then the supernatants resolved by SDS–PAGE.
GST pull down assay
Cells were cultured at a final concentration of 90% in p100 plates. After a quick wash with iced-cold PBS, cells were lysed with GST-Fish buffer (50mM Tris–HCl ph 7.4, 2mM MgCl2, 1% NP-40, 10% glycerol, 100mM NaCl, 1mg/ml leupeptin, 1mg/ml pepstatin, 1mg/ml aprotinin, 1mM PMSF, and 2mM DTT). The lysates were cleared by centrifugation in a pre-cooled rotor. One hundred fifty micrograms of total protein extract was mixed with 10 μg of GST-PED recombinant protein coupled to glutathione-sepharose beads and incubated 30 min at 4°C under agitation. Beads were then rinsed three times rapidly with 1ml of iced-cold GST-Fish buffer. The amounts of total Hsc70 was estimated by immunoblot against Hsc70.
Western blotting
Total proteins from A549 and HEK293 cells were extracted with RIPA buffer (0.15mM NaCl, 0.05mM Tris–HCl, pH 7.5, 1% Triton, 0.1% SDS, 0.1% sodium deoxycolate and 1% Nonidet P40). Fifty micrograms of sample extract were resolved on 10–15% SDS–polyacrylamide gels using a mini-gel apparatus (Bio-Rad Laboratories, Richmond, CA) and transferred to Hybond-C extra nitrocellulose. Membranes were blocked for 1 h with 5% non-fat dry milk in TBS containing 0.05% Tween-20, incubated over night with primary antibody, washed and incubated with secondary antibody, and visualized by chemiluminescence.
Immunofluorescence staining
Briefly, cells were fixed with 4%paraformaldehyde for 10 min, permeabilized and blocked in blocking solution (0.1 % Saponin, 10% FBS in PBS), followed by the incubation of primary and secondary antibody for 60 min each. Primary antibodies were used at the following dilutions: PED (1:100), Hsc70 (1:100), LAMP-2A (1:200) in 1:10 blocking solution. Antigen-antibody complexes were visualized with green fluorocrome (Alexa 488-conjugated phalloidin-BD Bioscience), and red fluorocrome (Alexa 459) diluted 1: 1000 in 1:10 blocking solution.
Animals
Adult male Wistar rats and C57BL/6 mice (Charles River Laboratories) fasted for 48h before sacrifice were used. Mice were treated and euthanized in compliance with Albert Einstein College of Medicine institutional guidelines and protocols
Lysosomal Isolation
Lysosomes from rat liver were isolated from a light mitochondrial-lysosomal fraction in a discontinuous metrizamide density gradient by the shorter method described previously [19]. Lysosomal integrity was verified after isolation by measuring β-hexosaminidase latency [25] and only preparations with more than 95% intact lysosomes were used.
Recombinant PED protein generation
GST-PED vector was transformed in BL21 cells. The cells were inducted with 0,5 mMol of IPTG (Qiagen, Milan Italy) for 4 hr. Cells were harvested and resuspended in PBS and then sonicated. 1% Triton X-100 was added at final concentration of 0.1% and the cells were left for 30 min in agitation on the wheel at 4°C. Lysates were then centrifuged at 12000 g for 10 min at 4°C and the a polyacrylamide analytic gel was loaded. The protein was isolated using GST TRAP column (GE Healthcare, Milan Italy). The protein was aliquoted and store at −80°C.
Measurement of CMA activity
PED was incubated in the MOPS buffer with untreated or protease inhibitor-treated lysosomes as described [26]. After incubation for 20min at 37°C, lysosomes were collected by centrifugation and samples were subjected to SDS–PAGE and immunoblotted with an antibody against PED. Uptake was calculated as the difference between the amount of substrate associated to lysosomes (protease inhibitor-treated lysosomes) and the amount of substrate bound to their membrane (untreated lysosomes).
Cell Viability quantification
Cells were plated in 96-well plates in triplicate and incubated at 37°C in a 5% CO2 incubator. Super Killer TRAIL was used at final concentration of 50 ng/ml for 24hr. Cell viability was evaluated with the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Milan Italy) according to the manufacturer's protocol. Metabolically active cells were detected by adding 20 μL of MTS to each well. After 2 h of incubation, the plates were analyzed in a Multilabel Counter (BioTek, Milan Italy).
DISCUSSION
In this manuscript we describe the identification of a new substrate for chaperone-mediated autophagy (CMA), the protein PED. We identified two KFERQ-like motifs in the amino acid sequene of PED. Interestingly, one of them is located within previously descrbed PED phosphorylation sites. So far, although it is clear that the chaperone protein Hsc70 is able to recognize the KFERQ motifs, little is known on how it is modulated. P rotein folding-unfolding may be important for that recognition [14]. Thus, iif the KFERQ motif is present in the protein core, the binding of Hsc70 may occur only when the protein unfolds. In other proteins the motif may be masked by protein-protein interactiosn. Very little is known on the effect of post-translational modifications in the ability of hsc70 to recognize CMA targeting motifs. Acetylation is the only post-translational modification described to date to modulate CMA of a protein [27]. PED is the first example where phosphorylation modifies the ability for Hsc70 to recognize KFERQ-like motifs.
PED exists in vivo as three isoforms, namely N, Pa and Pb, which correspond to the unphosphorylated, mono and diphosphorylated forms, respectively [12]. Phosphorylation occurs on two serine residues, Ser104 and Ser116. Ser104 is a PKC target [24]. The second site, Ser116, is the target of the CaMK-II and AKT [11,12,23].Several reports suggest that PED phosphorylation is determinant for its functions. PED-phosphorylation may lead to increased proliferation and apoptosis inhibition, thus promoting PED-tumor promoter function, whereas unphosphorylated PED binds to ERK1/2, blocking its nuclear translocation and cell proliferation [10], thus acting as tumor suppressor. Phosphorylation of Ser116 regulates the anti-apoptotic function of PEA-15 and modulates its targeting to the death inducing signaling complex (DISC) [1,11].
Several findings support the idea that PED may have a dual function, as pro-survival and tumorigenic in many types of cancer (skin, breast, ovarian, glioblastomas, and lung cancer), and tumor-suppressive in other types of cancer [28,29]. Thus, PED phosphorylation levels within the cells are of great importance for cell fate. Although most emphasis has been placed in the identification of the kinases that by actively phosphorylating PED may contribute to the switch from anti- to pro-oncogenic, the contribution of other cellular processes to the modulation of this switch has not been previously explored. In this work, we show for the first time that changes in the stability of both forms of the protein, and consequently in the balance between levels of both forms, may be mediated by their ability to undergo degradation via CMA.
PED function may be regulated by another Heat shock protein, Hsp27. Hayash et al. demonstrated that Hsp27 regulates PED phosphorylation status by modulating Akt activity and stability. Akt-mediated PED phosphorylation alters its binding specificity, resulting in its dissociation from ERK, leading to increase in proliferation, and association with FADD leading to apoptosis inhibition [30] In this study, we demonstrate that Hsc70 binds preferentially the non-phosphorylated mutant of PED, promoting its degradation via CMA. Thus, CMA preferentially degrades the tumor suppressor-non phosphorylated form of PED, leaving the cells with greater amounts of PED tumor-promoter type.
Recent reports describe that CMA is up-regulated in cancer, in particular in lung cancer [31]. We have observed that PED is overexpressed in lung cancer, and its expression correlates with histological grade and with apoptosis resistance [3].PED could be overexpressed in cancer cells to compensate for its faster degradation due to the upregulation of CMA. However, a more attractive explanation is that the upregulation of CMA observed in this type of cancer [31] may contribute to further increase the disbalance between the pro-oncogenic and anti-oncogenic variants of PED by taking care of the removal of a subset of PED that has “inhibitory” effect on tumorigenesis. We have demonstrated here that indeed, the anti-oncongenic variant is readily delivered for CMA degradation whereas phosphorylation protects the pro-oncogenic PED from this fate. Therefore, we speculate that CMA-increase in lung cancer may preferentially degrade the tumor-suppressor form of PED, and this may contribute to tumor progression. Further studies will aim to elucidate if the described anti-oncogenic effect of blocking CMA in cancer cell lines and tumors [31] is attained, at least in part, through an increase in the amount of the anti-oncogenic PED.
In conclusion, in this manuscript we identified PED as a new CMA substrate. In cancer cells, CMA removes the tumor suppressor form of PED thus contributing to increase the activity of PED tumor promoter type. Inhibitory modulation of CMA presents a possible therapeutic potential against tumorigenesis, at least in part by altering the cancer cellular ratio of the tumor promoter PED versus the tumor suppressor form of PED.
Supplementary Material
Acknowledgments
Grant Support: This work was partially supported by funds from Associazione Italiana Ricerca sul Cancro, AIRC to GC (grant n.ro 10620), MERIT (RBNE08E8CZ_002) to GC, POR Campania FSE 2007-2013 Project CRÉME to GC, Fondazione Berlucchi to GC, and NIH/NIA AG031782 and AG03872 to AMC C.Q. was supported by the “Federazione Italiana Ricerca sul Cancro” (FIRC) Post-Doctoral Research Fellowship and I.T. by a Fulbright Postdoctoral Fellowship.
References
- 1.Condorelli G, Vigliotta G, Cafieri A, Trencia A, Andalo P, et al. PED/PEA-15: an anti-apoptotic molecule that regulates FAS/TNFR1-induced apoptosis. Oncogene. 1999;18:4409–4415. doi: 10.1038/sj.onc.1202831. [DOI] [PubMed] [Google Scholar]
- 2.Renault F, Formstecher E, Callebaut I, Junier MP, Chneiweiss H. The multifunctional protein PEA-15 is involved in the control of apoptosis and cell cycle in astrocytes. Biochem Pharmacol. 2003;66:1581–1588. doi: 10.1016/s0006-2952(03)00514-8. [DOI] [PubMed] [Google Scholar]
- 3.Zanca C, Garofalo M, Quintavalle C, Romano G, Acunzo M, et al. PED is overexpressed and mediates TRAIL resistance in human non-small cell lung cancer. J Cell Mol Med. 2008;225:63–72. doi: 10.1111/j.1582-4934.2008.00283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Incoronato M, Garofalo M, Urso L, Romano G, Quintavalle C, et al. miR-212 increases tumor necrosis factor-related apoptosis-inducing ligand sensitivity in non-small cell lung cancer by targeting the antiapoptotic protein PED. Cancer Res. 2010;70:3638–3646. doi: 10.1158/0008-5472.CAN-09-3341. [DOI] [PubMed] [Google Scholar]
- 5.Stassi G, Garofalo M, Zerilli M, Ricci-Vitiani L, Zanca C, et al. PED mediates AKT-dependent chemoresistance in human breast cancer cells. Cancer Res. 2005;65:6668–6675. doi: 10.1158/0008-5472.CAN-04-4009. [DOI] [PubMed] [Google Scholar]
- 6.Garofalo M, Quintavalle C, Zanca C, De Rienzo A, Romano G, et al. Akt regulates drug-induced cell death through Bcl-w downregulation. Plos ONE. 2008;3:e4070. doi: 10.1371/journal.pone.0004070. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Kitsberg D, Formstecher E, Fauquet M, Kubes M, Cordier J, et al. Knock-out of the neural death effector domain protein PEA-15 demonstrates that its expression protects astrocytes from TNFalpha-induced apoptosis. J Neurosci. 1999;19:8244–8251. doi: 10.1523/JNEUROSCI.19-19-08244.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hao C, Beguinot F, Condorelli G, Trencia A, Van Meir EG, et al. Induction and Intracellular Regulation of Tumor Necrosis Factor-related Apoptosis-inducing Ligand (TRAIL) Mediated Apotosis in Human Malignant Glioma Cells. Cancer Research. 2001;61:1162–1170. [PubMed] [Google Scholar]
- 9.Garofalo M, Romano G, Quintavalle C, Romano MF, Chiurazzi F, et al. Selective inhibition of PED protein expression sensitizes B-cell chronic lymphocytic leukaemia cells to TRAIL-induced apoptosis. Int J Cancer. 2007;120:1215–1222. doi: 10.1002/ijc.22495. [DOI] [PubMed] [Google Scholar]
- 10.Formstecher E, Ramos JW, Fauquet M, Calderwood DA, Hsieh J-C, et al. PEA-15 Mediates Cytoplasmic Sequestration of ERK MAP Kinase. Developmental Cell. 2001;1:239–250. doi: 10.1016/s1534-5807(01)00035-1. [DOI] [PubMed] [Google Scholar]
- 11.Trencia A, Perfetti A, Cassese A, Vigliotta G, Miele C, et al. Protein kinase B/Akt binds and phosphorylates PED/PEA-15, stabilizing its antiapoptotic action. Mol Cell Biol. 2003;23:4511–4521. doi: 10.1128/MCB.23.13.4511-4521.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sulzmaier FJ, Opoku-Ansah J, Ramos JW. Phosphorylation is the switch that turns PEA-15 from tumor suppressor to tumor promoter. Small GTPases. 2012;3:173–177. doi: 10.4161/sgtp.20021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zanca C, Cozzolino F, Quintavalle C, Di Costanzo S, Ricci-Vitiani L, et al. PED regulates cell migration process in human Non Small Cell Lung Cancer cells through Rac1. j Cell Physiol. 2010;225:63–72. doi: 10.1002/jcp.22197. [DOI] [PubMed] [Google Scholar]
- 14.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends in Cell Biology. 2012;22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The Chaperone-Mediated Autophagy Receptor Organizes in Dynamic Protein Complexes at the Lysosomal Membrane. Molecular and Cellular Biology. 2008;28:5747–5763. doi: 10.1128/MCB.02070-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massey AC, Zhang C, Cuervo AM. Current Topics in Developmental Biology. Academic Press; 2006. Chaperone-Mediated Autophagy in Aging and Disease; pp. 205–235. [DOI] [PubMed] [Google Scholar]
- 17.Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1999;15:305–309. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- 18.Kaushik S, Cuervo AM. Chaperones in autophagy. Pharmacological Research. 2012;66:484–493. doi: 10.1016/j.phrs.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cuervo AM, Dice JF, Knecht E. A Population of Rat Liver Lysosomes Responsible for the Selective Uptake and Degradation of Cytosolic Proteins. Journal of Biological Chemistry. 1997;272:5606–5615. doi: 10.1074/jbc.272.9.5606. [DOI] [PubMed] [Google Scholar]
- 20.Cuervo AM, Terlecky SR, Dice JF, Knecht E. Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. Journal of Biological Chemistry. 1994;269:26374–26380. [PubMed] [Google Scholar]
- 21.Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem. 2000;275:27447–27456. doi: 10.1074/jbc.M001394200. [DOI] [PubMed] [Google Scholar]
- 22.Cuervo AM, Dice JF. Regulation of lamp2a levels in the lysosomal membrane. Traffic. 2000;1:570–583. doi: 10.1034/j.1600-0854.2000.010707.x. [DOI] [PubMed] [Google Scholar]
- 23.Condorelli G, Trencia A, Vigliotta G, Perfetti A, Goglia U, et al. Multiple members of the mitogen-activated protein kinase family are necessary for PED/PEA-15 anti-apoptotic function. J Biol Chem. 2002;277:11013–11018. doi: 10.1074/jbc.M110934200. [DOI] [PubMed] [Google Scholar]
- 24.Krueger J, Chou F-L, Glading A, Schaefer E, Ginsberg MH. Phosphorylation of Phosphoprotein Enriched in Astrocytes (PEA-15) Regulates Extracellular Signal-regulated Kinase-dependent Transcription and Cell Proliferation. Molecular Biology of the Cell. 2005;16:3552–3561. doi: 10.1091/mbc.E04-11-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Storrie B, Madden EA. Isolation of subcellular organelles. Methods Enzymol. 1990;182:203–225. doi: 10.1016/0076-6879(90)82018-w. [DOI] [PubMed] [Google Scholar]
- 26.Kaushik S, Cuervo AM. Methods in Enzymology. Academic Press; 2009. Chapter 19 Methods to Monitor Chaperone-Mediated Autophagy; pp. 297–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lv L, Li D, Zhao D, Lin R, Chu Y, et al. Acetylation Targets the M2 Isoform of Pyruvate Kinase for Degradation through Chaperone-Mediated Autophagy and Promotes Tumor Growth. Molecular Cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Funke V, Lehmann-Koch J, Bickeböller Ml, Benner A, Tagscherer KE, et al. The PEA-15/PED protein regulates cellular survival and invasiveness in colorectal carcinomas. Cancer Letters. 2013;335:431–440. doi: 10.1016/j.canlet.2013.02.053. [DOI] [PubMed] [Google Scholar]
- 29.Lee J, Bartholomeusz C, Krishnamurthy S, Liu P, Saso H, et al. PEA-15 unphosphorylated at both serine 104 and serine 116 inhibits ovarian cancer cell tumorigenicity and progression through blocking [beta]-catenin. Oncogenesis. 2012;1:e22. doi: 10.1038/oncsis.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayashi N, Peacock JW, Beraldi E, Zoubeidi A, Gleave ME, et al. Hsp27 silencing coordinately inhibits proliferation and promotes Fas-induced apoptosis by regulating the PEA-15 molecular switch. Cell Death Differ. 2012 2012 Jun;19(6):990–1002. doi: 10.1038/cdd.2011.184. doi: 101038/cdd2011184 Epub 2011 Dec 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, et al. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. 2011;3:109ra117. doi: 10.1126/scitranslmed.3003182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.