

Abstract
We developed a strategy for identifying positions in G protein-coupled receptors that are amenable to bioorthogonal modification with a peptide epitope tag under cell culturing conditions. We introduced the unnatural amino acid p-azido-l-phenylalanine (azF) into human CC chemokine receptor 5 (CCR5) at site-specific amber codon mutations. We then used strain-promoted azide–alkyne [3+2] cycloaddition to label the azF-CCR5 variants with a FLAG peptide epitope-conjugated aza-dibenzocyclooctyne (DBCO) reagent. A microtiter plate-based sandwich fluorophore-linked immunosorbent assay was used to probe simultaneously the FLAG epitope and the receptor using infrared dye-conjugated antibodies so that the extent of DBCO incorporation, corresponding nominally to labeling efficiency, could be quantified ratiometrically. The extent of incorporation of DBCO at the various sites was evaluated in the context of a recent crystal structure of maraviroc-bound CCR5. We observed that labeling efficiency varied dramatically depending on the topological location of the azF in CCR5. Interestingly, position 109 in transmembrane helix 3, located in a hydrophobic cavity on the extracellular side of the receptor, was labeled most efficiently. Because the bioorthogonal labeling and detection strategy described might be used to introduce a variety of different peptide epitopes or fluorophores into engineered expressed receptors, it might prove to be useful for a wide range of applications, including single-molecule detection studies of receptor trafficking and signaling mechanism.
G protein-coupled receptors (GPCRs) are heptahelical transmembrane (TM) proteins indispensable for mediating cellular activities in response to signals from various extracellular signals, ranging from light to small molecule or peptide ligands.1 The tool kit repertoire for studying GPCR structure–activity relationships has been expanding, especially in the context of live-cell or whole-cell imaging and tracking.2 In one important application, methodologies that facilitate incorporation of fluorophores into target GPCRs have allowed high-resolution single-molecule detection (SMD).3,4 Such studies can illuminate kinetic and dynamic information at the molecular level, which is usually averaged and lost in ensemble measurements. This is especially important for precisely understanding the assembly and function of GPCR “signalosomes”, which are complexes of GPCRs, ligands, and accessory proteins such as G proteins, GPCR kinases (GRKs), and arrestins.5 A long-term goal would be to image signalosomes in live cells.6,7
Fluorescent probes can be strategically introduced into GPCRs using either small organic fluorophores or large fluorescent proteins (FPs). Single-molecule tracking (SMT) of GPCRs bearing fluorescent probes via N- and C-terminal fusions of FPs, self-labeling proteins like HaloTag, SNAP-tag, and CLIP-tag, or antibodies labeled with quantum dots has been reported, albeit with limitations on sites of incorporation.4,8,9 The general methodology has been further developed in FRET-based GPCR sensors designed to study GPCR activation in live cells.10,11 Although fusion proteins can be genetically encoded and therefore targeted to any cellular locale, they add significant bulk to the GPCR. This could potentially alter conformation, functional dynamics, or both. Bioorthogonal chemical reactions are thus being developed to site-specifically introduce probes like fluorophores onto protein tags harboring desirable physical and chemical properties with minimal structural and functional perturbation to target GPCRs.12,13
The primary goal of such reactions is to achieve simple and fast reaction kinetics between the small tag on the target protein and the probe, with added flexibility in choosing probes and easy detection of the labeled protein. In early fluorescence studies with modified GPCRs, cysteine conjugation chemistry was frequently used to obtain site-specific labeling. For example, fluorescent conjugates have been formed by labeling cysteines in rhodopsin (Rho) with a pyrene moiety,14 and more recently with a bimane.15 A substituted-cysteine accessible method (SCAM) has been used to probe water accessible ligand binding surfaces in GPCRs by reacting substituted cysteines with methanosulfonate reagents.16 Cysteine residues can also be conjugated to biarsenical-functionalized fluorescent probes such as FlAsH and ReAsH when they are part of a short tetracysteine peptide tag.17 In fact, agonist-dependent conformational changes have been studied by FRET between FlAsH-bound internal sites and C-terminal YFP in muscarinic receptors,18 or small molecule Alexa fluorophore-labeled single cysteines in β2 adrenoreceptor.19
Another recent advancement in the field of bioorthogonal labeling has been the utilization of small, minimally perturbing unnatural amino acids (uaas) with chemical handles into GPCRs transiently expressed in mammalian cells. This technology utilizes a bioorthogonal “suppressor tRNA/aminoacyl tRNA synthetase” pair to incorporate site-specifically a desired uaa at a nonsense amber mutation position introduced by site-directed mutagenesis in target cDNA.20,21 We adapted the amber suppression technology to achieve heterologous expression of low-abundance GPCRs in mammalian cell culture by combining an engineered tyrosine synthetase originally developed in a yeast system22 with a novel chimera of human and Bacillus stearothermophilus tRNATyr.21 This approach provides vast flexibility in selecting a uaa that will possess the desired functional moiety required by a particular application.23 For instance, p-azido-l-phenylalanine (azF) has been used as an infrared (IR) probe to study GPCR conformational changes.24,25 Other applications utilize uaas as photoactivatable cross-linkers to map the binding sites of peptides, mAbs, or small molecules with GPCRs.26−31 In addition to GPCRs, the photoactivatable uaas have been applied to investigate the activation and inactivation dynamics of the ionotropic glutamate receptor family, including the N-methyl-d-aspartate (NMDA) receptor32 and the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) subtype receptor.33
In our recent work with human CC chemokine receptor 5 (CCR5), which is a coreceptor for human immunodeficiency virus-1 (HIV-1) cellular entry, we used the cell compatibility of the Staudinger ligation to identify azF-incorporated positions in CCR5 amenable to labeling in native cellular membranes.34,35 The azide-phosphine Staudinger ligation, although highly biocompatible,36 exhibits poor reaction stoichiometry in comparison to that of azide–alkyne cycloaddition reactions.37 Click reactions between azides and terminal alkynes are typically catalyzed by copper (copper-catalyzed azide–alkyne cycloaddition, CuAAC), which renders the reaction toxic to cells. Cu-stabilizing ligands like TBTA, THPTA, and BTTES are now often used to improve biocompatibility.38 Strain-promoted azide–alkyne [3+2] cycloaddition (SpAAC) reactions, on the other hand, utilize cyclic alkynes that react efficiently in the absence of catalysts to relieve the ring strain. These reactions are increasingly being explored in cell-based and in vivo applications.39
Here, we capitalize on the superior labeling stoichiometry and reaction rate of SpAAC reactions,37,40,41 and biocompatibility of cyclooctyne reagents to label expressed CCR5 in cell cultures. We describe a robust screening strategy for identifying positions in azF-modified CCR5 transiently expressed in mammalian cells that can be modified with a FLAG peptide epitope tag by facile chemical reaction with DBCO. We then develop a fluorophore-linked immunosorbent assay (ISA), using LI-COR technology to detect two fluorophore IR dyes simultaneously.42 We took advantage of this sensitive LI-COR-based dual-color readout to simultaneously quantify the extent of label incorporated at specific azF positions on CCR5 and the total receptor level. After screening site-specific FLAG-tagged CCR5 variants, we identified four relatively highly reactive sites amenable to direct DBCO-FLAG labeling, which is remarkable because two of them are located in the allosteric binding pocket for maraviroc in CCR5. We anticipate that our GPCR labeling and screening methods will facilitate future high-resolution tracking and imaging experiments in addition to fragment-based screening methods.
Materials and Methods
Materials
The anti-FLAG polyclonal antibody produced in rabbit was obtained from Sigma. 1D4 mAb was obtained from the National Cell Culture Center. Anti-CCR5 T21/8 mAb and T21/8-biotin were obtained from eBioscience. IRDye 800CW goat anti-mouse secondary antibody, IRDye 680LT goat anti-mouse IgG2a-specific secondary antibody, and IRDye 680RD streptavidin were purchased from LI-COR. azF was purchased from Chem-Impex International. DBCO-PEG4-maleimide was purchased from Click Chemistry Tools. FLAG-aza-dibenzocyclooctyne (DBCO-FLAG) was synthesized by the Rockefeller University Proteomics Resource Center using a reported protocol,43 by conjugating DBCO-PEG4-maleimide to the eight-residue FLAG peptide (DYKDDDDK) containing a C-terminal cysteine residue.
Plasmids and Site-Directed Mutagenesis
Plasmid pSVB.Yam carrying the gene encoding the chimera amber suppressor tRNA was derived from B. stearothermophilus Tyr-tRNACUA.21 The amino-acyl tRNA synthetase for azF without a C-terminal FLAG tag was described previously.21,34 The human CCR5 gene was in a pcDNA 3.1(+) plasmid and contained a C-terminal 1D4 epitope tag (TETSQVAPA). The amber mutations were introduced into CCR5 using a QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene).
Heterologous Expression and In-Culture Labeling of azF-CCR5 Variants in Mammalian Cells
Method A
When an on-cell ISA was performed, wt CCR5 or amber variants were expressed in HEK293T cells by transient transfection in six-well plates. Twenty-four hours post-transfection, cells were prepared for in-culture labeling as described previously.34 Forty-eight hours post-transfection, once the cells were adhered and ready for labeling in 96-well plates, cells were washed three times with 100 μL each of Dulbecco’s phosphate-buffered saline containing Ca2+ and Mg2+ (DPBS; Invitrogen) to remove any residual azF-containing media. Labeling reagent was prepared from a 20 mM stock of DBCO-FLAG diluted to a final working concentration of 100 μM in DPBS. Each well of the 96-well plate was treated with 60 μL of DBCO-FLAG incubated at 37 °C for 1 h, except for the control no-label-treated cells that were maintained in DPBS. Postreaction labeling buffer was removed, and cells in the 96-well plate were further subjected to an on-cell ISA.
Method B
When labeled samples were prepared for sandwich ISA experiments, wild-type (wt) CCR5 or amber variants were expressed in HEK293T cells by transient transfection in 10 cm dishes. Forty-eight hours post-transfection, the medium was aspirated and the cells were gently harvested in phosphate-buffered saline [PBS (Dulbecco’s phosphate-buffered saline without calcium or magnesium); Invitrogen] from the plate, pelleted at 1000g for 3.5 min using a tabletop centrifuge, and then resuspended in 60 μL of labeling medium (100 μM DBCO-FLAG) in a tube. Cells were returned to 37 °C for incubation with gentle nutation for 1 h. The cells were pelleted and washed with PBS to remove excess labeling reagent. Cells were then resuspended in 1 mL of buffer N {20 mM Tris-HCl (pH 7.0), 0.1 M (NH4)2SO4, 10% (v/v) glycerol, 0.07% cholesteryl hemisuccinate (CHS), 0.018% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 0.008% 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS), 0.33% n-dodecyl β-d-maltoside (DM), and 0.33% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS)} for lysis and receptor solubilization.44 The lysates were cleared by centrifugation 10000g for 10 min at room temperature (RT). Receptors solubilized in buffer N were expected to retain the correct folded conformation.45 Lysate was then further subjected to a sandwich ISA.
Detection of the Labeled Receptor by an On-Cell ISA
All treatments during the ISA were performed in blocking buffer [BB (0.5% BSA in DPBS)]. Prior to the assay, cells were washed three times with BB and then fixed with 100 μL/well of freshly prepared methanol-free paraformaldehyde for 20 min at RT. A 4% working stock was prepared from 16% paraformaldehyde (Pierce) in DPBS. Following fixation, cells were washed three times with BB followed by a 20 min blocking step. Incubation in primary antibody was conducted in 100 μL for 1.5 h on ice. The anti-CCR5 T21/8 mAb was used at a 1:1000 dilution in BB, and the anti-FLAG polyclonal antibody was used at a 1:3000 dilution. Postprimary antibody incubation cells were washed three times with BB followed by a secondary antibody incubation for 1 h at RT. Wells treated with the anti-FLAG polyclonal antibody were incubated with IRDye 800CW goat anti-rabbit secondary antibody (1:20000 dilution), and those treated with T21/8 were incubated with IRDye 680RD goat anti-mouse secondary antibody (1:20000 dilution). After several careful washes with BB, 50 μL of BB was added to each well and the plate was read on a LI-COR Odyssey Sa Infrared Imaging System with dual-color detection. The amount of receptor was determined in the 700 nm channel that detects the N-terminal signal, while the amount of labeled sample in the 800 nm channel detects the FLAG signal. The image of the plate was analyzed using Image Studio LI-COR. The integrated fluorescence intensity of each well was used to quantify the labeling efficiency and receptor cell surface expression.
Detection of the Labeled Receptor by a Sandwich ISA
Ninety-six-well plates (clear bottom, high binding EIA/RIA; Costar) were first pretreated with anti-1D4 or anti-CCR5 T21/8 mAb antibody (100 μL/well) at a concentration of 1 μg/mL in PBS overnight at 4 °C to prepare the plate for affinity capture. The next day, the wells were washed several times with PBS containing 0.05% Tween 20, followed by incubation with 1% bovine serum albumin (BSA) (200 μL/well) in PBS overnight at 4 °C. The following day, wells were washed with wash buffer [WB (0.1% BSA in PBS)]; 100 μL of lysate sample prepared after in-culture labeling of CCR5 variants was added to each well and incubated overnight at 4 °C to capture the receptor on the plate. The following day, the unbound lysate was washed away in WB and prepared for an ISA. In triplicate, wells were treated with 100 μL/well of the appropriate primary antibody, anti-CCR5 T21/8-biotin mAb (1:1000) or anti-1D4 mAb-biotin (1:1000), and incubated on ice for 1.5 h. Subsequently, wells were washed with WB and treated with 100 μL/well of the respective secondary detection reagent: IRDye 680RD streptavidin, IRDye 680RD goat anti-mouse secondary antibody, IRDye 680LT goat anti-mouse (IgG2a-specific) secondary antibody, or IRDye 800CW goat anti-rabbit secondary antibody for 1 h at RT. After several washes with WB, the plate was read on a LI-COR Odyssey imager.
Dual-Color Immunoblot Analysis of Solubilized and Purified Receptors
Cell pellets were lysed in buffer N to solubilize the expressed CCR5 variants with a C-terminal 1D4 tag. The CCR5 receptor was immunopurified by overnight incubation with 1D4-derivatized Sepharose resin at 4 °C. The resin was washed three times and eluted in buffer N. Lysate samples or 1D4-purified receptor samples were subjected to SDS–PAGE (NuPAGE Novex 4–12% Bis-Tris Gel) and then transferred to a PVDF Immobilon-FL Transfer Membrane (Millipore) for immunoblotting. The membrane was blocked in 1% BSA in Tris-buffered saline with 0.05% Tween 20 (TBST) for 1 h at RT. After being washed, the membranes were incubated with a mixture of primary antibodies, anti-1D4 mAb (1:2000), and anti-FLAG polyclonal antibody (1:3000) in 0.5% BSA in PBS on a shaker at 4 °C overnight. The blots were washed in PBS and incubated with a mixture of IRDye 680RD goat anti-mouse and IRDye 800CW goat anti-rabbit secondary antibodies in 1% BSA in TBST for 1 h. The membranes were scanned with a LI-COR Odyssey imager to visualize the 1D4 signal of CCR5 receptors (700 nm, red) and the FLAG signal from covalent modification (800 nm, green).
Results
On-Cell ISA for Detecting Epitope-Tagged azF-CCR5
We first designed a dual-color on-cell ISA to detect the peptide tag attached to GPCRs. This assay was designed to be a variation of our previously established two-step antibody detection strategy, which involved secondary antibodies conjugated to horseradish peroxidase (HRP).34 While HRP has the advantage of signal amplification, it provides only qualitative or semiquantitative results. To overcome the drawback of HRP, we utilized two spectrally distinct IRDye fluorescent secondary antibody/primary antibody pairs to achieve simultaneous detection of distinct epitope targets. We chose the anti-FLAG polyclonal antibody paired with IRDye 800CW goat anti-rabbit secondary antibody (detected in the 800 nm channel) to measure FLAG label-tagged cell surface receptor and an anti-CCR5 T21/8 mAb that binds the N-terminus of CCR5, combined with IRDye 680RD goat anti-mouse secondary antibody (detected in the 700 nm channel) to quantify cell surface receptor expression.46
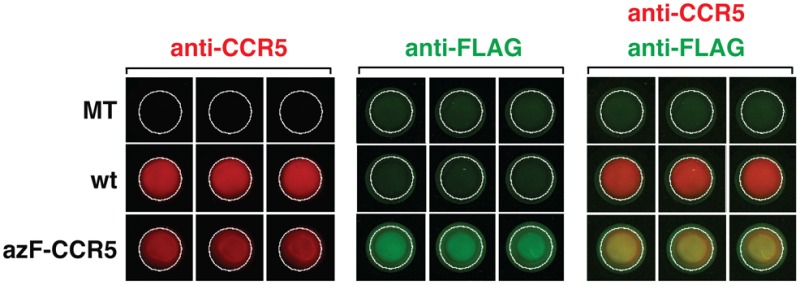
We incorporated azF into CCR5 at different positions within the extracellular (EC) loops, intracellular (IC) loops, and transmembrane (TM) domains by amber codon suppression technology.34 The azF-tagged CCR5 expressed at the cell surface was then bioorthogonally labeled with a reagent containing the eight-residue FLAG peptide epitope conjugated to a cyclooctyne reagent (DBCO-FLAG) using SpAAC chemistry (Figure 1A). We chose two CCR5 variants, N24azF and F96azF, to evaluate the on-cell ISA (Figure 1B, inset), with the mock-transfected (MT) cells and wt CCR5 cells as controls. In our previous work, we found that these two CCR5 variants are amenable to covalent modification by FLAG peptide under cell culturing conditions using a phosphine-FLAG reagent.34 We treated the transfected HEK293T cells in a 96-well microtiter plate with 100 μM DBCO-FLAG, washed away the excess label, and performed an on-cell ISA for the DBCO-FLAG-treated cells (dark gray bars, Figure 1B), with the untreated cells as the control (light gray bars, Figure 1B). When the FLAG signal was probed, we found that F96azF-CCR5 cells exhibited a 2-fold enhancement of their FLAG signal compared with those of the wt and MT controls, whereas the signal from N24azF-CCR5 cells was similar to those of the wt CCR5 and MT controls (Figure 1B). We also quantified the expression level of N24azF-CCR5 and F96azF-CCR5 on the cell surface (Figure S1 of the Supporting Information). After the normalization of the FLAG signal against the expression level, both N24azF-CCR5 and F96azF-CCR5 exhibited reactivity with DBCO-FLAG higher than those of the wt CCR5 and MT controls. We also noticed that even for the wt CCR5 and MT groups, the FLAG signal of the DBCO-FLAG-treated group was at least 2-fold stronger than that of the untreated group. Because the wt CCR5 cells and MT cells were not cultured in azF-supplemented medium, this finding suggests either nonspecific covalent labeling of cells by DBCO-FLAG or difficulty in washing away the excess labeling reagent.47
Figure 1.

Fluorophore-linked immunoassay for detecting azF-CCR site-specifically peptide epitope modified by SpAAC. (A) Scheme showing the in-culture labeling of the GPCR, CCR5, the uaa azF genetically encoded at a target site. SpAAC reaction is used to site-specifically attach the FLAG peptide epitope (green) conjugated to a DBCO (red) via a maleimide cross-linker (DBCO-FLAG). (B) Representative results of the on-cell ISA for measuring the FLAG signal with or without DBCO. HEK293T cells expressing N24azF or F96azF CCR5 variants were treated with 100 μM DBCO-FLAG (dark gray bars), and data for the untreated cells are also shown (light gray bars). Wild-type CCR5 (wt) and mock-transfected (MT) controls were tested under the same condition. Error bars represent the standard error of the mean of triplicate measurements. (C) Western blot analysis of labeled and purified azF-CCR5. In the left panel, the crude lysate sample was probed simultaneously with anti-FLAG polyclonal antibody and 1D4 monoclonal antibody to detect the extent of FLAG peptide epitope tagging (green) and full-length receptor expression (red), respectively. The receptor is visualized in the red channel and separates at 37 kDa. The right panel shows full-length CCR5 immunoaffinity purified with the C-terminal 1D4 engineered tag. The purified receptor separates at 37 kDa (red) as detected with the anti-1D4 antibody against the C-terminal 1D4 epitope tag, and the FLAG tag is visualized with the anti-FLAG pAb (green).
To further investigate our observation of azF-CCR5-independent background labeling, we analyzed the DBCO-FLAG-treated MT, wt CCR5, and F96azF-CCR5 cells by dual-color quantitative Western blotting (Figure 1C). We first analyzed the crude lysates of these cells after DBCO-FLAG treatment (Figure 1C, left panel). Our CCR5 constructs carry an engineered 1D4 C-terminal epitope, and thus, receptor expression was visualized using anti-1D4 mAb/IRDye 680RD goat anti-mouse IgG (red). The presence of the FLAG peptide was probed using anti-FLAG pAb/IRDye 800CW goat anti-rabbit IgG (green). In the crude lysates of wt CCR5 and F96azF-CCR5 cells, the expressed receptor was found as a major red band appearing at 37 kDa and some additional weaker bands, probably caused by dimerization and the immature form of CCR5. By contrast, in the lysates of MT, wt CCR5, and F96azF cells, the FLAG signals were shown as pervasive green bands spanning a wide range of molecular weights, demonstrating the presence of nonspecific covalent labeling of proteins by DBCO-FLAG independent of CCR5 or azF. To reveal the FLAG signal from labeled CCR5, we immunopurified the CCR5 receptor from the cell lysate using the 1D4 Sepharose. We analyzed the purified receptor using the dual-color Western blot (Figure 1C, right panel). The immunopurification step eliminated all the green bands found in the crude lysates of wt CCR5 cells. The purification product from F96azF-CCR5 cell lysate appeared in the Western blot as a sharp yellow band (FLAG+1D4 signal) slightly above a red band (1D4 signal) (Figure 1C, right panel, and Figure S2 of the Supporting Information), demonstrating the specific labeling of F96azF-CCR5 by DBCO-FLAG. The yellow band’s shifts toward higher molecular weights can be readily explained by the additional 1.7 kDa contributed by DBCO-FLAG to azF-CCR5. Overall, these results explained the background labeling for MT and wt CCR5 cells shown earlier (Figure 1B) and suggest that an immunopurification step would greatly facilitate the detection of specifically labeled receptors.
Multiplex Detection of FLAG-Tagged azF-CCR5 Using a Sandwich ISA
On the basis of the results of on-cell ISA and Western blot experiments, we further developed a microtiter plate-based sandwich ISA for multiplex detection of site-specifically labeled azF-CCR5 with improved sensitivity by incorporating an immunoaffinity enrichment mechanism into the assay scheme (Figure 2). It relies on the capture of full-length azF-CCR5 to the plate surface via the C-terminal 1D4 tag to achieve the separation of CCR5 from the vast majority of proteins.
Figure 2.
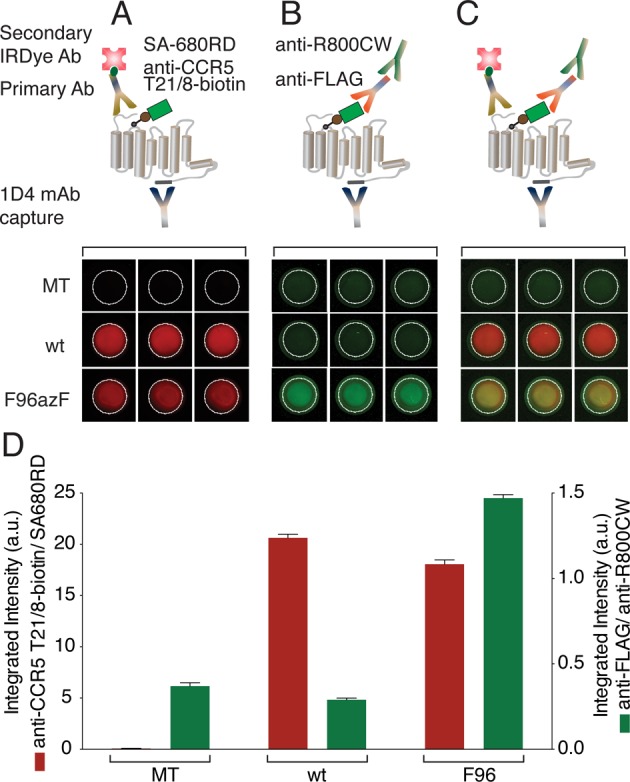
Multiplex detection using a sandwich fluorophore-linked immunosorbent assay. (A) The top panel shows the schematic diagram of the immunosorbent assay (ISA) for detecting the expression levels of wt and F96azF-CCR5. The receptor is captured to the plate surface using the C-terminally specific 1D4 mAb. The N-terminal epitope of CCR5 is probed using anti-CCR5 T21/8-biotin mAb followed by streptavidin coupled to IRDye 680RD (SA-680RD). The bottom panel shows the corresponding fluorescence image of the triplicate wells (pseudocolor, red, detected in the 700 nm channel). (B) The top panel shows the schematic diagram for detecting the labeling efficiencies of wt and F96azF-CCR5 with DBCO-FLAG. With the receptor immobilized to the plate surface with the C-terminally specific 1D4 mAb, the FLAG tag epitope of CCR5 is probed with anti-FLAG pAb followed by anti-rabbit IgG conjugated to IRDye 800CW (anti-R800CW). The bottom panel shows the corresponding fluorescence image of the triplicate wells (pseudocolor, green, detected in the 800 nm channel). (C) The top panel shows the schematic diagram of the ISA experiment for FLAG-labeled CCR5. The dual-color detection allowed simultaneous quantification of the receptor expression level and the labeling efficiency by DBCO-FLAG. The bottom panel shows the corresponding fluorescence image of the triplicate wells shown for both the N-terminal epitope signal (red, 700 nm channel) and FLAG signal (green, 800 nm channel). Wells containing wt CCR5 treated with DBCO-FLAG show a strong N-terminal epitope signal but a much weaker FLAG signal. Wells containing F96azF-CCR5 treated under the same reaction condition yielded strongly both the N-terminal epitope signal and FLAG signal, indicating the presence of expressed full-length receptor tagging with a FLAG epitope (merged, yellow). (D) Integrated intensities of the wells shown in panels A–C, plotted in arbitrary units (a.u.). The expression of wt CCR5 and F96azF-CCR5 (red bars) and the level of FLAG signal indicate specific labeling of F96azF-CCR5. Data from a representative experiment are presented, and error bars represent the standard error of the mean of triplicate measurements.
We treated the MT or CCR5 cells as described above. The lysates of these cells were applied to a 96-well microtiter plate precoated with anti-1D4 mAb. The unbound fraction was washed away to reduce the intensity of the nonspecific signal of DBCO-FLAG. We employed a streptavidin-coupled IRDye 680RD paired with the primary anti-CCR5 T21/8-biotin mAb to detect the presence of full-length receptor (Figure 2A). The resulting images of the wells (red) showed essentially background-free measurement of wt and F96azF-CCR5 expression levels based on the N-terminal epitope signal. The presence of FLAG tag was measured using an anti-FLAG pAb/IRDye 800CW goat anti-rabbit IgG pair (Figure 2B). The resulting fluorescent images of the wells (green) containing F96azF-CCR5 lysate showed a clear enhancement of the FLAG signal as compared with that of wells containing wt CCR5 lysate, confirming specific bioorthogonal labeling of F96azF-CCR5 with DBCO-FLAG.48 The merged images of the wells demonstrated simultaneous detection of the N-terminal epitope and the FLAG signal from FLAG-tagged F96azF-CCR5 (yellow, Figure 2C, bottom panel). The integrated fluorescence intensities of the wells are plotted in arbitrary units in Figure 2D. The results indicate that while wt and F96azF-CCR5 were expressed at similar levels, the azF variant showed an increase in the intensity of the FLAG signal of >4-fold, greater than the 2-fold difference observed by the on-cell ISA. The MT sample served as a negative control with no expression of CCR5 or label incorporation. The addition of an immunoaffinity enrichment step to the multiplex detection scheme allowed us to circumvent the time-consuming electrophoresis step and improved the throughput of the assay.
Screening of Site-Specific Bioorthogonally Labeled and Purified azF-CCR5 Variants
In earlier work, we used CCR5 as a model system to perform an accessibility screen for bioorthogonal epitope tagging with a FLAG-phosphine reagent via the Staudinger ligation.34 After testing several positions on CCR5, we concluded that the FLAG-phosphine reagent was cell-permeable because both EC and IC residues were accessible to the labeling reagent in live cells. The earlier study motivated us to employ our newly developed sandwich ISA to determine the accessibility of DBCO-FLAG to various sites on CCR5 by utilizing SpAAC reaction. We expressed 32 azF-CCR5 variants with the reactive uaa introduced at EC, TM, and IC positions (Figure 3A). The azF-CCR5 variants were treated with DBCO-FLAG and analyzed by the sandwich ISA. The fluorescence images of the wells for each variant, performed in triplicate, are shown in Figure 3B, and the integrated fluorescence intensities are plotted in Figure 3C. The results (from two sets of experiments, each performed in triplicate) showed comparable levels of receptor expression for the tested azF-CCR5 variants (Figure 3C, top panel, red bars). The accessibility screen indicated a few key sites as those being most amenable to epitope tagging by SpAAC (Figure 3C, bottom panel, green bars).
Figure 3.

Accessibility screen for bioorthogonal site-specific modification of azF-CCR5 variants detected by the multiplex sandwich ISA (C, inset). (A) CCR5 schematic highlighting positions of incorporation of azF with four distinctively labeled sites colored green. (B) Representative fluorescence image of the multiplex detection of DBCO-FLAG-treated azF-CCR5 samples in triplicate. (C) Quantification of the receptor expression level and labeling efficiency for the azF-CCR5 variants. In the top panel, the column graph shows the average integrated intensities from each set of wells representing the expression of wt CCR5 and azF-CCR5 variants obtained by probing with anti-CCR5 T21/8-biotin mAb followed by SA-680RD (700 nm channel, red). In the bottom panel, the column graphs the corresponding FLAG signals probed with the anti-FLAG pAb/anti-R800CW pair (800 nm channel, green), representing the extent of FLAG epitope tagging. All results are denoted as arbitrary units with error bars representing the standard error of the mean for triplicate measurements.
We calculate the ratio of FLAG tag incorporation to total receptor expression (Table 1). Four sites (N24 in the N-terminus, Y37 in TM1, F109 in TM3, and Y251 in TM6) in particular stood out with label/receptor ratios well above background, whereas no significant labeling (p > 0.01) could be observed at four other sites (F85, A233, F260, and F264). The label-to-protein ratio of F109azF-CCR5 was almost twice as large as that of Y251azF-CCR5, which displayed the next best labeling efficiency. We also used a dual-color Western blot to analyze three azF-CCR5 variants identified to be highly reactive with DBCO-FLAG (Y37, F109, and Y251) and F96azF-CCR5, which was used for initial optimization of the sandwich ISA (Figure S2 of the Supporting Information). The 1D4-immunopurified, labeled products of all the variants appeared in the blot as two juxtaposed bands corresponding to the unlabeled azF-CCR5 (red, bottom band) and the FLAG-tagged azF-CCR5 (yellow, top band), thus confirming specific labeling of azF-CCR5.
Table 1. Sandwich ISA Label:Receptor Ratiosa.
| topological position | azF | label:receptor ratio (arbitrary unit) | topological position | azF | label:receptor ratio (arbitrary unit) |
|---|---|---|---|---|---|
| wt | 0.011 ± 0.001 | ||||
| N-terminus | I23 | 0.056 ± 0.002 | ECL2 | Y176 | 0.029 ± 0.002 |
| N24 | 0.166 ± 0.006 | C178 | 0.070 ± 0.010 | ||
| K26 | 0.050 ± 0.004 | F182 | 0.080 ± 0.007 | ||
| I28 | 0.035 ± 0.001 | Y184 | 0.038 ± 0.005 | ||
| TM1 | Y37 | 0.146 ± 0.005 | Y187 | 0.057 ± 0.001 | |
| ICL1 | T65 | 0.022 ± 0.002 | F189 | 0.028 ± 0.001 | |
| TM2 | F85 (ns) | 0.019 ± 0.004 | ICL3 | L218 | 0.039 ± 0.002 |
| W86 | 0.068 ± 0.002 | T220 | 0.031 ± 0.002 | ||
| ECL1 | F96 | 0.095 ± 0.006 | C224 | 0.030 ± 0.002 | |
| TM3 | Y108 | 0.017 ± 0.001 | E227 | 0.025 ± 0.001 | |
| F109 | 0.489 ± 0.019 | A233 (ns) | 0.016 ± 0.001 | ||
| ICL2 | A129 | 0.028 ± 0.001 | TM5 | Y251 | 0.264 ± 0.022 |
| V130 | 0.060 ± 0.002 | ECL3 | F260 (ns) | 0.016 ± 0.002 | |
| ECL2 | F166 | 0.095 ± 0.011 | E262 | 0.042 ± 0.004 | |
| R168 | 0.052 ± 0.002 | F264 (ns) | 0.020 ± 0.003 | ||
| E172 | 0.049 ± 0.005 | ICL4 | G301 | 0.060 ± 0.009 |
These ratios are calculated from the integrated intensity detected in the 800 nm channel (anti-R800CW, green) for receptor expression by anti-CCR5 T21/8-biotin mAb to the integrated intensity in the 700 nm channel (SA-680RD, red). Error bars represent the mean standard error of multiple data sets. Residues F85, A233, F260, and F264 exhibited nonsignificant labeling (p > 0.01), whereas those highlighted in bold represent the more efficiently labeled. The positions are correlated to topological position on the predicted map of CCR5.
It is noteworthy that the positions tested on the IC loops exhibited an only marginal increase in the labeling ratios over the wt control (Figure 3 and Table 1), which suggests DBCO-FLAG, unlike phosphine-FLAG reagent, is largely membrane-impermeable. However, we wanted to further confirm that the low intensity of the FLAG signal from the IC loop variants was not an artifact embedded in the detection scheme. Namely, the C-terminal capture scheme in the sandwich ISA did not preclude detection of the FLAG tag possibly introduced into the IC side of CCR5. We addressed this question by swapping the role of N-terminally specific and C-terminally specific antibodies to capture receptor with the N-terminally specific anti-CCR5 T21/8 mAb (Figure S3A of the Supporting Information, inset). For multiplex detection, we utilized anti-1D4 mAb-biotin/IRDye 680RD streptavidin to measure receptor expression and anti-FLAG pAb/IRDye 800CW anti-rabbit IgG to measure FLAG tag incorporation. The results from this reverse capture/probing strategy (Figure S3 of the Supporting Information) exhibited trends similar to those observed in Figure 3, but drastically reduced signals (only ∼10%) for both labeling efficiency and receptor expression. The lower overall intensity of the signals suggests that anti-CCR5 T21/8 mAb is inferior to 1D4 mAb in capturing CCR5 to the well surface. Nevertheless, the results confirmed F109azF-CCR5 as the most robustly labeled mutant in the screen (Figure S3 of the Supporting Information). More importantly, the reverse capturing/detection strategy excludes the alternative explanation that the apparent lack of labeling on the IC surface of CCR5 is merely due to the sterical problem with detecting a peptide label conjugated to a cytoplasmic site of a receptor that is captured to the well surface with the cytoplasmic, C-terminal tail. In summary, our optimized multiplex ISA detection scheme illustrated in Figure 2 is the preferred approach to identifying suitable azF-CCR5 variants for modification with DBCO reagents.
Discussion
The word “bioorthogonal” was introduced in early 2000 to reference specific modifications performed on target proteins within the background milieu of other cellular proteins, with reasonably fast reaction rates. Site-specific introduction of probes into target proteins is a topic of intense interest for studying structure–activity relationships. We have established the site-specific incorporation of such probes by optimizing an amber suppression-based methodology for transiently expressing GPCRs site-specifically incorporating uaas in mammalian cell culture.21 One such probe is the uaa azF bearing an azido moiety that serves as a chemically reactive handle.25,37,49 The small size and its virtual absence from biological systems make azF a versatile probe for minimally perturbing cell-based applications. Azido can react with alkynes via click reactions such as CuAAC and SpAAC, and phosphines via Staudinger ligation, making it an attractive target for bioorthogonal modifications.
CuAAC has been successfully demonstrated in combination with the PRIME method for the site-specific labeling of lysine residues in genetically encoded peptide tags.50 It is a highly efficient reaction; however, the cellular toxicity of the Cu(I) catalyst requirement can be a problem for certain applications. A modified CuAAC reaction uses a copper-chelating azide, which slightly reduces the requirement for high copper concentrations.51 SpAAC by nature of its mechanism does not require copper catalysis. Because of its introduction, several cyclooctyne derivatives, including DIBO, DIFO, DBCO, BARAC, and DIMAC, have been developed to improve reaction kinetics and efficiency.12 Candidate cyclooctyne reagent selection can be guided by density functional theory (DFT) calculations that predict reactivity.52 Aside from the utility of the SpAAC reaction to label azF-modified sialic acids in vivo,53 it has also been successfully demonstrated in designing targeted therapeutics,54,55 live-cell imaging of newly synthesized proteins,56 synthesis of nanomaterial hybrids,57in vivo microPET,58,59 modifying viruses to study viral entry,60 and fluoro-switch click reactions in mammalian cells using fluorescent DIBO variants.61 Staudinger ligation between azido and phosphine groups is an alternative cell-compatible reaction that has been successfully used to probe biomolecules in cells,34,62 and in vivo.36 There is an interesting comparative analysis of reaction efficiency of FLAG peptide-tagged cyclooctyne derivatives and phosphines with azides in mouse models.43
In this study, one particular challenge was to characterize the reactivity of a bioorthogonal reaction in a set of weakly expressing transmembrane receptor variants with a single uaa substitution at different sites. In principle, for a conjugation reaction between a peptide tag and a target protein, the reaction efficiency can be analyzed by either Western blot- or microplate immunoassay-based methods. Western blot-based methods have the advantage of simultaneously detecting the protein size and the degree of covalent modification of the target protein. However, Western blotting can be a cumbersome process involving many variables and multiple steps, including gel electrophoresis, membrane transfer, and immunodetection. Moreover, only a relatively small number of samples can be analyzed each time by Western blotting. In contrast, enzyme- or fluorophore-linked ISA-based immunoassays performed in microtiter plates offer higher throughput. ISA-based immunoassays typically require smaller amounts of sample and proceed rapidly to immunodetection by omitting an intermediate separation step.
We have earlier described a strategy for detecting azF-CCR5 variants labeled with phosphine reagents using a whole-cell-based ELISA. In this work, we utilized an ISA-based detection strategy, which is a variation of a standard ELISA, in which fluorescence detection can occur by simply having either a primary or secondary antibody coupled to a fluorophore. Fluorescent antibodies provide high sensitivity of detection, and their ability to multiplex makes fluorescence detection the method of choice for many applications. In this respect, near-IR (NIR, 700–900 nm) imaging is worth mentioning as a recent advancement in fluorescence detection with reduced artifacts from autofluorescence and light scattering compared to visible dyes.63 For instance, NIR quantum dots have been used to image tumor vasculature in vivo with significantly more information compared to visible quantum dots (QDs).64 There are also several reports on GPCR-related assays with studies on oligomerization,65 internalization,42 and signaling.66 In contrast to an ELISA, a fluorophore-linked ISA allows simultaneous dual-color quantification of two different antigens. The dual-color fluorophore-linked ISA scheme presented here allows ratiometric analysis of the degree of labeling.
Here we applied the SpAAC chemistry to target specific azido-CCR5 variants with a peptide epitope-tagged cyclooctyne to bypass the known disadvantages in the Staudinger ligation such as insufficient reaction stoichiometry with azF and phosphine oxidation.67 We first tested the ability of DBCO-FLAG to react with azF at two positions, N24 and F96, that we previously identified to be reactive with FLAG-phosphine. In an on-cell ISA-based readout, we observed a small but reproducibly detectable enhancement in signal for F96azF-CCR5 over the background. However, we also observed an increase in signal with mock-transfected cells (MT) after incubation with the DBCO-FLAG reagent, indicating nonspecific background reactions (cf. Figure 1B). This finding was not entirely surprising because previous reports have demonstrated that cyclooctynes participate in undesirable background reactions via a radical-based thiol–yne addition mechanism to modify cysteine residues in proteins.47 We further showed that detecting labeled receptors that were immunopurified dramatically reduced the number of nonspecifically labeled background bands (cf. Figure 1C).
Our observations led us to develop a multiplex ISA that would detect DBCO-FLAG-labeled CCR5 variants immunocaptured on a microtiter plate. We exploited the dual-color detection capability of the LI-COR system to achieve simultaneous quantification of the receptor expression level and the labeling efficiency. The dual-color detection of a double-sandwich ISA allows identification of three epitopes on a single molecular entity at once. DBCO-FLAG labeling of the azF-CCR5 variants was first performed on live cells. Full-length CCR5 variants were subsequently “immunopurified” by being captured on a 1D4 mAb-coated microplate. This step enhanced the specific signal by reducing (1) the C-terminally truncated CCR5 receptor population that could arise naturally or as a result of inefficient amber suppression and (2) the nonspecifically labeled proteins other than CCR5 that appeared in the Western blot as multiple nonspecific bands (Figure 1C). We chose biotinylated anti-CCR5 T21/8 mAb that targets an N-terminal sequence of CCR5 to monitor the expression levels, and anti-FLAG pAb to probe the labeling efficiency. On the basis of prior knowledge and experience with the Staudinger–Bertozzi ligation,34 we used F96azF-CCR5 to conduct initial optimization for the multiplex detection method. This multiplex ISA-based detection strategy allowed estimation of the label-to-receptor ratio in a semi-high-throughput fashion.
Finally, we applied the multiplex ISA-based detection strategy to screen for azF-CCR5 variants that can be efficiently modified by DBCO-FLAG. Because of the inherent nature of the infrared fluorescence imaging system, we present only the ratio of labeling and expression in arbitrary units. Although obtaining an absolute labeling ratio is desirable and important, it is beyond the scope of this study and will be addressed in the future. We tested a panel of 32 sites in the IC loops, TM helices, and EC sites of CCR5 and found that only some residues within the TM helices exhibited high reactivity with DBCO-FLAG. In contrast, phosphine-FLAG exhibited preferred reactivity with sites located in the EC and IC region.34 A particularly interesting example that highlights the contrast was the signal from labeled F109azF in TM3, which was very weak in the reaction with phosphine-FLAG but very strong for the reaction with DBCO-FLAG compared with those of the other tested azF-CCR5 variants.
The different site-dependent selectivity of DBCO and phosphine reagents can be explained by several factors. First, triaryl-phosphine is ∼20-fold more hydrophobic than DBCO as estimated by predicted octanol/water partitioning coefficients41 and thus more likely to permeate the cell membrane and label the IC region. The significantly lower labeling efficiency of DBCO in the IC region of CCR5 suggests that DBCO is less membrane-permeable than phosphine. Second, it was suggested in earlier studies that the SpAAC reaction with a protein might be modulated by local variations in the partition coefficient of the hydrophobic DBCO group and probe accessibility.2,49 We previously observed dramatic 4–400-fold rate enhancements when reacting analogous DIBO cyclooctyne reagents with the GPCR rhodopsin.49 In the study presented here, we treated the azF-CCR5 variants with 100 μM DBCO-FLAG for 1 h at 37 °C. Assuming values of 0.1, 1, 10, and 100 M–1 s–1 for the second-order rate constants between DBCO-FLAG and azF, the corresponding values for the extent of reaction after 1 h would be 3.5, 30, 97, and 100%, respectively. It is known that DIBAC, a cyclooctyne very similar to DBCO, has relatively low reactivity in model reactions with a second-order rate constant of 0.36 M–1 s–1.41 Therefore, under our reaction conditions, we expect reactivity of DBCO-FLAG would not suffice to make the reaction reach completion, and the labeling efficiency should be very sensitive to modulation of the local environment on the protein surface. Third, the strain-promoted cycloaddition between cyclooctyne and azide and the Staudinger ligation between phosphine and azide involved reaction intermediates with very different configurations. Consequently, the sterical constraints on the protein surface might affect these two reactions in distinct ways. The sterical consideration might also explain the site selectivity of DBCO within the TM core of the receptor (i.e., the selectivity of site 109 over site 108). If the rotation of DBCO is restricted in the binding pocket, it can react with only a limited subset of sites that allow the alkyne group to be aligned with the azide in the correct orientation. Fourth, the DBCO reagent used in this study includes a long and flexible PEG4 linker between the FLAG epitope and the bioorthogonal reactive group, which facilitates recognition by the anti-FLAG antibody. It is possible that the shorter phosphine-FLAG reagents, even if reacted with the site deep in the binding pocket, cannot be efficiently detected because the epitope is not fully exposed to the antibody. Thus, the enhanced detectability of the DBCO-FLAG reagent makes it a superior probe for evaluating the extent of site-specific modification, as the readout is less dependent on the accessibility of the epitope.
The most reactive site identified in our screen, F109, was particularly interesting because it is located deep in the binding pocket of CCR5 (Figure 4, red). We previously demonstrated by photo-cross-linking that F109azF interacts directly with the allosteric CCR5 inhibitor, maraviroc,27 and later the contact was confirmed by the crystal structure of the CCR5–maraviroc complex.68 This opens up potential applications of DBCO-modified azF-CCR5 such as (1) altering the pharmacology of CCR5 by tethering a DBCO-linked peptide in the binding pocket to induce conformational changes in CCR5, (2) identifying CCR5 allosteric modulators to treat maraviroc-resistance HIV infection69 by providing a completely different binding space for new chemical groups tethered to DBCO, and (3) aiding the rational drug design that targets CCR5 through DBCO-linked fragment-based drug discovery (FBDD). Because the FBDD approaches have been widely employed as a complementary strategy to conventional high-throughput screening (HTS) methods,70 DBCO may serve as an anchor to keep chemical fragments in the maraviroc binding pocket, even if these chemical fragments do not possess a minimum of millimolar binding affinities required by the traditional FBDD approach. DBCO-linked fragments can also serve as a space filler to exclude therapeutic candidates that share a similar binding mechanism with maraviroc during the drug discovery process. Moreover, via adjustment of the linker length between DBCO and fragments of interest, it might be possible to identify interactions between the fragment and CCR5 that are topologically distinct from those of the maraviroc binding pocket of CCR5.
Figure 4.
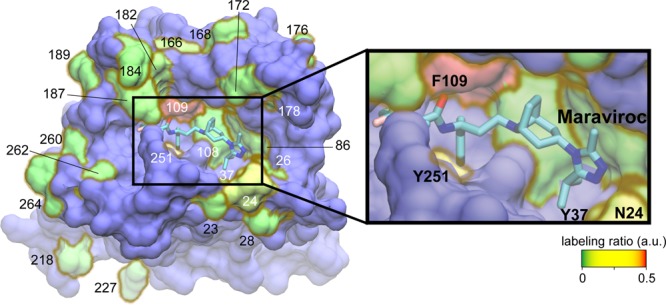
Surface representation of the CCR5–maraviroc complex (Protein Data Bank entry 4MBS(68)) with an inset showing a close-up of the binding pocket of maraviroc. Four sites (N24, Y37, F109, and Y251) identified to have high DBCO-FLAG reactivity are shown, and maraviroc is depicted as sticks (cyan, carbon; blue, nitrogen; red, oxygen). Site F96 initially used for optimization of the multiplex detection strategy is not visible in this view of the receptor. The color bar indicates the degree of DBCO-FLAG labeling reactivity from nonreactive sites (green) to highly reactive sites (red). Residues not tested in this study are color-coded in blue. This molecular graphic was prepared with VMD version 1.9.1.71
Of note, we also evaluated the performance of the multiplex ISA-based detection method with an N-terminal capture scheme (cf. Figure S3 of the Supporting Information). In a separate screening for the 32 azF-CCR5 variants described above (cf. Figure 3), we used N-terminally specific anti-CCR5 T21/8 mAb to capture the receptor to the plate surface and the C-terminally specific 1D4 mAb to estimate the receptor expression level. The goal was to assess whether the FLAG pAb accessibility was dependent on the orientation in which the receptor was captured to the plate. The reactivity profiles for the azF-CCR5 variants measured under the N-terminal capture scheme were similar to the earlier screening results under the N-terminal capture scheme, although the 1D4 signal, the FLAG signal, and the ratio of specific FLAG signal to background FLAG signal turned out to be lower (cf. Figure S3 of the Supporting Information). These results highlight the flexibility of the method with respect to the choice of antibody pairs for recognition and detection. Additionally, we have preliminary results that suggest successful use of alternative peptide epitope tags and GPCRs demonstrating the versatility of the labeling and detection strategy.
In summary, we report here a sandwich ISA-based CCR5 screening assay that identifies sites that, when substituted with azF, are efficiently modified with SpAAC reaction. This assay offers various advantages, including (1) the small quantities of samples required for detection and a low level of reagent consumption, (2) multiplex detection, (3) flexibility in combining the capture and detection strategies, and (4) higher throughput. The screening strategy described here should provide a universal platform for identifying suitable sites for bioorthogonal tagging of GPCRs, as a wide variety of epitope tags and antibodies are available. We envision the SpAAC reaction, with its superior labeling stoichiometry and expansive range of selectable reagents, to have broad applicability as a method for site-specifically introducing desired probes or handles into GPCRs. The labeling strategy can be further improved by minimizing the cross-reactivity with cysteine thiols, a goal of future work. The screening strategy described here should facilitate the preparation of stoichiometrically labeled receptors for single-molecule fluorescence studies.2
Acknowledgments
We thank Manija A. Kazmi for scientific assistance and helpful discussion during manuscript preparation. Peptide synthesis was performed by the Proteomics Resource Center of the Rockefeller University.
Glossary
Abbreviations
- azF
p-azido-l-phenylalanine
- BARAC
biarylazacyclooctynone
- BCN
bicyclo[6.1.0]nonyne
- BTTES
2-[4-({bis[(1-tert-butyl-1H-1,2,3-triazol-4-yl)methyl]amino}methyl)-1H-1,2,3-triazol-1-yl] ethyl hydrogen sulfate
- CCR5
human C–C chemokine receptor 5
- DIBO
dibenzocyclooctyne
- DBCO
aza-dibenzocyclooctyne
- DM
n-dodecyl β-d-maltoside
- DIFO
difluorinated cyclooctyne
- EC
extracellular
- ELISA
enzyme-linked immunosorbent assay
- DIMAC
dimethoxyazacyclooctyne
- FlAsH
fluorescein arsenical hairpin binder
- FRET
fluorescence resonance energy transfer
- GPCR
G protein-coupled receptor
- IC
intracellular
- IgG
immunoglobulin G
- mAb
monoclonal antibody
- ISA
immunosorbent assay
- ReAsH
resorufin arsenical hairpin binder
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SpAAC
strain-promoted azide–alkyne [3+2] cycloaddition
- TBTA
tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine
- THPTA
tris(3-hydroxypropyltriazolymethyl)amine
- TM
transmembrane
- uaa
unnatural amino acid
- wt
wild type.
Supporting Information Available
Supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Immunoassay Development, Siemens Healthcare Diagnostics, Inc., Tarrytown, NY 10591.
Author Contributions
S.N., S.R.-S., and M.P. contributed equally to this work.
S.R.-S. and H.T. were funded in part by National Institutes of Health Training Grant T32 DK-07313 and the Tri-Institutional Program in Chemical Biology, respectively. This work was also supported by the Danica Foundation, the Crowley Family Fund, and an International Research Alliance at the Novo Nordisk Foundation Center for Basic Metabolic Research through an unconditional grant from the Novo Nordisk Foundation to the University of Copenhagen.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Khoury E.; Clement S.; Laporte S. A. (2014) Allosteric and Biased G Protein-Coupled Receptor Signaling Regulation: Potentials for New Therapeutics. Front. Endocrinol. 5, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber T.; Sakmar T. P. (2014) Chemical Biology Methods for Investigating G Protein-Coupled Receptor Signaling. Chem. Biol. 21, 1224–1237. [DOI] [PubMed] [Google Scholar]
- Bockenhauer S.; Furstenberg A.; Yao X. J.; Kobilka B. K.; Moerner W. E. (2011) Conformational Dynamics of Single G Protein-Coupled Receptors in Solution. J. Phys. Chem. B 115, 13328–13338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D.; Rieken F.; Wagner J.; Sungkaworn T.; Zabel U.; Borzi A.; Cocucci E.; Zurn A.; Lohse M. J. (2013) Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc. Natl. Acad. Sci. U.S.A. 110, 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber T.; Sakmar T. P. (2011) Escaping the flatlands: New approaches for studying the dynamic assembly and activation of GPCR signaling complexes. Trends Pharmacol. Sci. 32, 410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T.; Li N.; Fang X. (2013) Single-molecule fluorescence imaging in living cells. Annu. Rev. Phys. Chem. 64, 459–480. [DOI] [PubMed] [Google Scholar]
- Yang H. (2010) Progress in single-molecule spectroscopy in cells. Curr. Opin. Chem. Biol. 14, 3–9. [DOI] [PubMed] [Google Scholar]
- Snaar-Jagalska B. E.; Cambi A.; Schmidt T.; de Keijzer S. (2013) Single-Molecule Imaging Technique to Study the Dynamic Regulation of GPCR Function at the Plasma Membrane. Methods Enzymol. 521, 47–67. [DOI] [PubMed] [Google Scholar]
- Xu X., Brzostowski J. A., and Jin T. (2009) Monitoring Dynamic GPCR Signaling Events Using Fluorescence Microscopy, FRET Imaging, and Single-Molecule Imaging. In Chemotaxis: Methods and Protocols (Jin T., and Hereld D., Eds.) pp 371–383, Humana Press Inc., Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Saini D. K., and Gautam N. (2010) Live Cell Imaging for Studying G Protein-Coupled Receptor Activation in Single Cells. In Analgesia: Methods and Protocols (Szallasi A., Ed.) pp 191–207, Humana Press Inc., Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Ward R. J.; Milligan G. (2014) Structural and biophysical characterisation of G protein-coupled receptor ligand binding using resonance energy transfer and fluorescent labelling techniques. Biochim. Biophys. Acta 1838, 3–14. [DOI] [PubMed] [Google Scholar]
- Ramil C. P.; Lin Q. (2013) Bioorthogonal chemistry: Strategies and recent developments. Chem. Commun. 49, 11007–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. (2009) Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed. 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrens D. L.; Khorana H. G. (1995) Structure and function in rhodopsin. Measurement of the rate of metarhodopsin II decay by fluorescence spectroscopy. J. Biol. Chem. 270, 5073–5076. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H.; Farrens D. L. (2013) A constitutively activating mutation alters the dynamics and energetics of a key conformational change in a ligand-free G protein-coupled receptor. J. Biol. Chem. 288, 28207–28216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liapakis G., Simpson M. M., and Javitch J. A. (2001) The Substituted-Cysteine Accessibility Method (SCAM) to Elucidate Membrane Protein Structure. In Current Protocols in Neuroscience, John Wiley & Sons, Inc., New York. [DOI] [PubMed] [Google Scholar]
- Spagnuolo C., Joselevich M., Leskow F., and Jares-Erijman E. (2011) Tetracysteine and Bipartite Tags for Biarsenical Organic Fluorophores. In Advanced Fluorescence Reporters in Chemistry and Biology III (Demchenko A. P., Ed.) pp 263–295, Springer, Berlin. [Google Scholar]
- Ziegler N.; Batz J.; Zabel U.; Lohse M. J.; Hoffmann C. (2011) FRET-based sensors for the human M1-, M3-, and M5-acetylcholine receptors. Bioorg. Med. Chem. 19, 1048–1054. [DOI] [PubMed] [Google Scholar]
- Granier S.; Kim S.; Fung J. J.; Bokoch M. P.; Parnot C. (2009) FRET-based measurement of GPCR conformational changes. Methods Mol. Biol. 552, 253–268. [DOI] [PubMed] [Google Scholar]
- Daggett K. A.; Sakmar T. P. (2011) Site-specific in vitro and in vivo incorporation of molecular probes to study G-protein-coupled receptors. Curr. Opin. Chem. Biol. 15, 392–398. [DOI] [PubMed] [Google Scholar]
- Ye S.; Kohrer C.; Huber T.; Kazmi M.; Sachdev P.; Yan E. C. Y.; Bhagat A.; RajBhandary U. L.; Sakmar T. P. (2008) Site-specific incorporation of keto amino acids into functional G protein-coupled receptors using unnatural amino acid mutagenesis. J. Biol. Chem. 283, 1525–1533. [DOI] [PubMed] [Google Scholar]
- Chin J. W.; Cropp T. A.; Anderson J. C.; Mukherji M.; Zhang Z. W.; Schultz P. G. (2003) An expanded eukaryotic genetic code. Science 301, 964–967. [DOI] [PubMed] [Google Scholar]
- Liu C. C.; Schultz P. G. (2010) Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem. 79, 413–444. [DOI] [PubMed] [Google Scholar]
- Ye S.; Zaitseva E.; Caltabiano G.; Schertler G. F. X.; Sakmar T. P.; Deupi X.; Vogel R. (2010) Tracking G-protein-coupled receptor activation using genetically encoded infrared probes. Nature 464, 1386–1390. [DOI] [PubMed] [Google Scholar]
- Ye S.; Huber T.; Vogel R.; Sakmar T. P. (2009) FTIR analysis of GPCR activation using azido probes. Nat. Chem. Biol. 5, 397–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunbeck A.; Huber T.; Sachdev P.; Sakmar T. P. (2011) Mapping the Ligand-Binding Site on a G Protein-Coupled Receptor (GPCR) Using Genetically Encoded Photocrosslinkers. Biochemistry 50, 3411–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunbeck A.; Huber T.; Abrol R.; Trzaskowski B.; Goddard W. A. III; Sakmar T. P. (2012) Genetically Encoded Photo-cross-linkers Map the Binding Site of an Allosteric Drug on a G Protein-Coupled Receptor. ACS Chem. Biol. 7, 967–972. [DOI] [PubMed] [Google Scholar]
- Grunbeck A.; Huber T.; Sakmar T. P. (2013) Mapping a Ligand Binding Site Using Genetically Encoded Photoactivatable Crosslinkers. Methods Enzymol. 520, 307–322. [DOI] [PubMed] [Google Scholar]
- Coin I.; Katritch V.; Sun T.; Xiang Z.; Siu F. Y.; Beyermann M.; Stevens R. C.; Wang L. (2013) Genetically encoded chemical probes in cells reveal the binding path of urocortin-I to CRF class B GPCR. Cell 155, 1258–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray-Saha S.; Huber T.; Sakmar T. P. (2014) Antibody epitopes on G protein-coupled receptors mapped with genetically encoded photoactivatable cross-linkers. Biochemistry 53, 1302–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Hansen L.; Park M.; Huber T.; Grunbeck A.; Naganathan S.; Schwartz T. W.; Sakmar T. P. (2014) Mapping Substance P Binding Sites on the Neurokinin-1 Receptor Using Genetic Incorporation of a Photoreactive Amino Acid. J. Biol. Chem. 289, 18045–18054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Riou M.; Yao C. A.; Carvalho S.; Rodriguez P. C.; Bensaude O.; Paoletti P.; Ye S. (2014) Genetically encoding a light switch in an ionotropic glutamate receptor reveals subunit-specific interfaces. Proc. Natl. Acad. Sci. U.S.A. 111, 6081–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippenstein V.; Ghisi V.; Wietstruk M.; Plested A. J. (2014) Photoinactivation of glutamate receptors by genetically encoded unnatural amino acids. J. Neurosci. 34, 980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganathan S.; Ye S.; Sakmar T. P.; Huber T. (2013) Site-Specific Epitope Tagging of G Protein-Coupled Receptors by Bioorthogonal Modification of a Genetically Encoded Unnatural Amino Acid. Biochemistry 52, 1028–1036. [DOI] [PubMed] [Google Scholar]
- Naganathan S.; Grunbeck A.; Tian H.; Huber T.; Sakmar T. P. (2013) Genetically-encoded molecular probes to study G protein-coupled receptors. J. Visualized Exp. 10.3791/50588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescher J. A.; Dube D. H.; Bertozzi C. R. (2004) Chemical remodelling of cell surfaces in living animals. Nature 430, 873–877. [DOI] [PubMed] [Google Scholar]
- Huber T.; Naganathan S.; Tian H.; Ye S.; Sakmar T. P. (2013) Unnatural amino acid mutagenesis of GPCRs using amber codon suppression and bioorthogonal labeling. Methods Enzymol. 520, 281–305. [DOI] [PubMed] [Google Scholar]
- Besanceney-Webler C.; Jiang H.; Zheng T.; Feng L.; Soriano del Amo D.; Wang W.; Klivansky L. M.; Marlow F. L.; Liu Y.; Wu P. (2011) Increasing the Efficacy of Bioorthogonal Click Reactions for Bioconjugation: A Comparative Study. Angew. Chem., Int. Ed. 50, 8051–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. (2011) From mechanism to mouse: A tale of two bioorthogonal reactions. Acc. Chem. Res. 44, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa T.; Ishii R.; Fukunaga R.; Kobayashi T.; Sakamoto K.; Yokoyama S. (2008) Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode Nε-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem. Biol. 15, 1187–1197. [DOI] [PubMed] [Google Scholar]
- Debets M. F.; van der Doelen C. W. J.; Rutjes F. P. J. T.; van Delft F. L. (2010) Azide: A Unique Dipole for Metal-Free Bioorthogonal Ligations. ChemBioChem 11, 1168–1184. [DOI] [PubMed] [Google Scholar]
- Miller J. W. (2004) Tracking G Protein-coupled Receptor Trafficking Using Odyssey Imaging, LI-COR Biosciences (www.licor.com). [Google Scholar]
- Chang P. V.; Prescher J. A.; Sletten E. M.; Baskin J. M.; Miller I. A.; Agard N. J.; Lo A.; Bertozzi C. R. (2010) Copper-free click chemistry in living animals. Proc. Natl. Acad. Sci. U.S.A. 107, 1821–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H.; Sakmar T. P.; Huber T. (2013) Site-specific labeling of genetically encoded azido groups for multicolor, single-molecule fluorescence imaging of GPCRs. Methods Cell Biol. 117, 267–303. [DOI] [PubMed] [Google Scholar]
- Knepp A. M.; Grunbeck A.; Banerjee S.; Sakmar T. P.; Huber T. (2011) Direct measurement of thermal stability of expressed CCR5 and stabilization by small molecule ligands. Biochemistry 50, 502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollok-Kopp B.; Schwarze K.; Baradari V. K.; Oppermann M. (2003) Analysis of ligand-stimulated CC chemokine receptor 5 (CCR5) phosphorylation in intact cells using phosphosite-specific antibodies. J. Biol. Chem. 278, 2190–2198. [DOI] [PubMed] [Google Scholar]
- van Geel R.; Pruijn G. J. M.; van Delft F. L.; Boelens W. C. (2012) Preventing Thiol-Yne Addition Improves the Specificity of Strain-Promoted Azide–Alkyne Cycloaddition. Bioconjugate Chem. 23, 392–398. [DOI] [PubMed] [Google Scholar]
- Lee B.; Sharron M.; Blanpain C.; Doranz B. J.; Vakili J.; Setoh P.; Berg E.; Liu G.; Guy H. R.; Durell S. R.; Parmentier M.; Chang C. N.; Price K.; Tsang M.; Doms R. W. (1999) Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in chemokine and coreceptor function. J. Biol. Chem. 274, 9617–9626. [DOI] [PubMed] [Google Scholar]
- Tian H.; Naganathan S.; Kazmi M. A.; Schwartz T. W.; Sakmar T. P.; Huber T. (2014) Bioorthogonal fluorescent labeling of functional G-protein-coupled receptors. ChemBioChem 15, 1820–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttamapinant C.; Sanchez M. I.; Liu D. S.; Yao J. Z.; Ting A. Y. (2013) Site-specific protein labeling using PRIME and chelation-assisted click chemistry. Nat. Protoc. 8, 1620–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttamapinant C.; Tangpeerachaikul A.; Grecian S.; Clarke S.; Singh U.; Slade P.; Gee K. R.; Ting A. Y. (2012) Fast, cell-compatible click chemistry with copper-chelating azides for biomolecular labeling. Angew. Chem., Int. Ed. 51, 5852–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon C. G.; Mackey J. L.; Jewett J. C.; Sletten E. M.; Houk K. N.; Bertozzi C. R. (2012) Reactivity of biarylazacyclooctynones in copper-free click chemistry. J. Am. Chem. Soc. 134, 9199–9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S. W.; Lee S.; Na J. H.; Yoon H. I.; Lee D. E.; Koo H.; Cho Y. W.; Kim S. H.; Jeong S. Y.; Kwon I. C.; Choi K.; Kim K. (2014) Cell Labeling and Tracking Method without Distorted Signals by Phagocytosis of Macrophages. Theranostics 4, 420–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedrow-Byers J. R.; Moore A. L.; Ganguly T.; Hopkins M. R.; Fulton M. D.; Benny P. D.; Berkman C. E. (2013) PSMA-targeted SPECT agents: Mode of binding effect on in vitro performance. Prostate 73, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman E. S.; Heibeck T. H.; Gill A.; Li X.; Murray C. J.; Madlansacay M. R.; Tran C.; Uter N. T.; Yin G.; Rivers P. J.; Yam A. Y.; Wang W. D.; Steiner A. R.; Bajad S. U.; Penta K.; Yang W.; Hallam T. J.; Thanos C. D.; Sato A. K. (2014) Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconjugate Chem. 25, 351–361. [DOI] [PubMed] [Google Scholar]
- Beatty K. E.; Fisk J. D.; Smart B. P.; Lu Y. Y.; Szychowski J.; Hangauer M. J.; Baskin J. M.; Bertozzi C. R.; Tirrell D. A. (2010) Live-cell imaging of cellular proteins by a strain-promoted azide-alkyne cycloaddition. ChemBioChem 11, 2092–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbo P.; Novoa S.; Biesinger M. C.; Workentin M. S. (2013) Interfacial strain-promoted alkyne-azide cycloaddition (I-SPAAC) for the synthesis of nanomaterial hybrids. Chem. Commun. 49, 3982–3984. [DOI] [PubMed] [Google Scholar]
- Lee D. E.; Na J. H.; Lee S.; Kang C. M.; Kim H. N.; Han S. J.; Kim H.; Choe Y. S.; Jung K. H.; Lee K. C.; Choi K.; Kwon I. C.; Jeong S. Y.; Lee K. H.; Kim K. (2013) Facile method to radiolabel glycol chitosan nanoparticles with 64Cu via copper-free click chemistry for MicroPET imaging. Mol. Pharmaceutics 10, 2190–2198. [DOI] [PubMed] [Google Scholar]
- Koo H.; Lee S.; Na J. H.; Kim S. H.; Hahn S. K.; Choi K.; Kwon I. C.; Jeong S. Y.; Kim K. (2012) Bioorthogonal copper-free click chemistry in vivo for tumor-targeted delivery of nanoparticles. Angew. Chem., Int. Ed. 51, 11836–11840. [DOI] [PubMed] [Google Scholar]
- Hao J.; Huang L. L.; Zhang R.; Wang H. Z.; Xie H. Y. (2012) A mild and reliable method to label enveloped virus with quantum dots by copper-free click chemistry. Anal. Chem. 84, 8364–8370. [DOI] [PubMed] [Google Scholar]
- Mbua N. E.; Guo J.; Wolfert M. A.; Steet R.; Boons G.-J. (2011) Strain-Promoted Alkyne–Azide Cycloadditions (SPAAC) Reveal New Features of Glycoconjugate Biosynthesis. ChemBioChem 12, 1912–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon E.; Bertozzi C. R. (2000) Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- Piruska A.; Nikcevic I.; Lee S. H.; Ahn C.; Heineman W. R.; Limbach P. A.; Seliskar C. J. (2005) The autofluorescence of plastic materials and chips measured under laser irradiation. Lab Chip 5, 1348–1354. [DOI] [PubMed] [Google Scholar]
- Allen P. M.; Liu W.; Chauhan V. P.; Lee J.; Ting A. Y.; Fukumura D.; Jain R. K.; Bawendi M. G. (2010) InAs(ZnCdS) quantum dots optimized for biological imaging in the near-infrared. J. Am. Chem. Soc. 132, 470–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel E.; Murray V. L.; Liu A. P. (2014) Multiplex Detection of Homo- and Heterodimerization of G Protein-Coupled Receptors by Proximity Biotinylation. PLoS One 9, e93646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkirk J. V.; Nottebaum L. M.; Ford I. C.; Santos M.; Malany S.; Foster A. C.; Lechner S. M. (2006) A novel cell-based assay for G-protein-coupled receptor-mediated cyclic adenosine monophosphate response element binding protein phosphorylation. J. Biomol. Screening 11, 351–358. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. (2004) A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- Tan Q.; Zhu Y.; Li J.; Chen Z.; Han G. W.; Kufareva I.; Li T.; Ma L.; Fenalti G.; Li J.; Zhang W.; Xie X.; Yang H.; Jiang H.; Cherezov V.; Liu H.; Stevens R. C.; Zhao Q.; Wu B. (2013) Structure of the CCR5 Chemokine Receptor–HIV Entry Inhibitor Maraviroc Complex. Science 341, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berro R.; Yasmeen A.; Abrol R.; Trzaskowski B.; Abi-Habib S.; Grunbeck A.; Lascano D.; Goddard W. A. III; Klasse P. J.; Sakmar T. P.; Moore J. P. (2013) Use of G-protein-coupled and -uncoupled CCR5 receptors by CCR5 inhibitor-resistant and -sensitive human immunodeficiency virus type 1 variants. J. Virol. 87, 6569–6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M.; Chessari G.; Tisi D.; Woodhead A. J. (2008) Recent developments in fragment-based drug discovery. J. Med. Chem. 51, 3661–3680. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: Visual molecular dynamics. J. Mol. Graphics 14, 27–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.