

Abstract
The rate at which genome sequencing data is accruing demands enhanced methods for functional annotation and metabolism discovery. Solute binding proteins (SBPs) facilitate the transport of the first reactant in a metabolic pathway, thereby constraining the regions of chemical space and the chemistries that must be considered for pathway reconstruction. We describe high-throughput protein production and differential scanning fluorimetry platforms, which enabled the screening of 158 SBPs against a 189 component library specifically tailored for this class of proteins. Like all screening efforts, this approach is limited by the practical constraints imposed by construction of the library, i.e., we can study only those metabolites that are known to exist and which can be made in sufficient quantities for experimentation. To move beyond these inherent limitations, we illustrate the promise of crystallographic- and mass spectrometric-based approaches for the unbiased use of entire metabolomes as screening libraries. Together, our approaches identified 40 new SBP ligands, generated experiment-based annotations for 2084 SBPs in 71 isofunctional clusters, and defined numerous metabolic pathways, including novel catabolic pathways for the utilization of ethanolamine as sole nitrogen source and the use of d-Ala-d-Ala as sole carbon source. These efforts begin to define an integrated strategy for realizing the full value of amassing genome sequence data.
The number of newly reported protein sequences inferred from genome sequencing continues to grow at a rate that severely outpaces the assignment of function through comparative genomics or direct biochemical analysis. This situation results in a large proportion of unannotated and misannotated protein sequences,1 precluding the discovery of novel enzymes activities and metabolic pathways important to (1) understanding the contributions of the gut microbiome to human health, (2) the realization of new chemical processes for industry, and (3) our understanding of critical environmental issues, including global nutrient cycles and the evolution of complex microbial communities. Accordingly, the development of strategies, tools, and infrastructure for enhanced functional assignment represents major challenges to the postgenomic biological community.
A range of computational, informatics, and experimental approaches, either individually or collectively, can be leveraged to streamline the discovery of new molecular function and metabolism, but the great expanse of chemical space and the vast array of biologically relevant transformations represent continuing and significant obstacles. These considerations are particularly relevant to the in vitro analysis of enzymes, which frequently exhibit high specificity, requiring a single unique compound or a very limited number of compounds, for meaningful rates of catalysis to be achieved. Even molecules closely related to the true substrate (e.g., substrate-derived fragments containing key reactive groups or molecules missing only a single functionality) often do not support significant turnover due to loss of binding energy and impaired transition state recognition. Thus, while enzymes are the major effectors of the chemical transformations underlying metabolism, they present significant challenges for high-throughput functional annotation due to their often stringent requirements for cognate substrates.
In contrast, solute binding proteins (SBPs) for transport systems possess features that make them particularly amenable to large-scale functional annotation. The first step in a catabolic pathway is frequently the passage of a metabolite across the cellular membrane by SBP-dependent transport machinery. In many cases, the transporter genes are colocated or coregulated with genes encoding the enzymes responsible for catabolism of the transported molecule. Many transport systems utilize an SBP, located either in the periplasm (Gram negative) or tethered to the outer membrane (Gram positive), for capture of the first reactant in a pathway and its subsequent delivery to transmembrane components that direct translocation to the cytosol. These SBPs exhibit relatively high binding affinities (i.e., high nanomolar to low micromolar) and are composed of two alpha-beta domains, joined by flexible segments, which undergo a venus flytrap-like closure upon binding cognate small molecule ligands.2,3 Thus, SBPs perform a strictly biophysical binding function and do not suffer from the complications associated with chemistry. The ability to identify the initial reactant (or a closely related molecule) for a catabolic pathway provides an immediate toe-hold by placing significant constraints on the regions of chemical space that need to be considered and, in conjunction with knowledge of colocalized and coregulated genes, begins to define details of the in vivo biochemical transformations operating within the metabolic pathway.
Three SBP-dependent transport systems have been described: (1) the TRipartite ATP-independent Periplasmic transporters (TRAPs), (2) the ATP-binding cassette transporters (ABCs), and (3) the tripartite tricarboxylate transporters (TTTs). The TRAP systems are typically composed of a large transmembrane subunit (DctM, 12 membrane helices), a small transmembrane subunit (DctQ, 4 transmembrane helices), and an SBP (DctP, ∼320 residues) and drive transport by coupling to an electrochemical gradient.4 Of particular note is a highly conserved arginine within the ligand binding sites of the TRAP SBPs responsible for the family’s preference for organic acids.
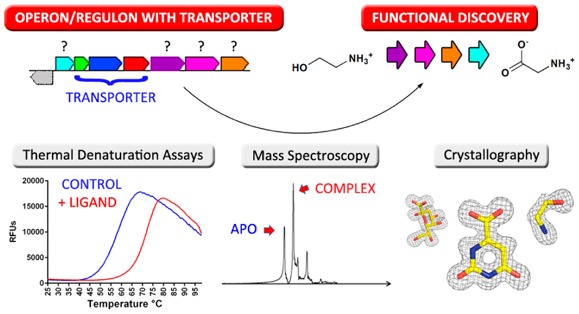
To exploit the favorable properties of the SBPs, the Enzyme Function Initiative, a multi-institutional consortium devoted to the development of strategies for enzyme functional annotation,5 has implemented a high-throughput differential scanning calorimetry (DSF) platform for the discovery of new ligands for SBPs. DSF measures fluorescence from an environmentally sensitive dye, whose emission properties change upon interaction with unfolded protein, allowing the rapid and efficient evaluation of thermally induced unfolding. This unfolding process is characterized by the midpoint of the unfolding transition (Tm), which is expected to increase (i.e., higher Tm) upon binding of ligands that result in a more stable protein–ligand complex.6
All previously identified ligands for TRAPs were organic acids (24 ligands), which simplified the construction of our initial screening library (189 compounds, including amino acids, acid sugars, and other carboxylate-containing small metabolites). We screened 158 TRAP SBPs by DSF and identified 89 positive DSF hits (i.e., ΔTm > 5 °C), resulting in the assignment of a ligand for 71 isofunctional clusters (10–120 sequence similarity network, ∼60% sequence identity), including 40 new ligands for TRAP SBP family members.
These studies also resulted in 60 high-resolution crystal structures of 46 unique TRAP sequences, with 51 containing bound ligands; 29 of these represent a subset of the newly identified ligands for the TRAP SBP family. Remarkably, a number of TRAP SBPs that were negative in the DSF screen yielded structures exhibiting adventitiously bound ligands derived from metabolites in the expression host. These ligands represent a wide range of chemotypes not present in the original screening library and, in conjunction with genome context considerations, support the existence of numerous new metabolic functions. One of these ligands was unambiguously identified as ethanolamine, which is a particularly surprising TRAP SBP ligand because it does not contain an acidic functionality typical of this family. Subsequent microbiology experiments resulted in the discovery and characterization of a novel catabolic pathway enabling the use of ethanolamine as a sole nitrogen source. These efforts, as well as complementary mass spectrometric approaches, represent elements of a general strategy for the efficient discovery of new metabolites, new protein–ligand interactions, and new metabolism utilizing the entire metabolome of the expression host as the metabolite library.
Experimental Procedures
Generation of TRAP SBP SSNs
In total, 8240 TRAP SBP sequences in the InterPro7 family IPR018389 were used to build the TRAP SBP sequence similarity networks. We used InterPro 41.0 (released on February 13, 2013), the most up-to-date release at the time of our study. The web server EFI-EST (refer to http://efi.igb.illinois.edu/efi-est/index.php), inspired by sequence similarity network creating program Pythoscape,8 was used to perform all-by-all blast analyses for the 8240 sequences and generate the full network for the TRAP SBP family. The 10–120 and 10–80 SSNs were generated by applying e-value cutoffs 10–120 and 10–80 to the full network, respectively. Those sequences which exhibit 100% sequence identity but are from unique organisms are represented by a single node in the networks.
TRAP SBP Cloning and Protein Purification
TRAP SBPs were amplified from genomic DNA by PCR using KOD hot start DNA polymerase (Novagen). The conditions were as follows: 2 min at 95 °C, followed by 40 cycles of 30 s at 95 °C, 30 s at 66 °C, and 30 s at 72 °C. The amplified fragment was cloned into the N-terminal TEV cleavable 6×-His-tag containing vector pNIC28-Bsa49 or the C-terminal TEV cleavable 10×-His-tag vector pNYCOMPS-LIC-TH10-ccdB10 by ligation-independent cloning.11 For those TRAPs cloned into pNIC28-Bsa4, the periplasmic signal sequence, as predicted by SignalP,12 was not included in the final cloned product. Vectors containing the cloned target were transformed into Escherichia coli BL21 (DE3) containing the pRIL plasmid (Stratagene) and used to inoculate a 20 mL culture of 2× YT containing 50 μg mL–1 kanamycin and 34 μg mL–1 chloramphenicol. The culture was allowed to grow overnight at 37 °C in a shaking incubator. The overnight culture was used to inoculate 2 L of ZYP-5052 autoinduction media.13 The expression culture was placed in a LEX48 airlift fermenter and incubated at 37 °C for 4 h and then at 22 °C overnight (16–20 h). Culture was harvested, pelleted by centrifugation at 6000g, and stored at −80 °C. Cells were resuspended in lysis buffer (20 mM HEPES, pH 7.5, 20 mM imidazole, 500 mM NaCl, 5% glycerol, and 5 mM MgCl2) and lysed by sonication. Lysate was clarified by centrifugation at 35 000g and loaded onto a 1 mL HisTrap Ni-NTA column (GE Healthcare) using an AKTAxpress FPLC (GE Healthcare). The column was washed with 10 column volumes of lysis buffer and eluted in buffer containing 20 mM HEPES, pH 7.5, 500 mM NaCl, 500 mM imidazole, 5% glycerol, and 5 mM MgCl2 directly onto a HiLoad S200 16/60 PR gel filtration column equilibrated with buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, 5% glycerol, 5 mM MgCl2, and 5 mM DTT. Eluted protein was analyzed by SDS-PAGE, snap frozen in liquid nitrogen, and stored at −80 °C.
TEV Protease Preparation
Engineered TEV protease (eTEV) was expressed from plasmid pMHTDelta238 as described by Blommel and Fox.14 eTEV was purified by Ni-NTA chromatography, diluted to 10 mg mL–1, and stored at −80 °C in Ni-NTA elution buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 300 mM imidazole) supplemented with 50% glycerol (w/v).
Differential Scanning Fluorimetry
SYPRO Orange was purchased from Invitrogen (Carlsbad, CA) as a 5000× stock concentration. Liquid stocks of library components (100 or 10 mM) were prepared in H2O and stored at −20 °C. Final DSF reaction mixtures (20 μL final volume) composed of 10 μM protein, 1 mM ligand, and 5× SYPRO Orange in DSF buffer (100 mM HEPES, pH 7.5, 150 mM NaCl) were distributed in 384-well PCR plates with 8 control wells (query protein with no ligand). The fluorescence intensities were measured using an Applied Biosystems 7900HT fast real-time PCR system with excitation at 490 nm and emission at 530 nm. The samples were heated from 22 to 99 °C at a rate of 3 °C min–1, with each sample present in duplicate. The midpoint of the unfolding transition (Tm) was obtained from fitting the melting curve to a Boltzmann equation.15 ΔTm for each specific ligand was calculated as the difference of the Tm values measured with a ligand (average of 2 measurements) and without ligand (average of 8 control wells).
Crystallization and Structure Determination
Protein samples for crystallization were rapidly thawed in a 37 °C water bath and stored on ice. Engineered TEV protease was added in a 1:80 ratio followed by incubation at 4 °C for 2 h. Buffer was exchanged to 20 mM HEPES, pH 7.5, 5 mM DTT by centrifugal concentration and concentrated to a final protein concentration of 30–60 mg mL–1. Proteins were screened against the commercial screens MCSG1, MCSG2, and MCSG4 (MICROLYTIC) utilizing sitting drop vapor diffusion in 96-well Intelliplates (Art Robbins) and incubated at 18 °C. In general, the protein was screened at 3 concentrations (∼40, 20, and 10 mg mL–1) with the inclusion of 10 mM of the ligand eliciting the greatest ΔTm in the DSF screen. Cryoprotectants were added to 20% when needed, and crystals were flash-cooled by plunging directly into liquid nitrogen. Details of crystallization and cryo-protection are provided in Supporting Information Excel 2.
Initially, native sulfur-methionine proteins were prepared for structure determination by molecular replacement (MR). If MR failed, then native crystals were subjected to rapid soaks (30 s to 5 min, 200 mM to 1 M) of NaI or IC3 (5-amino-2,4,6-triiodoisophthalic acid), and structures were determined by SAD phasing using Cu Kα radiation from an RU300 generator, with data collected on a R-Axis IV+2 detector, at 100 K. Finally, as required, targets were prepared as selenomethionone (Se-MET) derivatized materials to support structure determination by single anomalous dispersion (SAD); data from Se-MET crystals were collected at 100 K on beamline 31-ID (Lilly-CAT; Advanced Photon Source) using a wavelength of 0.97929 Å. Data were integrated using iMOSFLM16 and scaled using SCALA17 or, alternatively, processed using HKL3000.18 The heavy atom substructure and initial phases were calculated utilizing PHENIX19 or HKL3000.18 Initial models were obtained with a variety of density fitting programs, typically, ArpWarp,20 buccaneer,21 or PHENIX autobuild.22 Iterative cycles of manual rebuilding within the molecular graphics program COOT,23 and refinement against the data within PHENIX were performed until convergence was achieved. Only at this point were ligands fit to the remaining difference density. During the final refinement cycles, TLS refinement24 was performed with TLS ranges as determined within PHENIX. Stereochemistry of the models was examined within the program MOLPROBITY.25 Of 60 structures determined, 28 were by molecular or isomorphous replacement, 9 by rapid soak SAD, and 23 by Se-MET SAD. Details of the data collection, structure determination, and refinement statistics are listed in Supporting Information Excel 2.
X-FTMS of Co-purified Ligands
For extraction of adventitiously bound ligands, protein samples were washed three times with 0.2 M ammonium acetate (pH 6.0) using a filter with a 10 kDa MWCO (Millipore) and concentrated to >50 mg mL–1. One-hundred ninety microliters of methanol was added to 10 μL of concentrated protein solution, and the mixture vortexed for 1 min and centrifuged at 4 °C for 20 min (16 000g). The resulting supernatant was used for subsequent analysis. For each injection, 50 μL was loaded onto a 1.0 × 50 mm C18 column (Phenomenex, CA). After desalting with solvent A (5% acetonitrile, 0.1% formic acid) for 5 min, bound extracted ligands were eluted with a 30 min gradient composed of 5 to 100% solvent B (95% acetonitrile, 0.1% formic acid). The effluent was directly delivered into a 12T QFT-ICR-MS (Agilent Technologies, Inc.) for mass analysis. A Shimadzu HPLC, with two LC-20AD pumps, was used to generate a gradient with 50 μL min–1 flow rate.
ESI-FTMS of Co-purified Ligands
All experiments were performed under the optimum conditions found for the detection of noncovalent complexes on a Bruker Solarix 12 T electrospray ionization source Fourier transform mass spectrometer. Mass spectra were recorded in the positive ion mode with a mass range from 50 to 6000 m/z for broadband low-resolution acquisition. Each spectrum was an average of 32 transients (scans) composed of 1 or 2 megabyte data points. All aspects of pulse sequence control and data acquisition were controlled by Solarix control software. A bovine carbonic anhydrase II (bCAII) sulfanilamide complex was used as a control for optimization.26−28 bCAII (29 089 Da, Sigma-Aldrich) was dissolved in ammonium acetate (10 mM, pH 7) to generate a stock solution (34 μM). Sulfanilamide (172 Da, Sigma-Aldrich) was dissolved in methanol to make a stock solution (5.8 mM). bCAII (100 μL, 3.4 μM) in ammonium acetate (10 mM) was mixed with the sulfanilamide (10 μL, 581 μM) and incubated for 1 h at room temperature (20 °C). The inhibitor/protein ratio was 16:1. TRAP SBPs were buffer-exchanged to 10 mM ammonium acetate, pH 7, using size-exclusion chromatography prior to analysis. The Δm/z for the unbound protein and the protein–ligand complex ions was multiplied by the charge state (z) to directly afford the MW of the bound ligand, using the following equation: MWligand = Δm/z × z.
Cloning, Expression, and Purification of VanX and Csal_0679
The gene encoding VanX, the d-Ala-d-Ala dipeptidase from Chromohalobacter salexigens DSM3043 (UniProt AC Q1QZT4), was PCR-amplified from genomic DNA and inserted into NdeI/BamHI digested pET15b (Novagen). The gene encoding the putative l-glutamine synthetase, Csal_0679 from C. salexigens DSM3043 (UniProt ID Q1QZR8), was PCR-amplified from genomic DNA and inserted into NdeI/BlpI digested pET-15b (Novagen). The resulting constructs were transformed into E. coli BL21 (DE3) for expression.
Expression and purification of hexahistidine-tagged VanX was executed using a chelating Sepharose fast flow (Amersham Biosciences) column charged with Ni2+ as previously described.29 The protein was concentrated to 7.1 mg mL–1, flash-frozen using liquid nitrogen, and stored at −80 °C prior to use. For expression of Csal_0679, cells were grown at 37 °C with agitation at 220 rpm in 1 L of LB supplemented with 100 μg/mL ampicillin to OD600 of ca. 0.6 and induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The culture was allowed to grow an additional 20 h at 20 °C before the cells were harvested by centrifugation. The cells were resuspended in 50 mL of binding buffer (5 mM imidazole, 0.3 M NaCl, 5 mM MgCl2, and 20 mM Tris-HCl, pH 7.9) and lysed by sonication. The lysate was cleared by centrifugation, and the supernatant containing the His-tagged protein was loaded onto a column containing 8 mL of Ni-NTA resin (Qiagen) previously equilibrated with binding buffer. After equilibration of the Ni-NTA resin with the supernatant on a rocking platform for 30 min, the flow through was discarded and the column was washed with 50 mL of wash buffer I (25 mM imidazole, 0.3 M NaCl, 5 mM MgCl2, and 20 mM Tris-HCl, pH 7.9) followed by 30 mL of wash buffer II (60 mM imidazole, 0.3 M NaCl, 5 mM MgCl2, and 20 mM Tris-HCl, pH 7.9). His-tagged protein was eluted with buffer III (250 mM imidazole, 0.3 M NaCl, 5 mM MgCl2, and 20 mM Tris-HCl, pH 7.9). Homogenous peak fractions were determined by SDS-PAGE gel electrophoresis, combined, and dialyzed against 20 mM Tris-HCl, pH 7.9, 5 mM MgCl2, and 15% glycerol. The dialysate was concentrated by centrifugation at 4 °C with an Amicon Ultra centrifugal filter unit (30 000 NMWL, Merck Millipore Ltd.) to a final concentration of ca. 12 mg mL–1. The homogeneous protein was flash-frozen dropwise into liquid nitrogen and stored at −80 °C.
Kinetic Measurements for VanX and Csal_0679
For kinetic measurements of purified VanX, a 200 μL reaction containing 50 mM deuterated-Tris, pH 7.9, 10 mM MgCl2, 1 μM VanX, and 10 mM d-Ala-d-Ala was prepared in ddH2O and incubated at 37 °C for 16 h (a control reaction was also prepared without VanX). Reactions were lyophilized for 24 h and resuspended in 800 μL of D2O at pD 7.9. 1H NMR spectra were recorded with a Unity INOVA 500NB instrument and analyzed with NUTS software.
γ-Glutamyl amide synthetase activity of Csal_0679 was assayed by measuring formation of ADP from ATP, where production of ADP was followed by the decrease of absorbance of NADH at 340 nm at 25 °C due to oxidation of NADH via substrate/product coupled pyruvate kinase and lactate dehydrogenase. The reaction mixture contained variable concentrations of tested substrate and defined concentration of co-substrate, 50 mM Tris-HCl buffer (pH 7.9), 10 mM KCl, 15 mM MgCl2, 5 mM ATP, 2.5 mM PEP, 0.16 mM NADH, 8 units of pyruvate kinase/lactate dehydrogenase from rabbit muscle (Sigma-Aldrich), and 1.2 × 10–6 M Csal_0679 in a final volume of 200 μL. Measurements of kinetic parameters for glutamate were made at 20 mM ethanolamine or l-alaninol, respectively. Kinetic constants for ethanolamine or l-alaninol were measured at 50 mM glutamate.
Bacterial Growth Conditions
Bacterial strains were grown aerobically at 30 °C (Agrobacterium tumefaciens C58, Roseobacter denitrificans OCh114) or 37 °C (C. salexigens DSM3043, E. coli) with shaking at 225 rpm and were routinely cultured in Luria–Bertani (Difco) supplemented with 1 M NaCl for C. salexigens or Marine Broth 2216 (Difco) for R. denitrificans OCh114. For gene expression analyses and carbon or nitrogen utilization studies, strains were cultured in M9 minimal medium (per liter: 12.8 g of Na2HPO4·7H2O, 3.0 g of KH2PO4, and 0.5 g of NaCl) supplemented with 1 mM MgSO4, 100 μM CaCl2, the following trace metals (per liter: 0.003 mg of CuSO4·5H2O, 0.025 mg of H3BO3, 0.007 mg of CoCl2·6H2O, 0.016 mg of MnCl2·4H2O, 0.003 mg of ZnSO4·7H2O, and 0.3 mg of FeSO4·7H2O) and vitamins (33 μM thiamine, 41 μM biotin, and 10 nM nicotinic acid). Minimal medium was supplemented with 0.001% yeast extract for R. denitrificans OCh114 or with 1.5 M NaCl for C. salexigens. Carbon sources (d-ala-d-ala, KDO, or glucose) and nitrogen sources (NH4Cl, glycine, or ethanolamine) were provided at 10 or 20 mM, as indicated.
Growth curves were carried out using the Bioscreen C instrument as previously described.30 Briefly, each well in the Bioscreen C plate containing the appropriate minimal medium was inoculated with a normalized 1:100 dilution of washed starter culture. Cells were grown at the appropriate temperature with continuous shaking at medium amplitude, and absorbance (OD600) was recorded every 30 min for 48–96 h.
Gene Expression Analysis
Cultures of C. salexigens DSM3043 or R. denitrificans OCh114 were grown in the appropriate minimal medium until early exponential phase. Cells were pelleted by centrifugation (4750g for 5 min at 4 °C), washed, and resuspended in the appropriate minimal medium supplemented with the following carbon or nitrogen sources: 10 mM d-Ala-d-Ala or 10 mM d-glucose as the sole carbon source for the Csal_0660 genome neighborhood in C. salexigens, 20 mM glucose or 20 mM KDO as the sole carbon source for the RDI_0742 genome neighborhood in R. denitrificans, or 20 mM NH4Cl or 20 mM ethanolamine as the sole nitrogen source for the Csal_0679 genome neighborhood in C. salexigens. Cells were harvested at early exponential phase for C. salexigens or at 1 and 24 h postinoculation for R. denitrificans. At the time of cell harvest, one volume of RNAprotect bacteria reagent (Qiagen) was added to two volumes of each actively growing culture. Samples were mixed by vortexing and incubated for 5 min at room temperature. Cells were pelleted by centrifugation (4750g for 5 min at 4 °C), and cell pellets were stored at −80 °C until further use.
RNA isolation was performed in an RNase-free environment at room temperature using the RNeasy mini kit (Qiagen) per the manufacturer’s instructions. Cells were disrupted according to the enzymatic lysis protocol in the RNAprotect Bacteria Reagent Handbook (Qiagen); lysozyme (Thermo-Pierce) was used at 15 mg mL–1. RNA concentrations were determined by absorption at 260 nm using a Nanodrop 2000 (Thermo), and absorption ratios, A260/A280 and A260/A230, were used to assess sample integrity and purity. Isolated RNA was stored at −80 °C until further use.
Reverse transcription (RT) PCRs were performed with 300 ng of total isolated RNA using the ProtoScript first strand cDNA synthesis kit (NEB) or the Transcriptor first strand cDNA synthesis kit (Roche), per the manufacturer’s instructions. Primers for quantitative real-time (qRT) PCR were designed using the Primer3 or Roche primer tool; amplicons were 100–200 bp in length. Primer sequences are provided in Table S1. Primers were 18 to 27 nucleotides in length and had a theoretical Tm of 55–60 °C. Primer efficiency was determined to be at least 90% for each primer pair. qRT-PCRs were carried out in 96-well plates using the Roche LightCycler 480 II instrument with the LightCycler 480 SYBR Green I master mix (Roche) per the manufacturer’s instructions. Minus-RT controls were performed to verify the absence of genomic DNA in each RNA sample for each gene target. Gene expression data were expressed as crossing threshold (CT) values by the 2–ΔΔCT (Livak) method,31 using the 16S rRNA gene as a reference. Each qRT-PCR was performed in triplicate, and fold-changes are the averages of at least three biological replicates.
Gene Disruption
The ΔDctP and ΔVanX knockouts in C. salexigens DSM3043 were made using overlap extension PCR as previously described.30 Primers are given in Table S1. For disruption of genes in the putative ethanolamine utilization operon in C. salexigens, the genomic region ∼1000 bp upstream and downstream of Csal_0678 and Csal_0679 was amplified from C. salexigens genomic DNA using Pfu Ultra high-fidelity DNA polymerase (Thermo) with primers Csal0678_Fwd and Csal0678_Rev or primers Csal0679_Fwd and Csal0679_Rev (Table S1). The Csal_0678 genomic fragment was inserted into EcoRI/HindIII digested pK19mobsacB (ATCC 87097) to generate pCsal0678KO. To disrupt the Csal_0678 coding region in pCsal0678KO, the nonpolar gentamicin resistance cassette from p34s-Gm was inserted into the single BamHI site in Csal_0678, yielding pCsal0678KOGm, the final plasmid used for gene disruption. The Csal_0679 genomic fragment was inserted into EcoRI/HindIII digested pK19mobsacB to generate pCsal0679KO. To disrupt the Csal_0679 coding region in pCsal0679KO, the nonpolar gentamicin resistance cassette from p34s-Gm was inserted into the single PstI site in Csal_0679, yielding pCsal0679KOGm, the final plasmid used for gene disruption.
Gene disruption plasmids pCsal0678KOGm or pCsal0679KOGm were electroporated into E. coli WM6029 (obtained from W. Metcalf at the University of Illinois at Urbana–Champaign), and plasmids were introduced into C. salexigens by biparental mating following established protocols.30 Cointegrates (single-crossover insertions) were selected by resistance to kanamycin and gentamicin. After selection on 10% sucrose medium, double-crossover events were identified by resistance to gentamicin and sensitivity to kanamycin and confirmed by genomic PCRs.
Ethanolamine Pathway: Metabolomics
Cell preparation. LC-FTMS metabolomics of whole cell extracts was carried out with samples of A. tumefaciens C58 fed with ethanolamine, following the procedure of Zhao et al.32 Cells grown in NH4Cl minimal medium were diluted 1:100 into 200 mL of minimal medium with 20 mM ethanolamine as the sole source of nitrogen and grown to an OD600 of 0.6 (approximately 18 h). Cells were harvested by centrifugation (4000g, 10 min, 4 °C), washed, and resuspended in minimal medium without a nitrogen source. The cell suspension was then depleted of catabolic metabolites by incubation at 30 °C for 30 min before transferring it to ice. Cell density was adjusted to OD600 = 6.0, and 1 mL aliquots were prepared on ice. Twenty millimolar ethanolamine was added to half of the samples followed by incubation at 30 °C. At time points of 0, 2, 5, 15, and 30 min, samples were pelleted by centrifugation (16 000g for 1 min), the supernatant was removed, and cell pellets were flash-frozen in liquid nitrogen. Samples were stored at −80 °C prior to analysis.
Metabolomics analysis followed the procedure of Erb et al.33 Metabolites were extracted directly from cell pellets with 0.375 mL of 10 mM ammonium bicarbonate (pH 9.2) in 90% acetonitrile followed by 15 min of vortexing at room temperature. Cell extracts were cleared of debris via two rounds of centrifugation at 16 000g before analysis. Samples were applied to a custom 11T LTQ-FT mass spectrometer (Thermo-Fisher Scientific) with an Agilent 1200 HPLC system equipped with a Sequant Zic-HILIC column (2.1 mm × 150 mm) previously equilibrated with extraction buffer (solvent B). Solvent A was 10 mM ammonium bicarbonate, pH 9.2. One-hundred microliters of each extracted sample was injected for three separate chromatographic runs. Samples were eluted with a 200 μL min–1 flow rate using the following elution profile: 100% B for 17 min, a linear gradient from 100 to 40% B over 3 min, and another linear gradient from 40 to 100% B over 15 min. Data were collected at a resolution of 50 000 with full scan set to m/z 100–1000, and duplicate samples were individually analyzed in either positive or negative ion mode. Data analysis was performed with the Qualbrowser application of Xcalibur (Thermo-Fisher Scientific).
Results
Sequence Similarity Networks
Sequence similarity networks (SSNs) are a simple and powerful tool for visualizing relationships among sequences in protein (super)families.34 We examined the SSN for 8240 TRAP SBP sequences in the IPR018389 InterPro family7 using Cytoscape35 at an e-value of 10–120, which corresponds to a median sequence identity of ∼60% (Figure 1). At this stringency, the majority of the experimentally annotated SBP sequences fall within isofunctional clusters; therefore, this limit (10–120) was used to assign cluster numbers, with the cluster number becoming an associated property of the sequences in the cluster. We also visualized the SSN at the less stringent e-value of 10–80 (corresponding to median sequence identity ∼42%), which resulted in merging of many of the clusters at 10–120 into larger groups whose cognate ligands can be assigned to a more general chemotype (e.g., uronic acids, aldonic acids,).
Figure 1.

TRAP SBP ligands and structures prior to this study. TRAP SBP SSN network at an e-value of 10–120. In the network, each node is labeled with its cluster number, and each color represents a unique function (see Table S2 for ligand to color mapping). Only a small number of sequences have their functions annotated and/or have their structures determined. The known functions prior to our study are shown by the larger colored nodes. Sequences with PDB structures are shown as diamonds with red borders, labeled with the PDB ID. If there is more than one PDB structure for a sequence, then only one is listed.
Prior to this study, 24 SBP ligands were known, which mapped to 17 clusters in the 10–120 SSN (Figures 1 and S1). Even fewer determined structures (9 sequences from 8 clusters) had been determined, with two (2HPG from cluster 32 and 3U65 from cluster 517) having no assigned function (Figure 1). In total, there were 1796 sequences in 17 clusters for which at least one sequence possessed a functional annotation, i.e., only 21.8% of the total 8240 TRAP SBP sequences had a homologue (10–120 or better) with an annotated function, and only 789 sequences (9.6% of the total 8240 SBP sequences) had a homologue (10–120 or better) with a determined structure.
Target Selection and Cloning
We selected 304 TRAP SBP target sequences that did not cluster with existing experimentally determined functions or X-ray structures. These targets were cloned and expressed as either a C-terminally His6-tagged protein with intact periplasmic secretion signal or an N-terminally His10-tagged protein lacking the periplasmic secretion signal (i.e., cytoplasmic localization). Of the 258 targets where cloning attempts were made in both formats, the N-terminally tagged constructs (226 clones in total, no periplasmic sequence) yielded 112 targets with significant expression of highly soluble material (49%) in small-scale cultures (i.e., 0.75 mL), whereas the C-terminally tagged constructs (216 clones in total, periplasmic secretion) yielded 96 targets with significant expression of highly soluble material (44%). These two expression strategies were highly complementary, as 52 and 43 of the TRAP SBPs exhibited significant expression in only the N- or C-terminally tagged vectors, respectively. Of the 248 successfully cloned SBP targets, 171 targets (69%) were highly soluble (i.e., >20 mg mL–1 per liter of culture) on the basis of small-scale expression evaluation; ultimately, large-scale purifications (>5 mg) were successful for 158 (64%) of the cloned SBPs.
DSF of Trap Solute Binding Proteins
DSF was used to screen these targets with a 189 compound library composed of (1) known TRAP SBP ligands (e.g., ectoine, pyroglutamate, malonate, sialic acid); (2) an extensive sublibrary of acid sugars, including 3–6 carbon aldonic (e.g., gluconate), aldaric (e.g., glucarate), and uronic (e.g., glucuronate) acids; (3) a range of aromatic acids (e.g., benzoic acid, coumarate); and (4) a range of l- and d-amino acids (Table 1). The majority of the targets exhibited reproducible two-state thermal denaturation curves. All of the thermal transitions were within the temperature range of 25–95 °C, with >90% of the curves exhibiting Tm > 45 °C and low initial fluorescence values, indicative of well-folded proteins with a propensity to crystallize.36 Visual inspection of individual results, in combination with genome neighborhood analysis and structural results, suggested that ΔTm > 5 °C was an appropriate (and conservative) cutoff to capture the majority of the true positives, while minimizing the false positives. Cases with ΔTm < 5 °C were noted, and attempts were made to utilize the information if corroboratory data (genome neighborhood, homologous proteins with similar hits) were available.
Table 1. Ligands in the DSF Screena.
| Aldoses | Aldonic Acids | Aldaric Acids | Amino Acids | Monosaccharide Amines |
| d+l-threose | d+l-glycerate | d+l-tartrate | d+l-alanine | d-glucosamine |
| d+l-erythrose | d+l-threonate | meso-tartrate | d+l-serine | N-acetylglucosamine |
| d+l-arabinose | d+l-erythronate | meso-xylarate | d+l-threonine | N-acetylneuraminate |
| d+l-lyxose | d+l-xylonate | meso-ribarate | d+l-valine | N-acetylmuramate |
| d+l-xylose | d+l-ribonate | d+l-arabinarate | d+l-leucine | |
| d+l-ribose | d+l-lyxonate | meso-allarate | l-isoleucine | Phenolic Acids |
| d+l-galactose | d+l-arabinonate | meso-galactarate | d+l-phenylalanine | benzoate |
| d+l-glucose | d+l-talonate | d+l-altrarate | d+l-proline | 3-hydroxybenzoate |
| d+l-mannose | d+l-mannonate | d+l-mannarate | d+l-methionine | 4-hydroxybenzoate |
| d+l-idose | d+l-idonate | d+l-idarate | d+l-tryptophan | 3,4-dihydroxybenzoate |
| d+l-gulose | d+l-gluconate | d+l-glucarate | d+l-tyrosine | phenylacetate |
| d+l-talose | d+l-allonate | d+l-aparagine | 3,4-dihydroxylphenylacetate | |
| d+l-altrose | d+l-gulonate | Dicarboxylic Acids | d+l-glutamine | (R,S)-mandelate |
| d+l-allose | d+l-altronate | malonate | d+l-histidine | benzoylformate |
| d+l-fucose | d+l-galactonate | maleate | d+l-aspartate | p-coumarate |
| d+l-fuconate | fumarate | d+l-glutamate | caffeate | |
| Uronic Acids | l-rhamnonate | succinate | d+l-arginine | vanillate |
| d+l-galacturonate | d-gluconate-6P | d+l-malate | d+l-lysine | hydrocinnamate |
| d+l-glucuronate | l-galactonate-6P | 3,4-dihydroxyhydrocinnamate | ||
| d+l-mannuronate | d-glycerate-3P | Monocarbox. Acids | Dipeptides | syringate |
| d+l-iduronate | 6-deoxy-l-talonate | Adipate | l-ala-d-glu | sinapate |
| d+l-guluronate | suberate | d-ala-d-ala | gallate | |
| d+l-alluronate | Keto Acids | tetradecanoate | trans-ferulate | |
| d+l-taluronate | pyruvate | N-caproate | Amino Acid Deriv. | |
| d+l-altruronate | (d,l)-lactate | pyroglutamate | ||
| Polyols | 3OH-proline | |||
| Oligosaccharides | Osmolytes | d-arabitol | 4OH-proline | |
| fructose | glycinebetaine | d-mannitol | 2,6-diaminopimelate | |
| lactose | prolinebetaine | d-meso-ribitol | 5-aminolevulinate | |
| trehalose | 4OH-prolinebetaine | d-meso-galactitol | ||
| raffinose | ectoine | meso-xylitol | ||
| 5-hydroxyectoine | myo-inositol |
Compounds listed as d+l were screened as individual stereoisomers. (d,l)-lactate and (R,S)-mandelate were screened as the racemic mixture.
In the initial DSF screen, a total of 82 targets yielded a ligand hit of 5 °C or better, with 61 targets yielding a ligand hit >10 °C. An additional 7 targets yielded a ligand hit >5 °C in subsequent DSF screening utilizing ligands not in the original screen (Figure 2 and Supporting Information Excel 1). These 89 targets map to 71 unique SSN clusters at 10–120 and 39 unique SSN clusters at 10–80 (Figure 3 and Table S2). These cluster ranged in size from 335 sequences to 1 (singeltons) (Figure 3 and Table S3). The hits were diverse, with the most frequent top hits being aldonic acids (23 instances, e.g., l-gulonate), uronic acids (22 instances, e.g., d-galacturonate), aromatic acids (13 instances, e.g., benzoic acid), and small dicarboxylates (11 instances, e.g., malonate, fumarate). The aldaric acids (e.g., d-glucarate) were never the top hit, but they were observed to bind to SBPs that also bound uronic and aldonic acids. Surprisingly, despite the presence of a carboxylate functionality, the l/d-amino acids were not ligands for the SBPs. As expected, those ligands without a carboxylate moiety, such as the aldoses (e.g., d-glucose), disaccharides (e.g., sucrose), and polyols (e.g., xylitol), yielded no DSF hits.
Figure 2.

Schematic of TRAP SBP ligands determined in this study. These ligands were determined either by DSF and/or were co-purified (CO) ligands observed by crystallography. Co-purified ligands that were confirmed by X-FTMS or ESI-FTMS are marked by (MS). Those ligands that are novel for the TRAP SBP family, as defined by this study, are indicated by an asterisk.
Figure 3.

Annotated 10–120 TRAP SBP SSN network with data from this study. Targets are colored by ligand(s). The ligands determined prior to this study are shown by the smaller colored nodes (also visualized in Figure 1), whereas those determined here are shown by larger colored nodes. Sequences with PDB structures are shown as diamonds with red borders, labeled with the PDB ID. See Table S2 for a mapping of ligand, cluster number, and color and Table S3 for the number of sequences that map to those clusters.
Some targets hit a single compound, while others hit on a number of related compounds. Targets with multiple hits afforded insights in stereochemical preferences and selectivity. For example, the top DSF hit for BH2673 was d-gluconate (ΔTm = 9.8 °C), but BH2673 was also stabilized by four other aldonic acids (ΔTm’s ranging from 2.6 to 8.1 °C) (Figure 4). As the ligand library contains all stereoisomers of the 4, 5, and 6 carbon aldonic acids and these other variants were not stabilizing ligands (for example l-gluconate ΔTm = 0.2 °C), these data suggest that the carboxylate and the stereochemistry of the hydroxyls at positions 2 and 5 are the most critical to aldonic acid recognition by BH2673.
Figure 4.

DSF of the TRAP SBP BH2673 from Bacillus halodurans. Denaturation of BH2673 as a function of temperature as observed by increase in fluorescence of the indicator dye SYPRO Orange, which binds nonspecifically to hydrophobic surfaces. At higher temperatures, the intrinsic fluorescence degrades due to the formation of protein aggregates and dye dissociation. Ligands and their calculated ΔTm’s are shown. The light blue circles map where the stereochemistries of the C2–C5 hydroxyls of the weaker hits differ from that of the top hit, d-gluconate.
The DSF results were important not only for determining SBP ligand specificity but also provided ligands for subsequent co-crystallization studies to define the determinants responsible for binding specificity.
Structural Characterization
Of the 100 proteins subjected to crystallization trials, 64% produced diffraction-sized crystals, with those setup with a DSF ligand crystallizing modestly better (44 of 62, 71%) than those setup without a ligand (20 of 38, 53%). A total of 60 structures (42 unique SBPs) were determined with an average resolution of 1.7 Å (Table 2 and Supporting Information Excel 2). This significantly enhances the structural coverage of the TRAP SBPs, with 2437 sequences exhibiting 40% or better identity to the newly determined structures.
Table 2. Crystal Structures Determined of TRAP Solute Binding Proteins.
| UniProt | locus tag | cluster 10–80 (10–120) | PDB | res (Å) | ligand | UniProt | locus tag | cluster 10–80 (10–120) | PDB | res (Å) | ligand |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Q9HVH5 | PA4616 | 1(3) | 4NF0 | 1.85 | l-malate | Q5LSJ5 | SPO1773 | 18(279) | 4PAF | 1.6 | 3,4-dihydroxybenzoate |
| Q2IUT5 | RPB_3329 | 1(3) | 4O94 | 2 | succinate | Q5LSJ5 | SPO1773 | 18(279) | 4PAI | 1.4 | 3-hydroxybenzoate |
| A3QCW5 | Shew_1446 | 1(3) | 4O7M | 1.5 | l-malate | Q5LSJ5 | SPO1773 | 18(279) | 4PBH | 1.2 | benzoic Acid |
| A3QCW5 | Shew_1446 | 1(3) | 4OA4 | 1.6 | succinate | A3PQU4 | Rsph17029_3620 | 20(6) | 4PE3 | 1.35 | APO (Zn) |
| Q8ECK4 | SO_3134 | 1(3) | 4MX6 | 1.1 | succinate | Q21XD7 | Rfer_1840 | 24(35) | 4MCO | 1.6 | malonate (SPG1) |
| D1AZL7 | Sdel_0447 | 1(9) | 4OVS | 1.8 | succinate | Q21XD7 | Rfer_1840 | 24(35) | 4MEV | 1.8 | malonate (SPG2) |
| Q48AL6 | CPS_0129 | 2(15) | 4PET | 1.9 | calcium-pyruvate | A1U4I5 | Maqu_2829 | 26(639) | 4PFI | 2.3 | APO |
| Q0KC03 | H16_A1328 | 5(12) | 4P8B | 1.3 | (S)-2-acetolactate | Q7WJQ1 | BB2442 | 29(350) | 4P56 | 1.9 | (R,S)-mandelate |
| Q7WGZ0 | BB3421 | 7(98) | 4NQ8 | 1.5 | R-pantoate | B7LRA7 | EFER_1530 | 30(269) | 4P1E | 1.9 | APO |
| A6X7C5 | Oant_4429 | 7(100) | 4P47 | 1.3 | APO (C-terminus) | Q1QZT7 | Csal_0660 | 36(104) | 4N8G | 1.5 | d-Ala-d-Ala |
| Q12CD8 | Bpro_1871 | 7(123) | 4PDH | 1.8 | d-erythronate | Q311Q1 | Dde_1548 | 36(541) | 4NGU | 2.5 | d-Ala-d-Ala |
| A1WPV4 | Veis_3954 | 7(123) | 4P9K | 1.4 | d-erythronate | Q16C67 | RD1_0742 | 39(107) | 4PF6 | 1.75 | KDO |
| A1WPV4 | Veis_3954 | 7(123) | 4PAK | 1.2 | R-pantoate | Q8RE65 | FN1258 | 45(87) | 4PFB | 2.7 | sn-glycerol 3-phosphate |
| Q12HD7 | Bpro_0088 | 7(219) | 4PDD | 1.7 | d-erythronate | C6BWG5 | Desal_0342 | 45(277) | 4N6K | 1.2 | diglycerol phosphate |
| A5E8D2 | BBta_0128 | 9(4) | 4N8Y | 1.5 | d-galacturonate | C6C297 | Desal_3247 | 52(171) | 4N6D | 1.7 | APO (C-terminus) |
| Q128M1 | Bpro_3107 | 9(4) | 4MIJ | 1.1 | d-galacturonate | Q315G1 | Dde_0634 | 52(1677) | 4NAP | 2.3 | d-tryptophan |
| Q128M1 | Bpro_3107 | 9(4) | 4MHF | 1.46 | d-glucuronate | Q315G1 | Dde_0634 | 52(1677) | 4PGN | 1.8 | 3-indole pyruvate |
| A8AR30 | CKO_04899 | 9(44) | 4NG7 | 2.3 | APO | Q315G1 | Dde_0634 | 52(1677) | 4PGP | 2.25 | 3-indole acetate |
| Q0B2F6 | Bamb_6123 | 9(65) | 4LN5 | 2.1 | APO | A3T0D1 | NAS141_03721 | 64(267) | 4NX1 | 1.6 | d-taluronate |
| Q0B2F6 | Bamb_6123 | 9(65) | 4N15 | 1.65 | d-glucuronate | A3T0D1 | NAS141_03721 | 64(267) | 4OVP | 1.7 | d-manuronate |
| Q0B2F6 | Bamb_6123 | 9(65) | 4N17 | 1.5 | d-galacturonate | C9MHP2 | HICG_00826 | 66(223) | 4PBQ | 1.65 | l-gulonate |
| C7RDZ3 | Apre_1383 | 9(161) | 4N91 | 1.7 | d-glucuronate | C6BW16 | Desal_2161 | 67(153) | 4NN3 | 1.4 | orotic acid |
| Q1QUN2 | Csal_2479 | 9(186) | 4P1L | 1.7 | d-glucuronate | Q16BC9 | RD1_1052 | 71(97) | 4PC9 | 1.3 | d-mannonate |
| Q1QUN2 | Csal_2479 | 9(186) | 4P3L | 1.8 | d-glucuronate | Q16BC9 | RD1_1052 | 71(97) | 4PCD | 1.7 | l-galactonate |
| Q160Z9 | RD1_3994 | 9(319) | 4OVQ | 1.5 | d-glucuronate | Q122C7 | Bpro_4736 | 74(614) | 4MNC | 1.05 | benzoylformate (SPG1) |
| A3T0C3 | NAS141_03681 | 9(452) | 4PF8 | 1.5 | d-galacturonate | Q122C7 | Bpro_4736 | 74(614) | 4MNI | 1.9 | benzoylformate (SPG2) |
| A7IKQ4 | Xaut_3368 | 9(790) | 4OVR | 1.65 | d-galacturonate | Q7WPG5 | BB0719 | 154(353) | 4N4U | 1.57 | APO |
| Q2IWM2 | RPB_2686 | 14(1) | 4OAN | 1.35 | (S)-2-acetolactate | B8J100 | Ddes_1525 | 157(244) | 4NHB | 1.9 | sn-glycerol 3-phosphate |
| Q1QZR9 | Csal_0678 | 16(33) | 4UAB | 1.4 | ethanolamine | A3PQL6 | Rsph17029_3541 | 209(378) | 4PFR | 2.6 | APO |
| A6X5V3 | Oant_3902 | 17(49) | 4OVT | 1.8 | l-fuconate | ||||||
| A6VKP1 | Asuc_0158 | 17(197) | 4O8M | 1.7 | l-galactonate |
As observed previously, the SBPs exhibit a highly conserved fold, with two alpha-beta domains (domain 1 (residues ∼1–143 and 236–241) and domain 2 (residues ∼149–232)) connected by two adjoined coil linkers and a C-terminal helical wrapper (Figure S2).37 The relative orientation of the two domains is dictated by the reorganization/bending of both the large abutting α helix and the two adjoined coil linkers. The highly conserved arginine, responsible for the superfamily’s preference for organic acids, is located at the end of strand 6 in domain 2. Typically, domain 2 and the adjoining coil linkers participate in the majority of the interactions with the ligand carboxylate and atoms proximal to the carboxylate, whereas domain 1 is responsible for recognition of the more distal portions of the ligand. Eighty percent of the structures were determined to be in a closed form, with the bound ligands being fully inaccessible to solvent and exhibiting an average thermal factor typically 20–50% lower than that of the overall structure. Crystals produced in the presence of a DSF ligand always yielded the structure of the anticipated SBP–ligand complex. Nine SBPs crystallized in the absence of a DSF-identified ligand yielded APO structures, only one of which was in a closed conformation appropriate for molecular modeling (Bamb_6123, PDB ID 4LN5). Notably, the structures of 10 additional SBPs, crystallized in the absence of added small molecules, revealed fortuitously bound ligands derived from the E. coli expression host, none of which were represented in the original DSF screening library (see below).
Overall, these structures identify the determinants responsible for specificity, stereoselectivity, and promiscuous recognition within the TRAP SBP family. Here, we detail a select set of the DSF and structural results and highlight an emerging strategy, which leverages DSF, crystallography, and mass spectrometry, for the discovery of new metabolites and new metabolism.
Platform Validation with Known SBP Ligands from Known Clusters
While SBPs with known ligands were not the targets of this study, two targets with high sequence identity to experimentally annotated proteins served to validate the DFS platform. An SBP from Sinorhizobium meliloti (SM_b20036, cluster 100) was previously reported to bind d-quinic acid with high affinity (Kd = 5.9 nM).38 A homologue (SeqID = 78%) from cluster 100, Oant_4429 from Ochrobactrum anthropi, which had no hits in the standard DSF screen (quinic acid, shikimic acid not in screen), was significantly stabilized by d-quinic acid (ΔTm = 21.1 °C) and not by the related molecule shikimic acid (ΔTm = −0.4 °C).
A second TRAP transport system from S. meliloti (Sma0250-Sma0252) was shown to be involved in d-gluconate utilization by gene knockout and complementation studies (cluster 43).39 A homologue (SeqID = 88%) from A. tumefaciens (Atu2744) gave multiple DSF hits with 6-carbon aldonic acids such as d-gluconate and d-allonate (both ΔTm = 7.5 °C). These two examples served as initial controls to validate the experimental methods.
Known TRAP Ligands, Novel Clusters
Several known SBP ligands, including malate, succinate, fumarate, pyruvate, lactate, ectoine, malonate, and pyroglutamate, were mapped in this study to previously uncharacterized clusters, some of which are only distantly related to the previously characterized SBPs. Here, we discuss three select examples.
The protein SMa0157 from S. meliloti (cluster 17, 29 seq (sequences)) has been identified as a malonate SBP by gene knockouts of chromosomally colocated malonate catabolic genes.40 In this study, Mrad2831_2909 from Methylobacterium radiotolerans and Rfer_1840 from Rhodoferax ferrireducens, both from cluster 35 (40 seq), had DSF hits on malonate and succinate. The structure of Rfer_1840 was determined in complex with malonate (PDB ID 4MCO). Despite only moderate sequence identity between cluster 17 and 35 (∼38%), the majority of the malonate binding determinates are conserved (data not shown), and several genes annotated as putative malonate catabolic genes are chromosomally colocated with Rfer_1840.
TRAPs specific for the transport of the osmoprotectants ectoine and 5-hydroxyectoine have previously been described from Silicibacter pomeroyi and Halomonas elongata (both from cluster 13, 27 seq).41−44 In this study, Bsel_1187 from Bacillus selenitireducens (cluster 1038, 1 seq), BH3390 from Bacillus halodurans (cluster 389, 2 seq), and Csal_3140 from C. salexigens (cluster 254, 5 seq) all had DSF hits on an ectoine/5-hydroxyectoine mixture. Deconvolution of the ectoine/5-hydroxyectoine mix indicated that ectoine afforded a 6–8 °C greater stabilization than 5-hydroxyectoine. These organisms all possess homologues of genes involved in ectoine biosynthesis, although not in the genome context of these newly discovered ectoine TRAP transporters, which share low sequence identity (32–37% SeqID) with the previously characterized transporters.
Desal_3221 from Desulfovibrio salexigens (cluster 946), BBta_4928 from Bradyrhizobium strain BTAi1 (cluster 124, 2 seq), Rho54_34890 from Rhodobacter sphaeroides (cluster 190, 8 seq), and Rsc2166 from Ralstonia solanacearum (cluster 126, 12 seq) gave DSF hits only with pyroglutamate. The two known pyroglutamate SBPs,45 both from Bordetella pertussis (BP1891 from cluster 31 and BP1887 from cluster 166, 54 total seq), exhibit low sequence identity (∼30%) to these newly characterized pyroglutamate SBPs. Chromosomally adjacent to Rsc2166 is a gene annotated as a pyroglutamyl peptidase (Rsc2165), which we anticipate would catalyze the release of N-terminal pyroglutamyl groups from pyroglutamate-capped polypeptides; also, chromosomally adjacent to Desal_3221 (Desal_3219–20) are genes annotated as the A and B subunits of 5-oxoprolinase, which would cleave pyroglutamate to form glutamate.
Selected Novel TRAP SBP Ligands
This work identified 40 new TRAP SBP ligands; we highlight a few examples that were identified by DSF.
d-Glucuronate/d-Galacturonate SBPs
d-Galacturonate is the main monomeric constituent of pectins present in the primary cell wall of plants. d-Glucuronate is common in carbohydrate chains of proteoglycans in animals and is also found in hemicelluloses in plant cell walls. Many bacteria can utilize d-galacturonate and/or d-glucuronate using one of the several known variants of hexuronic acid catabolic pathways.46 In E. coli, hexuronates are taken up via secondary transporters from the major facilitator (MFS) gluconate:H+ symporter (GntP) families; however, the mechanisms of hexuronate uptake in other bacteria were largely unknown prior to this study.
SBPs from clusters 5, 161, and 585 hit only on d-glucuronate (84 associated seq), whereas proteins from clusters 4, 30, 44, 47, 65, 186, 310, 319, 452, and 790 hit on both d-glucuronate and d-galacturonate (502 associated seq). Comparative genomics reconstruction of catabolic pathways and regulons that included SBPs from these clusters is consistent with their DSF-determined ligand specificities. For example, prior to this study, the d-glucuronate-specific SBPs Asuc_0146 from Actinobacillus succinogenes (cluster 5) and Apre_1383 from Anaerococcus prevotii (cluster 161) were predicted (but not experimentally confirmed) to be coregulated with genes from the d-glucuronate catabolic pathway via transcriptional regulators from the LacI and GntR families, respectively.47,48 The DSF results provide the first direct biochemical demonstration that TRAP SBPs recognize hexuronic acids.
A similar analysis of Bpro_3107 from Polaromonas sp. (cluster 4), BBta_0128 from Bradyrhizobium sp. (cluster 4), Pput_1203 from Pseudomonas putida (cluster 47), and Bamb_6123 from Burkholderia ambifaria (cluster 65) is consistent with these TRAPs belonging to predicted regulons and/or operons encoding activities for catabolic pathways that use a uronate dehydrogenase and a lactone hydrolase to produce the diacid sugars, meso-galactarate and d-glucarate. These are diverse pathways; a representative predicted pathway for uronic acid utilization by Polaromonas sp. is shown in Figure 5A. Further analysis to confirm the predicted uronic acid catabolic enzymes downstream of the SBPs from these clusters is in progress.
Figure 5.

Functional implications from d-glucuronate/d-galacturonate TRAP SBPs. (A) Genome context of Bpro_3107, a TRAP SBP shown to bind d-galacturonate and d-glucuronate by DSF, and the encoded catabolic pathway predicted to convert these ligands to central metabolites. The newly annotated uronate dehydrogenase (Udh) and lactonase (UxuL) are shown in indigo and yellow, respectively. Gene annotations are listed as obtained from KEGG. The TctC proteins are the SBP component of the TTT family of transporters, whose colocalization within this operon/regulon suggests alternative entry points into the pathway, perhaps at galactarate or glucarate. (B) Interactions of the related TRAP SBP, Apre_1383 with d-glucuronate. Hydrogen bonds are shown as dashed lines, and hydrophobic contacts are represented by an arc with spokes radiating toward the ligand atoms that they contact. (C) Interactions of the TRAP SBP Bamb_6123 with d-galacturonate. Residues that interact with the ligand in an analogous fashion within panels B and C are shown in bold.
A total of 13 structures from eight different clusters were determined, most in complex with the cyclic form of either d-glucuronate and/or d-galacturonate. These structures revealed several conserved features, including (1) coordination of the uronic acid carboxylate by the conserved arginine (Arg169, Bamb_6123 numbering), (2) coordination by a second arginine (Arg149) of one of the endocyclic oxygen lone pairs and a carboxylate oxygen, and (3) hydrogen bonding from an asparagine (Asn209) to the second endocyclic oxygen lone pair and to O1 when it is in the alpha conformation (Figure 5B,C). Since these data are associated with a large number of sequence-related TRAP SBPs (586 seq) and catabolic pathways, the uronic acid DSF and structural results have the largest annotation reach of this study.
6-Carbon Aldonic Acid SBPs
Several SBPs from clusters 27, 197, 223, and 403 (80 associated seq) were stabilized by either l-gulonate or l-galactonate. On the basis of these DSF hits, a comparative genomics reconstruction resulted in the annotation of two nonorthologous clusters of Zn-dependent l-gulonate 5-dehydrogenases (Cog1063). We predict that the SBP proteins HICG_00826 from Haemophilus influenzae and MHA_0480 from Mannheimia hemolytica (cluster 223) belong to an l-gulonate catabolic pathway/regulon that contains a previously unannotated l-gulonate 5-dehydrogenase (HiGul5DH/MhGul5DH, l-gulonate to d-fructuronate) and all downstream enzymes for utilization of d-fructuronate (Figure 6A). Gul5DH activity was first observed in E. coli in 1980; however, the gene/protein was never identified.49 Recently, we purified and annotated an authentic Gul5DH from C. salexigens, CsGul5DH;30 however, CsGul5DH is from a nonorthologous Cog1063 cluster (SeqID 33% to HiGul5DH), although it is part of an analogous pathway for the degradation of l-gulonate.
Figure 6.

Functional implications from l-galactonate/l-gulonate TRAP SBPs. (A) Genome context of HI0052, a TRAP SBP shown to bind l-gulonate by DSF, and the encoded catabolic pathway that would convert l-gulonate to glycerol 3-phosphate and pyruvate. The newly annotated l-gulonate 5-dehydrogenase is shown in pink. Gene annotations are listed as obtained from KEGG. (B) Genome context of Asuc_0158, a TRAP SBP shown by DSF to bind l-galactonate, and the encoded catabolic pathway that would convert l-galactonate to glycerol 3-phosphate and pyruvate. The newly annotated l-galactonate 5-dehydrogenase is shown in pink. (C) Interactions of the related TRAP SBP, HICG_00826 with l-gulonate. Hydrogen bonds are shown as dashed lines, and hydrophobic contacts are represented by an arc with spokes radiating toward the ligand atoms that they contact. (D) Interactions of the TRAP SBP Asuc_0158 with l-galactonate. Residues that interact with the ligand in an analogous fashion within panels C and D are shown in bold.
In contrast, the l-galactonate DSF hit and comparative genomics reconstruction of Asuc_0158 from Actinobacillus succinogenes (Cluster 197) and related SBPs suggests an l-galactonate catabolic pathway/regulon that includes a previously unannotated l-galactonate 5-dehydrogenase (AsLga5DH, l-gulonate to d-tagaturonate), also from Cog1063, and the downstream enzymes for utilization of d-tagaturonate (Figure 6B). Subsequently, an orthologous Lga5DH from E. coli (YjjN, EcLga5DH, 66% SeqID) was reported to have Gal5DH activity, consistent with the proposed pathway.51 The structures of complexes of HICG_00826 with l-gulonate (1.65 Å resolution) and Asuc_0158 with l-galactonate (1.7 Å resolution) demonstrate the two divergent aldonic acid SBPs coordinate the α-hydroxy-acid group similarly with two arginines (Arg168 and Arg148, HICG_00826 numbering) and an asparagine (Asn208), while a spatially conserved glutamate (Glu73) coordinates the C3 and C4 hydroxyls in HICG_00826 and the C4 and C5 hydroxyls in Asuc_0158 (Figure 6C,D). As with the hexuronic SBP example, DSF and comparative genomics reconstructions allows for the prediction of annotations and novel pathways for explicit experimental validation.
d-Ala-d-Ala SBP
Several SBPs from clusters 104 (29 seq) and 541 (2 seq) have a putative VanX (d-alanyl-d-alanine dipeptidase) in their genomic context (Figure 7A). The SBPs Csal_0660 (cluster104), PA3779 (cluster104), and Dde_1548 (cluster 541) exhibited significant and highly specific stabilization by d-Ala-d-Ala (13.5–15.7 °C). The structures of the complexes of Csal_0660 and Dde_1548 to d-Ala-d-Ala were determined to 1.5 and 2.5 Å resolution, respectively, and revealed similar recognition elements for the dipeptide. In particular, all dipeptide hydrogen-bond donors and acceptors are satisfied, with the carboxylate of the d-Ala-d-Ala forming an ionic interaction with the conserved arginine (Figure 7B). In addition to the presence of VanX in the immediate genome neighborhood of Csal_0660 and Dde_1548, Dde_1548 has an alanine racemase (Dde_1547) and alanine dehydrogenase (Dde_1553) in its genome neighborhood, as would be expected for the conversion of d-Ala-d-Ala to NH3+, pyruvate, and reduced NADH/NADPH.
Figure 7.

Functional implications from d-Ala-d-Ala TRAP SBPs. (A) Genome environment of the three TRAP SBPs that had DSF hits on the dipeptide d-Ala-d-Ala. Genes putatively assigned for the transport and catabolic degradation of d-Ala-d-Ala are shown in color. (B) Interactions of Csal_0660 with d-Ala-d-Ala. Hydrogen bonds are shown as dashed lines, and hydrophobic contacts are represented by an arc with spokes radiating toward the ligand atoms that they contact. (C) 1H NMR verification of CsVanX (Csal_0663) dipeptisase activity on d-Ala-d-Ala. The control spectrum is show on the bottom, and the reaction is shown on top (glycerol from the enzyme prep is present between 3.6 and 3.4 ppm). Insets show magnifications of the control and reaction peaks. (D) Fold change in transcript measured by qRT-PCR for Csal_0660 genome neighborhood related genes when C. salexigens is grown on d-Ala-d-Ala versus d-glucose as a carbon source. (E) Growth curves of wild-type C. salexigens versus a deletion mutant of the d-Ala-d-Ala TRAP SBP (ΔCsDctP) or deletion mutant of the d-Ala-d-Ala dipeptidase (ΔCsVanX).
The VanX found in the genome context of Csal_0660 is only 33% identical to the most similar VanX of confirmed activity. Using purified Csal_0663, d-alanyl-d-alanine dipeptidase activity was verified by 1H NMR (Figure 7C). On the basis of this observed activity and genome context, we propose that the VanX found in C. salexigens is utilized in d-Ala-d-Ala metabolism rather than its canonical role in vancomycin resistance.52,53 Although this function has been previously suggested for a subset of VanX homologues, growth on d-Ala-d-Ala has been observed only when E. coli VanX and an associated cluster of dipeptide permease genes were overexpressed on a high-copy plasmid, but not in wild-type E. coli.54 In contrast, we demonstrated that C. salexigens can utilize d-Ala-d-Ala as sole carbon source when VanX expression is driven by the wild-type chromosomal gene. Upon a shift to d-Ala-d-Ala as carbon source, qRT-PCR analysis of the Csal_0660 genome neighborhood showed 50–150-fold upregulation of the TRAP transporter genes and the VanX gene (Figure 7D). Furthermore, C. salexigens mutants harboring deletions of either the C. salexigensd-Ala-d-Ala SBP or VanX were unable to utilize d-Ala-d-Ala as a carbon source (Figure 7E). These observations verify the physiological role of this TRAP transporter and VanX as the primary means of d-Ala-d-Ala metabolism in C. salexigens.
SBPs with Co-purified Ligands
The above examples demonstrate the significant insights that DSF-based screening can provide for ligand discovery and, in particular, highlight the value of SBPs for defining new metabolic function. Our screening library, however, does not contain all potential metabolites/ligands. Of particular interest in this regard are the 10 SBP structures that revealed adventitiously bound ligands derived from the expression host, which provides a means for overcoming the limited diversity of the DSF screening library. These adventitiously bound ligands were not represented in the library and include (R)-pantoate, 3-deoxy-d-manno-oct-2-ulosonic acid, orotic acid, glycerol 3-phosphate, diglycerol phosphate, 2-acetolactate, indole pyruvate, and ethanolamine. Several of these crystallographically identified ligands were confirmed by two high-resolution Fourier transform mass spectrometry (FTMS) approaches. The first utilized soluble ligands generated by methanol extraction of the protein–ligand complexes (X-FTMS). The second couples FTMS with soft ionization techniques to preserve the integrity of the complexes and allow for the indirect determination of the ligand mass by measuring the mass of the intact protein–ligand complex (ESI-FTMS).26−28
3-Deoxy-d-manno-oct-2-ulosonic Acid (KDO) SBP
The 1.75 Å resolution structure of a previously uncharacterized SBP, RD1_0742 (cluster 107, 16 seq) from R. denitrificans (no DSF hits), revealed electron density suggestive of 3-deoxy-d-manno-oct-2-ulosonic acid (KDO) (Figure 8), an integral component of the oligosacharide core of the lipopolysaccharide (LPS) outer membrane of all Gram-negative bacteria. High-resolution mass spectroscopy of the methanol-extracted ligands from E. coli-expressed RD1_0742 yielded a molecular weight consistent with KDO (Figure S3). A colocated gene (RD1_0744; Figure 8), annotated as a putative dihydrodipicolinate synthase (dapA), is predicted by association, therefore, to be a KDO aldolase, which produces d-arabinose and pyruvate as products. This reaction is analogous to that catalyzed by another dapA family member, N-acetylneuraminate (NANA) aldolase, which catalyzes the transformation of NANA to N-acetyl-d-mannosamine and pyruvate.55 A member of the PfkB sugar kinase family (RD1_0745) is divergently transcribed from the putative KDO aldolase and is by association, therefore, proposed to phosphorylate d-arabinose produced in the KDO cleavage reaction, which is isomerized to d-ribulose-5-phosphate. KDO did not support growth; however, cells exposed to supplemented KDO exhibited upregulation of the PfkB sugar kinase and KDO aldolase ∼10–15-fold, as measured by qRT-PCR, consistent with the hypothesis that these enzymes are functionally linked to the TRAP transporter, although for some purpose other than central metabolism (Figure S4).
Figure 8.

TRAP SBP co-purified ligands. Omit maps for adventitiously bound ligands contoured at 3 RMSD and the associated TRAP SBPs genomic environment. The genome environment for Dde_0634 was substituted with that of the related TRAP transporter from Geobacter sulfurreducens (KN400_2073-75), for which more genes were colocated. Gene annotations are those found in KEGG. Co-purified ethanolamine is shown in Figure 9.
sn-Glycerol 3-Phosphate, Diglycerol Phosphate SBPs
The 1.9 Å resolution structure of Ddes_1525 from Desulfovibrio desulfuricans (Cluster 244, no DSF hits) exhibited electron density consistent with sn-glycerol 3-phosphate (G3P) (Figure 8). Examination of purified Ddes_1525 by ESI-FTMS yielded a protein–ligand complex mass consistent with that of G3P (Figure S5). The immediate genome neighborhood of Ddes_1525 contains proteins annotated as a 3-component anaerobic glycerol-3-phosphate-dehydrogenase (GlpABC, Ddes_1529–31) and a HAD family phosphatase (Ddes_1528), consistent with Ddes_1525 being involved in the transport and utilization of G3P (Figure 8).
The 1.2 Å resolution structure of an unrelated TRAP SBP (<25% seqID), Desal_0342 from D. salexigens (cluster 277, DSF no hits, 4 seq), revealed electron density consistent with diglycerol phosphate (Figure 8), which was confirmed by mass spectroscopy of methanol extracted ligands (Figure S3). DGP accumulates under salt stress in the archaeon Archaeoglobus fulgidus and has been shown to have protein thermostabilization properties.56,57 Although D. salexigens has not been reported to utilize DGP as an osmoprotectant, D. salexigens has an absolute requirement for at least 0.5% NaCl (w/v) for growth and can grow at NaCl concentrations exceeding 10%.58 Therefore, we hypothesize that DGP may be used by D. salexigens as an osmolyte. Directly adjacent to Desal_0342 on the genome is an annotated glycerophosphoryl diester phosphodiesterase (Desal_0339), which could hydrolyze DGP to glycerol and glycerol 3-phosphate (Figure 8), suggesting that DGP could be used as a source of carbon or phosphate under nutrient-limiting conditions.59,60 Interestingly, DGP has not been reported as a known metabolic intermediate of E. coli, although clearly it was available for interaction with Desal_0342. Mass spectrometry of small molecule extracts of the E. coli expression host (BL21(DE3) RIL), without an expression plasmid, revealed a species of molecular mass equivalent to that of DGP (Figure S3). We speculate that DGP may be released from glycerophospholipids by the action of nonspecific phospholipases upon sonication; however, we cannot rule out the existence of specific metabolic pathways involving DGP.
Homology modeling of the sequence-related protein FN1258 (SeqID = 37.5%) from Fusobacterium nucleatum (cluster 87, no DSF hits, 32 seq) based on the structure of Desal_0342 suggested that the cognate ligand of FN1256 was G3P instead of DGP. Indeed, X-FTMS on methanol extracted ligands derived from E. coli-expressed FN1258 exhibited a peak consistent with that of G3P (Figure S3). A species of similar mass was found by ESI-FTMS of the complex (Figure S5). Subsequently, the structure of FN1258 was determined to 2.7 Å resolution from co-crystals containing glycerol 3-phosphate.
The structures of these three G3P/DGP SBPs reveal the basis for their specificity (Figure S6). In all three cases, the phosphate moieties are coordinated by two arginines, Arg168 (the conserved arginine, Desal_0342 numbering) and Arg154. Additional ionic interactions involving Lys32 in FN1258 and Lys37 in Ddes_1525 are consistent with FN1258 and Ddes_1525 favoring the G3P dianion over the DGP monoanion (confirmed by DSF with G3P and DGP, Supporting Information Excel 1). However, FN1258 and Ddes_1525 bind G3P in entirely different orientations, with the G3P glycerol moiety in FN1258 lying near the opening of the “venus fly trap” and making interactions similar to those in Desal_0342, whereas the glycerol of the G3P of Ddes_1525 is buried in a nearly orthogonal pose at the bottom of the “venus fly trap”.
2-Acetolactate SBP
The structures of RPB_2686 from Rhodopseudomonas palustris (1.3 Å resolution, cluster 1, no DSF hits, 142 seq) and H16_A1328 from Ralstonia eutropha (1.35 Å resolution, cluster 12, no DSF hits, 83 seq) revealed residual electron density consistent with bound 2-acetolactate (Figure 8), a precursor in the biosynthesis of the branched chain amino acids. Despite sharing only 31% sequence identity, these two proteins exhibit remarkably similar binding modes for 2-acetolactate (see Figure S7 for details). Repeated tests to confirm the copurification of 2-acetolactate by MS were unsuccessful using methanol-extracted ligands; however, a mass consistent with a 2-acetolactate–protein complex was observed by ESI-FTMS of RPB_2686 (Figure S5). The relatively high resolution of the structure determination and the ESI-FTMS of the complex strongly support the current assignment of (S)-2-acetolactate; however, further in vivo work is required to ascertain the TRAPs physiological role, as the genome neighborhoods appear to lack strong functional indications. For example, the genome neighborhood of H16_A1328 includes genes putatively annotated for the degradation of poly(3-hydroxyalkanoates), whereas that of RpB_2686 only has an Acyl-CoA synthetase in context (Figure 8).
Orotic Acid SBP
The structure of Desal_2161 (cluster 153, 6 seq) from Desulfovibrio salexigens was determined to 1.4 Å resolution, with clear electron density for a co-purified ligand similar to that of 3,5-benzoic acid (Figure 8). However, the structure revealed potential H-bonds involving Asn212OD1 and Glu37OE2 to the putative carbon atoms at endocyclic positions 2 and 4, suggesting that the ligand was most likely orotic acid. Although MS of Desal_2161 small molecule extracts did not show orotic acid, a ligand–protein complex mass consistent with that of bound orotic acid was found by ESI-FTMS (Figure S5). DSF analysis yielded ΔTm’s of 15.6 °C for orotic acid and 12.1° for dihydroorotic acid. Interestingly, the genomic environment of Desal_2161 suggests the presence of both catabolic and anabolic pathways for orotic acid (see Figure 8 for gene environment).
Indole Acids SBP
d- and l-amino acids did not result in significant hits in the DSF screen for any SBPs; however, d-tryptophan (d-Trp) gave modest stabilization (4.1–4.9 °C) to three related proteins from two clusters (167 and 171, 12 total seq), suggesting the weak recognition of a noncognate ligand. The structure of Dde_0634 from D. desulfuricans bound to d-Trp (10 mM in co-crystallization) was determined to 2.3 Å resolution. The conserved arginine (Arg173NH1) is hydrogen-bonded to only one of the d-Trp carboxylate oxygens and forms an apparently unfavorable interaction with the d-Trp α-amino group (Arg173NH2, 3.5 Å) (Figure S8). This structure resulted in a second round of DSF experiments with ligands possessing enhanced hydrogen-bonding potential, including 3-indolelactate (3-ILA, ΔTm = 18.8 °C), 3-indolepyruvate (3-IPA, ΔTm = 15.7 °C), and 3-indoleacetate (IAA, ΔTm = 13.3 °C), in which the incompatible amine was replaced by an oxygen, resulting in greater thermal stabilizations, suggestive of stronger binding.
Concurrently, it was observed that isomorphous crystals could be grown without added ligand, allowing for an apo structure to be determined to 1.8 Å resolution. Clear density was observed for co-purified 3-IPA (Figure 8), which was confirmed by mass spectrometry of the methanol-extracted ligand (Figure S5). In contrast to the d-Trp complex, the conserved arginine in the adventitious Dde_0634 3-IPA complex and a Dde_0634 3-ILA complex generated by co-crystallization were within proper H-bonding distance to the keto/carboxylate group of the ligands (∼2.9 Å, see Figure S8 for details). The genome environment of the related TRAP transporter from Geobacter sulfurreducens (KN400_2073-75) is consistent with this cluster of transporters binding indole acids (see genome environment Figure 8). This work shows that the identification of even weakly binding, noncognate ligands, when combined with structural analysis, can rapidly lead to the identification of the correct ligand and substantial functional insight.
(R)-Pantoate, d-Erythronate SBPs
The structure of BB3421 from Bordetella bronchiseptica was determined to 1.5 Å resolution (cluster 98, no DSF hits, 21 seq) with residual electron density in the ligand binding site that was initially assigned as (R)-pantoate (Figure 8). R-Pantoate was not observed by MS of methanol-extracted BB3421 ligands, but a ligand–protein complex mass consistent with bound R-pantoate was observed using ESI- FTMS (Figure S5).
A homologous protein, Veis_3954 (Seq ID 46%, cluster 123, 12 seq), from Verminephrobacter eiseniae had a DSF hit with d-erythronate (ΔTm = 11.2 °C), a 4-carbon sugar similar to (R)-pantoate but with a hydroxyl at C3 rather than the dimethyl functionality of (R)-pantoate. The structure of Veis_3954 co-crystallized with d-erythronate was determined at 1.4 Å resolution, revealing residual electron density immediately proximal to C3, suggesting that an additional small molecule had co-purified with Veis_3954, which, by analogy with BB3421, was postulated to be (R)-pantoate (Figure 8). Mass spectrometry of methanol-extracted Veis_3954 ligands indicated that (R)-pantoate (Figure S3) had co-purified with Veis_3954, and a crystal structure with exogenously supplied (R)-pantoate exhibited electron density consistent with only (R)-pantoate. When DSF was performed on these proteins with (R)-pantoate (not in original screen), only BB3421 was specific for (R)-pantoate, whereas Veis_3954 and two other related proteins (Bpro_0088 and Bpro_1873) gave significant hits on both d-erythronate and (R)-pantoate. There are several different gene contexts for the (R)-pantoate/d-erythronate SBPs, although, at present, none are readily interpretable in terms of a catabolic pathway. Although they are not conclusive in terms of the physiologically relevant SBP ligands, the DSF and crystallographic results substantially limit the number of possible starting molecules to test against candidate enzymes in putative catabolic pathways.
Ethanolamine SBP
The structure of Csal_0678 from Chromohalobacter salexigens (cluster 33, no DSF hits, 68 seq) was determined to 1.4 Å resolution with residual density suggestive of a molecule composed of 4 heavy-atoms, such as ethylene glycol (Figure 9A,B). Subsequently, the electron density was assigned as ethanolamine by examining a series of 4 and 5 atom small molecules by DSF. Csal_0678 was stabilized by ethanolamine (ΔTm = 11.7 °C) and dl-alaninol (ΔTm = 9.9 °C), but not by cysteamine (ΔTm = 1.6 °C), R- and S-1-amino-2-propanol (ΔTm = 0.8–1.2 °C), or ethylene glycol (ΔTm = 0.5 °C). As little as 15 μM ethanolamine caused stabilization of 5.6 °C, with >80% of the maximal stabilization occurring at 100 μM ligand (Figure 9C). The polar atoms of the co-purified ethanolamine were assigned on the basis of hydrogen-bonding potential (see Figure 9B for details).
Figure 9.

Functional implications from ethanolamine TRAP SBP. (A) 3 RMSD omit map for a co-purified ethanolamine ligand bound to the TRAP SBP Csal_0678 from Chromohalobacter salexigens. (B) Binding interactions of ethanolamine with Csal_0678. The amine is coordinated by the carbonyl of Trp215 and the side chains of Glu220 and Asp155, whereas the ethanolamine oxygen is coordinated by Tyr241 and Glu220. In Csal_0678, the highly conserved TRAP SBP arginine is replaced by phenylalanine (Phe177), and the position typically occupied by the ligand carboxylate is occupied by the indole group of Trp215. (C) Ligand concentration vs thermal denaturation stabilization of Csal_0678 for various 4 and 5 atom ligands similar to ethanolamine. (D) Genomic environment of the ethanolamine utilization pathway of Chromohalobacter salexigens and Agrobacterium tumefaciens. (E) Details of the chemical transformation of ethanolamine to glycine and the genes previous annotations.
Genes immediately adjacent to the ethanolamine SBP include other components of the TRAP transport system (Csal_0676 and Csal_0677) as well as genes annotated as l-glutamine synthetase (Csal_0679), aldehyde dehydrogenase (Csal_0680), alcohol dehydrogenase (Csal_0681), and an N-formylglutamate amidohydrolase (Csal_0675) (Figure 9D). When ethanolamine was assigned as the cognate ligand of Csal_0678, a possible pathway was readily constructed for the utilization of ethanolamine as a source of nitrogen, leading to functional predictions for the adjacent genes (Figure 9E). Strikingly, these genes are completely distinct from those of the canonical ethanolamine utilization pathway (EUT);61 genomic analyses confirmed that C. salexigens lacks all canonical EUT genes. We propose a catabolic pathway for ethanolamine utilization, terminating in glycine, a source of cellular nitrogen for many bacterial species (Figure 9E). We note that a similar role for l-glutamate as a molecular handle (γ-glutamylation followed by hydrolysis) has been described previously for the degradation of isopropylamine62 and putrescine.63
To confirm this novel pathway for ethanolamine catabolism, we utilized a combination of in vitro enzymology, in vivo microbiology, and metabolomics. C. salexigens was able to utilize ethanolamine or glycine as the sole source of nitrogen (Figure S9); growth on ethanolamine led to upregulation of each of the genes in the proposed ethanolamine utilization pathway (Figure S9). C. salexigens mutants carrying genetic disruptions in either Csal_0678 (SBP component of TRAP transport system) or Csal_0679 (l-glutamine synthetase) were unable to grow with ethanolamine as the sole nitrogen source (Figure S9). Purified Csal_0679 showed robust activity on ethanolamine (using a lactate-dehydrogenase/pyruvate kinase-based coupled-enzyme assay with l-glutamate provided at physiological concentration) with a specificity constant (kcat/KM) of 1.1 × 104 M–1 s–1 (kcat = 0.38 s–1, KM = 34 μM); the specificity constant of l-alaninol was 3× lower (3.4 × 103 M–1 s–1) (Table 3).
Table 3. Kinetic Constants for Csal_0679 a Putative Ethanolamine Gamma-Glutamylase.
| substrate | co-substrate | KM (mM) | kcat (s–1) | kcat/KM (M–1 s–1) |
|---|---|---|---|---|
| l-glutamate | ethanolamine, ATP | 7.8 ± 1 | 0.40 ± 0.02 | 51 |
| ethanolamine | l-glutamate, ATP | 0.034 ± 0.004 | 0.38 ± 0.009 | 1.1 × 104 |
| l-glutamate | l-alaninol, ATP | 5.6 ± 1 | 0.33 ± 0.02 | 59 |
| l-alaninol | l-glutamate, ATP | 0.10 ± 0.01 | 0.35 ± 0.01 | 3.4 × 103 |
We used targeted liquid chromatography Fourier transform mass spectrometry (LC-FTMS) to identify each of the predicted metabolites in the proposed pathway. However, because C. salexigens exhibited relatively slow growth on ethanolamine, we identified another organism with the same pathway that more effectively metabolized ethanolamine. Atu2142, annotated as an l-glutamine synthetase from A. tumefaciens C58, is a close homologue of Csal_0679 (65% SeqID). The genome neighborhood of Atu2142 is nearly identical to that of Csal_0679 (Figure 9D), and, importantly, A. tumefaciens C58 also lacks the canonical EUT pathway for ethanolamine utilization. Interestingly, annotations indicate that A. tumefaciens C58 may utilize an amino acid permease family member (Pfam- PF13520) for ethanolamine uptake (Atu2147) rather than a TRAP transport system. A. tumefaciens C58 was able to utilize ethanolamine or glycine as the sole nitrogen source and exhibited a growth rate on ethanolamine nearly three-times that of C. salexigens (Figure S9). In cell extracts of A. tumefaciens C58 grown with ethanolamine as the sole nitrogen source, we identified each of the predicted metabolites in the proposed pathway (Figure S10), including γ-glutamyl-ethanolamide and γ-glutamyl-glycine.
Discussion
To facilitate the annotation of an ever-growing number of protein sequences, we are focusing on transport systems that utilize a SBP to capture cognate ligands. As these transporters are often coregulated and/or colocated with catabolic and anabolic pathways for these ligands, they provide an immediate toe-hold into the possible functions and intermediates of the associated genes and pathways. We screened 158 TRAP SBPs by differential scanning fluorometry (DSF), identifying 89 ligand-stabilized targets supporting annotations of 2084 TRAP SBPs in 71 isofunctional clusters in the SSN at an e-value of 10–120. As validation of the DSF hits and to define the determinants responsible for ligand recognition, we determined 60 high-resolution crystal structures of 46 unique TRAP sequences, of which 51 contained bound ligands. While the majority of these complexes utilized ligands identified by DSF, 10 structures revealed adventitiously bound ligands derived from the expression host, which persisted through purification and crystallization. These fortuitously bound ligands represent a wide range of chemotypes and all are previously uncharacterized ligands for the TRAP SBP family. High-resolution FTMS approaches provided additional validation for a selected group of these ligands.
The DSF and structural results were leveraged in several ways: (1) These combined efforts discovered 40 novel SBP ligands, which are of considerable use for future annotation efforts. (2) When coupled with genome neighborhood analysis, they demonstrated the utilization of d-Ala-d-Ala as a carbon source by C. salexigens. (3) When coupled with regulon analysis, they allowed the description of novel pathways and activities for the utilization of the uronic acids d-glucuronate and d-galacturonate and the aldonic acids l-gulonate and l-galactonate. These ligands were not previously demonstrated to be bound by TRAP SBPs, but they now appear to be two of the major classes of TRAP SBP ligands. (4) When coupled with crystallography and molecular modeling, they allowed the prediction of the specificity of a distantly related SBP of unknown function (G3P with FN1258 (SeqID = 37.5%) from F. nucleatum). (5) When coupled with crystallography, they demonstrated how a weak DSF hit could be leveraged to identify physiological ligands (indole 3-acetate, indole-3-pyruvate for Dde_0634 from D. desulfuricans). (6) They demonstrated how the coupling of ligand discovery with microbiology, enzymology, and metabolomics enables the characterization of previously unknown pathways, such as the ethanolamine utilization pathway.
These studies also highlight limitations of current metabolite and metabolism discovery approaches, as experimental and computational approaches for metabolite discovery suffer from our incomplete understanding of metabolomes and the complexities associated with the synthesis of many important metabolites, i.e., we can study only those metabolites that are known to exist and which can be made in sufficient quantities for experimentation. Undoubtedly, the major reason for the lack of a DSF hit is that neither the cognate ligand nor a ligand sufficiently similar to it is present in the screening library. Prior to this work, all known TRAP SBP ligands were organic anions with carboxylate or, in one case, sulfate (taurine) functionalities. The ligand library was constructed with the same restrictions. However, having discovered both phosphorylated substrates (e.g., sn-glycerol 3-phosphate) and positively charged substrates (ethanolamine; pKa = 9.50), it is clear that future DSF screening of the TRAP SBPs should include a much wider range of substrates, although it will never be possible to include all conceivable physiologically relevant ligands. Ideally, approaches are needed that enable entire metabolomes from targeted organisms to be used as the screening libraries.
An intriguing (partial) solution to these challenges is suggested by the 10 SBP targets whose structures were determined with co-purified ligands, derived from the E. coli expression host, which exhibited sufficient affinity to persist through purification and crystallization. In this way, the expressed proteins were able to sample the entire E. coli metabolome for engagement of a tight binding (and presumably physiologically relevant) ligand. Notably, several of the captured ligands, such as (S)-2-acetolactate, (R)-pantoate, and diglycerol phosphate, are not currently commercially available, complicating their use in standard library-based screening approaches. The discovery of adventitious ligands is not limited to these SBPs; we have estimated that ∼5% of all structures determined in a high-throughput environment contain organic ligands derived from the expression host, including a wide range of protein families harboring nucleotides, amino acids, carbohydrates, and lipids,64 which provide immediate insight into biological function. Importantly, these proteins, as well as the SBPs that captured E. coli-derived ligands, originated from a range of Archaea and Gram-negative and -positive bacteria.
A particularly informative example of a crystallographically identified ligand was our previous discovery of carboxy-SAM as a novel metabolite that modulates tRNA function.64 The structure of CmoA, a member of the canonical S-adenosyl methionine (SAM)-dependent methyltransferase superfamily, revealed a ligand in the catalytic site consistent not with SAM but with carboxy-SAM (Cx-SAM), a previously unknown metabolite. Subsequent analyses defined the unique biosynthetic mechanism for the CmoA-mediated conversion of SAM to Cx-SAM and the utilization of Cx-SAM for conversion of 5-hydroxyuridine (ho5U) into 5-oxyacetyluridine (cmo5U) at the wobble position of tRNAs for the efficient translational decoding in Gram-negative bacteria.64−67
Although high-resolution structure determination is an effective read-out for bound ligands, it is limited, as, based on our experience, the majority of purified proteins generated in a high-throughput environment (∼80–90%) fail to yield a structure (http://sbkb.org/); thus, large numbers of purified proteins harboring adventitious ligands go undetected due to the lack of diffraction-quality crystals. As we have demonstrated, ESI-FTMS offers a highly effective alternative that permits purified proteins to be screened for tightly bound adventitious ligands selected from the E. coli metabolome, without the need for crystallization (Figure S5). Some proteins will recognize species-specific metabolites that are not present in E. coli but that can be provided by incubation with lysates derived from the organism of origin or other naturally occurring complex media. In this fashion, metabolomes appropriate for each specific protein can be examined. This approach affords unique opportunities, as these complete metabolomes (1) contain previously unknown metabolites (e.g., Cx-SAM), (2) contain metabolites that are not readily available (e.g., acetolactate, pantoate, and diglycerol phosphate), and (3) are composed of components that, by definition, are physiologically relevant. Finally, given the capabilities of ESI-FTMS to detect modest-to-weak binding ligands,26−28 this approach is likely to support the identification of modest-to-weak binding ligands derived from entire metabolomes of the expression host or lysates.
In summary, we have demonstrated two components of an integrated strategy for the discovery of new metabolites, new protein–ligand interactions and new metabolism. First, the use of library screening approaches to identify SBP ligands, and thus the likely initial reactant in a metabolic pathway, places important constraints on the regions of chemical space that need to be considered. The coupling of these results with informatics (operon and regulon) and additional experimental data (biochemistry, genetics, and metabolomics) provides clear paths to the discovery and characterization of new metabolic pathways. To circumvent the limitations of library-based methods, we described crystallographic- and MS-based strategies that allow for the use of the entire metabolomes of culturable organisms as the screening libraries. Together, these approaches begin to define strategies to realize the full value of accruing genome sequences data.
Acknowledgments
We gratefully acknowledge Jaeheon Lee-he of the University of Illinois, who applied the ethanolamine metabolomics samples on the LTQ-FT mass spectrometer, and Rafael Toro and Rahul Bohsle for maintenance of the AECOM crystallization facility and assistance and advice on crystallization experiment assembly.
Glossary
Abbreviations
- ABCs
ATP binding cassette transporters
- DSF
differential scanning fluorometry
- EUT
ethanolamine utilization pathway
- RMSD
root-mean-squared deviation
- SBPs
solute binding proteins
- TRAP
tripartite ATP-independent periplasmic transporters
- TTTs
tripartite tricarboxylate transporters
- Seq
sequences
- SeqID
sequence identity
Supporting Information Available
Primers used in this study; schematic of previously known TRAP SBP ligands; cluster numbering, coloring, and ligand associations for the 10–120 SSN network; table listing number of proteins in each cluster; ribbon diagram and schematic of typical SBP fold; X-FTMS spectra; KDO qRT-PCR transcriptional data; ESI-FTMS spectra; TRAP SBP-glycerol phosphate ligand interactions; TRAP SBP–(S)-2-acetolactate interactions; TRAP SBP–indole acid interactions; A. tumefaciens C58 and C. salexigens ethanolamine growth characteristics; LC-FTMS spectra of ethanolamine utilization intermediates and products. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinates and structure factors for the structures described in this article (Supporting Information Excel 2) have been deposited with the Protein Data Bank. This article describes functional characterization of proteins with the following UniProt accession ACs. TRAP SBPs: A0R7B0, A1U4I5, A1WPV4, A2SM19, A3PGB2, A3PLM5, A3PM25, A3PQ75, A3PQL6, A3PQU4, A3PQW9, A3QCW5, A3S8F6, A3SZC9, A3T0C3, A3T0D1, A3T293, A4FJU4, A5E8D2, A5EDR3, A5ELA2, A5ES37, A5ET65, A5VZQ4, A6VKM9, A6VKP1, A6VL03, A6VL04, A6VL97, A6VNI1, A6VPN6, A6VQM4, A6X5V3, A6X6D9, A6X7C5, A7IKQ4, A7JQX0, A7JT39, A7JT40, A8AER5, A8AMU5, A8AR30, A9CEX5, A9CHF0, B1G080, B1M416, B4EQJ2, B5BJH4, B7LPS3, B7LPY8, B7LRA7, B8J100, B8JG16, B9JHY6, B9JQU7, C6BRL2, C6BW16, C6BWG5, C6C271, C6C297, C7RDZ3, C7REM1, C9MHP2, D1AZL7, D3PMY1, D6Y196, D7AL34, E3ILJ0, Q0B2F6, Q0K2V0, Q0KC03, Q120S7, Q122C7, Q122T2, Q123A7, Q125D4, Q128M1, Q129N6, Q129N9, Q12BP4, Q12CD8, Q12E02, Q12G29, Q12HD7, Q15TH9, Q160Z9, Q16AR2, Q16BC9, Q16C67, Q1QSK0, Q1QSS4, Q1QT87, Q1QUN2, Q1QWQ9, Q1QWS3, Q1QYM9, Q1QZJ6, Q1QZM6, Q1QZR9, Q1QZT7, Q1R0T9, Q1R0U2, Q1R0X4, Q21VB6, Q21XD7, Q2IUT5, Q2IWM2, Q2J0K9, Q2JZM5, Q2T0D2, Q2T6L7, Q2W7Z6, Q2W961, Q311Q1, Q312S0, Q315G1, Q3IWM7, Q3J410, Q3SLJ1, Q485W8, Q48AL6, Q5LKW4, Q5LKY0, Q5LQ89, Q5LSJ5, Q5LTE9, Q5LTG0, Q5LVG4, Q7CRP6, Q7CS07, Q7N9X9, Q7W8F5, Q7WGZ0, Q7WHX9, Q7WJ47, Q7WJQ1, Q7WK32, Q7WM76, Q7WPG5, Q7WQQ0, Q8ECK4, Q8RE65, Q8XXE8, Q9A8A8, Q9HVH5, and Q9HXL6. VanX: Q1QZT4. γ-Glutamyl amide synthetase: Q1QZR8.
Author Contributions
¶ M.W.V., N.A.-O., S.Z., and B.S.F. contributed equally to this work.
This research was supported by the U.S. National Institutes of Health (U54GM093342 and U54GM094662). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under contract no. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
The authors declare the following competing financial interest(s): M.P.J. is a consultant to Schrödinger LLC, which developed or licensed some of the software used in this study.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Schnoes A. M.; Brown S. D.; Dodevski I.; Babbitt P. C. (2009) Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol. 5, e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam R.; Saier M. H. Jr. (1993) Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 57, 320–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berntsson R. P.; Smits S. H.; Schmitt L.; Slotboom D. J.; Poolman B. (2010) A structural classification of substrate-binding proteins. FEBS Lett. 584, 2606–2617. [DOI] [PubMed] [Google Scholar]
- Kelly D. J.; Thomas G. H. (2001) The tripartite ATP-independent periplasmic (TRAP) transporters of bacteria and archaea. FEMS Microbiol. Rev. 25, 405–424. [DOI] [PubMed] [Google Scholar]
- Gerlt J. A.; Allen K. N.; Almo S. C.; Armstrong R. N.; Babbitt P. C.; Cronan J. E.; Dunaway-Mariano D.; Imker H. J.; Jacobson M. P.; Minor W.; Poulter C. D.; Raushel F. M.; Sali A.; Shoichet B. K.; Sweedler J. V. (2011) The enzyme function initiative. Biochemistry 50, 9950–9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantoliano M. W.; Petrella E. C.; Kwasnoski J. D.; Lobanov V. S.; Myslik J.; Graf E.; Carver T.; Asel E.; Springer B. A.; Lane P.; Salemme F. R. (2001) High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screening 6, 429–440. [DOI] [PubMed] [Google Scholar]
- Hunter S.; Jones P.; Mitchell A.; Apweiler R.; Attwood T. K.; Bateman A.; Bernard T.; Binns D.; Bork P.; Burge S.; de Castro E.; Coggill P.; Corbett M.; Das U.; Daugherty L.; Duquenne L.; Finn R. D.; Fraser M.; Gough J.; Haft D.; Hulo N.; Kahn D.; Kelly E.; Letunic I.; Lonsdale D.; Lopez R.; Madera M.; Maslen J.; McAnulla C.; McDowall J.; McMenamin C.; Mi H.; Mutowo-Muellenet P.; Mulder N.; Natale D.; Orengo C.; Pesseat S.; Punta M.; Quinn A. F.; Rivoire C.; Sangrador-Vegas A.; Selengut J. D.; Sigrist C. J.; Scheremetjew M.; Tate J.; Thimmajanarthanan M.; Thomas P. D.; Wu C. H.; Yeats C.; Yong S. Y. (2012) InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 40, D306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber A. E. II; Babbitt P. C. (2012) Pythoscape: a framework for generation of large protein similarity networks. Bioinformatics 28, 2845–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky P.; Bray J.; Cooper C. D.; Marsden B. D.; Mahajan P.; Burgess-Brown N. A.; Gileadi O. (2010) High-throughput production of human proteins for crystallization: the SGC experience. J. Struct. Biol. 172, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancia F.; Love J. (2010) High-throughput expression and purification of membrane proteins. J. Struct. Biol. 172, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanidis C.; de Jong P. J. (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen J. D.; Nielsen H.; von Heijne G.; Brunak S. (2004) Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340, 783–795. [DOI] [PubMed] [Google Scholar]
- Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expression Purif. 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Blommel P. G.; Fox B. G. (2007) A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expression Purif. 55, 53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen F. H.; Berglund H.; Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- Battye T. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. 62, 72–82. [DOI] [PubMed] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr. 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Gopal K.; Grosse-Kunstleve R. W.; Hung L. W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Pai R. K.; Read R. J.; Romo T. D.; Sacchettini J. C.; Sauter N. K.; Storoni L. C.; Terwilliger T. C. (2004) Recent developments in the PHENIX software for automated crystallographic structure determination. J. Synchrotron Radiat. 11, 53–55. [DOI] [PubMed] [Google Scholar]
- Morris R. J.; Perrakis A.; Lamzin V. S. (2003) ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol. 374, 229–244. [DOI] [PubMed] [Google Scholar]
- Cowtan K. (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. 62, 1002–1011. [DOI] [PubMed] [Google Scholar]
- Terwilliger T. C.; Grosse-Kunstleve R. W.; Afonine P. V.; Moriarty N. W.; Zwart P. H.; Hung L. W.; Read R. J.; Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. 64, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Isupov M. N.; Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D57, 122–133. [DOI] [PubMed] [Google Scholar]
- Davis I. W.; Murray L. W.; Richardson J. S.; Richardson D. C. (2004) MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu H.; Pham N. B.; Quinn R. J. (2008) Direct screening of natural product extracts using mass spectrometry. J. Biomol. Screening 13, 265–275. [DOI] [PubMed] [Google Scholar]
- Vu H.; Roullier C.; Campitelli M.; Trenholme K. R.; Gardiner D. L.; Andrews K. T.; Skinner-Adams T.; Crowther G. J.; Van Voorhis W. C.; Quinn R. J. (2013) Plasmodium gametocyte inhibition identified from a natural-product-based fragment library. ACS Chem. Biol. 8, 2654–2659. [DOI] [PubMed] [Google Scholar]
- Maresca A.; Temperini C.; Vu H.; Pham N. B.; Poulsen S. A.; Scozzafava A.; Quinn R. J.; Supuran C. T. (2009) Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J. Am. Chem. Soc. 131, 3057–3062. [DOI] [PubMed] [Google Scholar]
- Wichelecki D. J.; Balthazor B. M.; Chau A. C.; Vetting M. W.; Fedorov A. A.; Fedorov E. V.; Lukk T.; Patskovsky Y. V.; Stead M. B.; Hillerich B. S.; Seidel R. D.; Almo S. C.; Gerlt J. A. (2014) Discovery of function in the enolase superfamily: d-mannonate and d-gluconate dehydratases in the d-mannonate dehydratase subgroup. Biochemistry 53, 2722–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichelecki D. J.; Vendiola J. A.; Jones A. M.; Al-Obaidi N.; Almo S. C.; Gerlt J. A. (2014) Investigating the physiological roles of low-efficiency d-mannonate and d-gluconate dehydratases in the enolase superfamily: pathways for the catabolism of l-gulonate and l-idonate. Biochemistry 53, 5692–5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K. J.; Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Kumar R.; Sakai A.; Vetting M. W.; Wood B. M.; Brown S.; Bonanno J. B.; Hillerich B. S.; Seidel R. D.; Babbitt P. C.; Almo S. C.; Sweedler J. V.; Gerlt J. A.; Cronan J. E.; Jacobson M. P. (2013) Discovery of new enzymes and metabolic pathways by using structure and genome context. Nature 502, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb T. J.; Evans B. S.; Cho K.; Warlick B. P.; Sriram J.; Wood B. M.; Imker H. J.; Sweedler J. V.; Tabita F. R.; Gerlt J. A. (2012) A RubisCO-like protein links SAM metabolism with isoprenoid biosynthesis. Nat. Chem. Biol. 8, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson H. J.; Morris J. H.; Ferrin T. E.; Babbitt P. C. (2009) Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS One 4, e4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P.; Markiel A.; Ozier O.; Baliga N. S.; Wang J. T.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupeux F.; Rower M.; Seroul G.; Blot D.; Marquez J. A. (2011) A thermal stability assay can help to estimate the crystallization likelihood of biological samples. Acta Crystallogr. 67, 915–919. [DOI] [PubMed] [Google Scholar]
- Fischer M.; Zhang Q. Y.; Hubbard R. E.; Thomas G. H. (2010) Caught in a TRAP: substrate-binding proteins in secondary transport. Trends Microbiol. 18, 471–478. [DOI] [PubMed] [Google Scholar]
- Mauchline T. H.; Fowler J. E.; East A. K.; Sartor A. L.; Zaheer R.; Hosie A. H.; Poole P. S.; Finan T. M. (2006) Mapping the Sinorhizobium meliloti 1021 solute-binding protein-dependent transportome. Proc. Natl. Acad. Sci. U.S.A. 103, 17933–17938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele T. T.; Fowler C. W.; Griffitts J. S. (2009) Control of gluconate utilization in Sinorhizobium meliloti. J. Bacteriol. 191, 1355–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. M.; Wang Y. B.; Jie S.; Yu A. Y.; Luo L.; Yu G. Q.; Zhu J. B.; Wang Y. Z. (2010) Identification of a TRAP transporter for malonate transport and its expression regulated by GtrA from Sinorhizobium meliloti. Res. Microbiol. 161, 556–564. [DOI] [PubMed] [Google Scholar]
- Lecher J.; Pittelkow M.; Zobel S.; Bursy J.; Bonig T.; Smits S. H.; Schmitt L.; Bremer E. (2009) The crystal structure of UehA in complex with ectoine-A comparison with other TRAP-T binding proteins. J. Mol. Biol. 389, 58–73. [DOI] [PubMed] [Google Scholar]
- Kuhlmann S. I.; Terwisscha van Scheltinga A. C.; Bienert R.; Kunte H. J.; Ziegler C. (2008) 1.55 Å structure of the ectoine binding protein TeaA of the osmoregulated TRAP-transporter TeaABC from Halomonas elongata. Biochemistry 47, 9475–9485. [DOI] [PubMed] [Google Scholar]
- Tetsch L.; Kunte H. J. (2002) The substrate-binding protein TeaA of the osmoregulated ectoine transporter TeaABC from Halomonas elongata: purification and characterization of recombinant TeaA. FEMS Microbiol. Lett. 211, 213–218. [DOI] [PubMed] [Google Scholar]
- Grammann K.; Volke A.; Kunte H. J. (2002) New type of osmoregulated solute transporter identified in halophilic members of the bacteria domain: TRAP transporter TeaABC mediates uptake of ectoine and hydroxyectoine in Halomonas elongata DSM 2581(T). J. Bacteriol. 184, 3078–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucktooa P.; Antoine R.; Herrou J.; Huvent I.; Locht C.; Jacob-Dubuisson F.; Villeret V.; Bompard C. (2007) Crystal structures of two Bordetella pertussis periplasmic receptors contribute to defining a novel pyroglutamic acid binding DctP subfamily. J. Mol. Biol. 370, 93–106. [DOI] [PubMed] [Google Scholar]
- Rodionova I. A.; Scott D. A.; Grishin N. V.; Osterman A. L.; Rodionov D. A. (2012) Tagaturonate-fructuronate epimerase UxaE, a novel enzyme in the hexuronate catabolic network in Thermotoga maritima. Environ. Microbiol. 14, 2920–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravcheev D. A.; Khoroshkin M. S.; Laikova O. N.; Tsoy O. V.; Sernova N. V.; Petrova S. A.; Rakhmaninova A. B.; Novichkov P. S.; Gelfand M. S.; Rodionov D. A. (2014) Comparative genomics and evolution of regulons of the LacI-family transcription factors. Front. Microbiol. 5, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvorova I. A.; Tutukina M. N.; Ravcheev D. A.; Rodionov D. A.; Ozoline O. N.; Gelfand M. S. (2011) Comparative genomic analysis of the hexuronate metabolism genes and their regulation in gammaproteobacteria. J. Bacteriol. 193, 3956–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper R. A. (1980) The pathway for l-gulonate catabolism in Escherichia coli K-12 and Salmonella typhimurium LT-2. FEBS Lett. 115, 63–67. [DOI] [PubMed] [Google Scholar]
- Kuivanen J.; Richard P. (2014) The yjjN of E. coli codes for an l-galactonate dehydrogenase and can be used for quantification of l-galactonate and l-gulonate. Appl. Biochem. Biotechnol. 173, 1829–1835. [DOI] [PubMed] [Google Scholar]
- Lessard I. A.; Walsh C. T. (1999) VanX, a bacterial d-alanyl-d-alanine dipeptidase: resistance, immunity, or survival function?. Proc. Natl. Acad. Sci. U.S.A. 96, 11028–11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Wright G. D.; Walsh C. T. (1995) Overexpression, purification, and characterization of VanX, a d-, d-dipeptidase which is essential for vancomycin resistance in Enterococcus faecium BM4147. Biochemistry 34, 2455–2463. [DOI] [PubMed] [Google Scholar]
- Lessard I. A. D.; Pratt S. D.; McCafferty D. G.; Bussiere D. E.; Hutchins C.; Wanner B. L.; Katz L.; Walsh C. T. (1998) Homologs of the vancomycin resistance d-Ala-d-Ala dipeptidase VanX in Streptomyces toyocaensis, Escherichia coli and Synechocystis: attributes of catalytic efficiency, stereoselectivity and regulation with implications for function. Chem. Biol. 5, 489–504. [DOI] [PubMed] [Google Scholar]
- Lilley G. G.; Barbosa J. A.; Pearce L. A. (1998) Expression in Escherichia coli of the putative N-acetylneuraminate lyase gene (nanA) from Haemophilus influenzae: overproduction, purification, and crystallization. Protein Expression Purif. 12, 295–304. [DOI] [PubMed] [Google Scholar]
- Martins L. O.; Huber R.; Huber H.; Stetter K. O.; Da Costa M. S.; Santos H. (1997) Organic solutes in hyperthermophilic archaea. Appl. Environ. Microbiol. 63, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamosa P.; Burke A.; Peist R.; Huber R.; Liu M. Y.; Silva G.; Rodrigues-Pousada C.; LeGall J.; Maycock C.; Santos H. (2000) Thermostabilization of proteins by diglycerol phosphate, a new compatible solute from the hyperthermophile Archaeoglobus fulgidus. Appl. Environ. Microbiol. 66, 1974–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postgate J. R.; Campbell L. L. (1966) Classification of Desulfovibrio species, the nonsporulating sulfate-reducing bacteria. Bacteriol Rev. 30, 732–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux M. J.; Huang Y.; Wang D. N. (2004) Glycerol-3-phosphate transporter of Escherichia coli: structure, function and regulation. Res. Microbiol. 155, 623–629. [DOI] [PubMed] [Google Scholar]
- Boos W. (1998) Binding protein-dependent ABC transport system for glycerol 3-phosphate of Escherichia coli. Methods Enzymol. 292, 40–51. [DOI] [PubMed] [Google Scholar]
- Garsin D. A. (2010) Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat. Rev. Microbiol. 8, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Azevedo Wasch S. I.; van der Ploeg J. R.; Maire T.; Lebreton A.; Kiener A.; Leisinger T. (2002) Transformation of isopropylamine to l-alaninol by Pseudomonas sp. strain KIE171 involves N-glutamylated intermediates. Appl. Environ. Microbiol. 68, 2368–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara S.; Oda S.; Kato K.; Kim H. G.; Koyanagi T.; Kumagai H.; Suzuki H. (2005) A novel putrescine utilization pathway involves gamma-glutamylated intermediates of Escherichia coli K-12. J. Biol. Chem. 280, 4602–4608. [DOI] [PubMed] [Google Scholar]
- Kim J.; Xiao H.; Bonanno J. B.; Kalyanaraman C.; Brown S.; Tang X.; Al-Obaidi N. F.; Patskovsky Y.; Babbitt P. C.; Jacobson M. P.; Lee Y. S.; Almo S. C. (2013) Structure-guided discovery of the metabolite carboxy-SAM that modulates tRNA function. Nature 498, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerwoniec A.; Dunin-Horkawicz S.; Purta E.; Kaminska K. H.; Kasprzak J. M.; Bujnicki J. M.; Grosjean H.; Rother K. (2009) MODOMICS: a database of RNA modification pathways. 2008 update. Nucleic Acids Res. 37, D118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasvall S. J.; Chen P.; Bjork G. R. (2004) The modified wobble nucleoside uridine-5-oxyacetic acid in tRNAPro(cmo5UGG) promotes reading of all four proline codons in vivo. RNA 10, 1662–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasvall S. J.; Chen P.; Bjork G. R. (2007) The wobble hypothesis revisited: uridine-5-oxyacetic acid is critical for reading of G-ending codons. RNA 13, 2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.