

Abstract
Assembly of polymerase chain reactions at room temperature can sometimes lead to low yields or unintentional products due to mispriming. Mutation of isoleucine 707 to leucine in DNA polymerase I from Thermus aquaticus substantially decreases its activity at room temperature without compromising its ability to amplify DNA. To understand why a conservative change to the enzyme over 20 Å from the active site can have a large impact on its activity at low temperature, we solved the X-ray crystal structure of the large (5′-to-3′ exonuclease-deleted) fragment of Taq DNA polymerase containing the cold-sensitive mutation in the ternary (E–DNA–ddNTP) and binary (E–DNA) complexes. The I707L KlenTaq1 ternary complex was identical to the wild-type in the closed conformation except for the mutation and a rotamer change in nearby phenylalanine 749, suggesting that the enzyme should remain active. However, soaking out of the nucleotide substrate at low temperature results in an altered binary complex made possible by the rotamer change at F749 near the tip of the polymerase O-helix. Surprisingly, two adenosines in the 5′-template overhang fill the vacated active site by stacking with the primer strand, thereby blocking the active site at low temperature. Replacement of the two overhanging adenosines with pyrimidines substantially increased activity at room temperature by keeping the template overhang out of the active site, confirming the importance of base stacking. These results explain the cold-sensitive phenotype of the I707L mutation in KlenTaq1 and serve as an example of a large conformational change affected by a conservative mutation.
When comparing homologous proteins, changes to another member of a group of amino acids with similar chemical side chain characteristics (e.g., hydrophobic, polar and uncharged, polar and charged, aromatic) are often dismissed as inconsequential. Two hydrophobic amino acids, leucine and isoleucine, have the same molecular weight, atomic composition, and volume. These two amino acids differ only in the position of a branching methyl group in the side chain, leucine at the γ carbon and isoleucine at the β carbon.
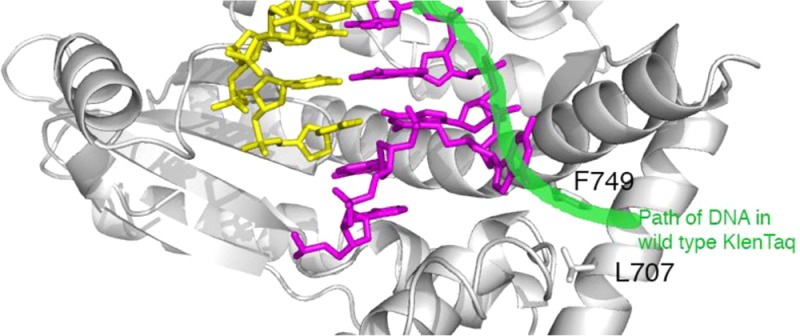
It is thus remarkable that mutation from isoleucine to leucine at position 707 in DNA polymerase I from Thermus aquaticus resulted in an unusual cold-sensitive phenotype. DNA polymerase I from T. aquaticus, or Taq DNA polymerase, is one of the best characterized DNA polymerases and the first thermostable enzyme to be employed for the polymerase chain reaction (PCR).1 One problem with Taq DNA polymerase as a PCR enzyme is that it remains active at low temperatures, leading to nonspecific priming.1,2 The amplification of nontarget sequences due to mispriming can lead to spurious products and low yields of the target sequence. This can be avoided by decreasing the activity of Taq DNA polymerase at low temperatures during reaction assembly and prior to heating. Kermekchiev and colleagues3 randomly mutated the large (5′-to-3′ exonuclease-deleted) fragment of Taq DNA polymerase (Klenow fragment of Taq DNA polymerase, or KlenTaq1) and screened mutants for low activity at 37 °C while retaining high activity at 68 °C. One mutation, isoleucine 707 to leucine, resulted in a large increase in the ratio of activity at high versus low temperature and a dramatic improvement in amplification of difficult PCR targets.3 According to the crystal structure of the wild-type KlenTaq1,4 this conservative mutation was located near the exterior of the enzyme and over 20 Å away from the polymerase active site (Figure 1A). It was not clear how this mutation, termed Cs3C, could selectively alter the polymerase mechanism at low temperature.
Figure 1.
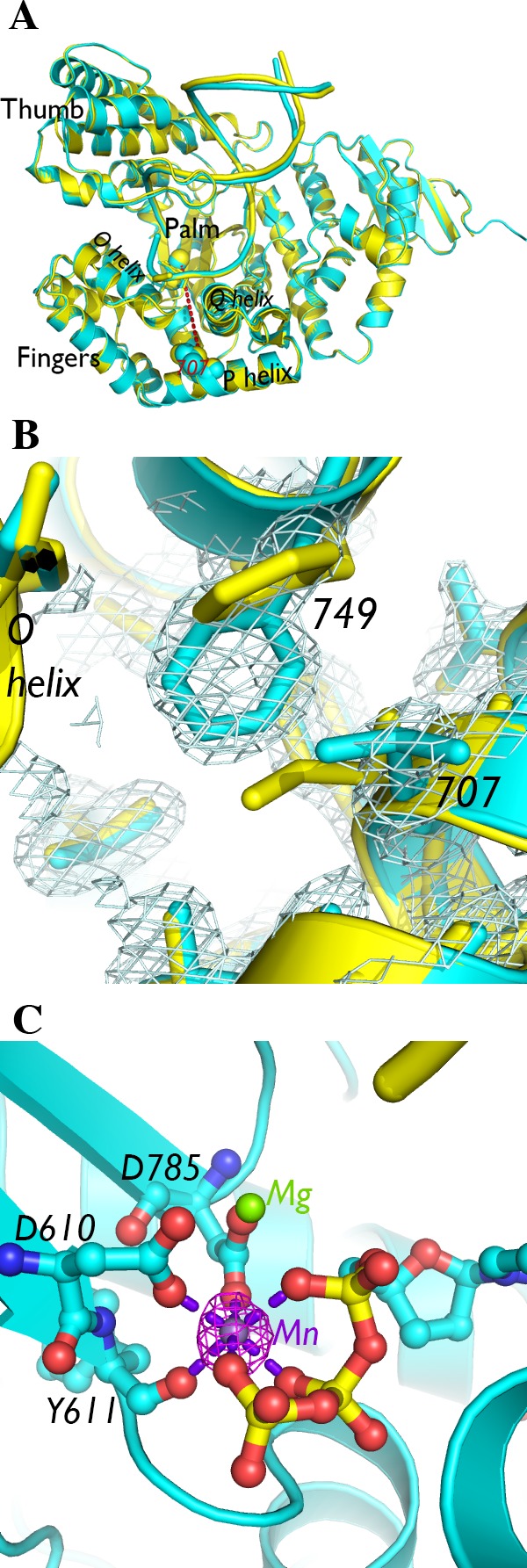
Crystal structure of I707L KlenTaq1–DNA–dNTP ternary complex. (A) The crystal structure of the I707L mutant ternary complex (cyan) superimposed on the wild-type KlenTaq1 ternary complex (3KTQ; yellow). Residue 707 is connected to an active site divalent cation with a red dashed line corresponding to ∼24 Å long. (B) The 2Fo – Fc map of the mutant ternary complex near the residue 707 mutation is shown at 1.0σ. (C) The active site, consisting of conserved aspartic acids and a ddCTP, contains two metal ions, one of which is associated with strong anomalous difference density (displayed at 4.0σ in magenta) around a divalent cation site. The manganese(II) atom is connected to nearby oxygen atoms with purple dashed lines corresponding to ∼2.1 Å long. The interatomic distances between the magnesium(II) atom and surrounding oxygen atoms are also ∼2.1 Å.
DNA polymerase I enzymes employ a complex induced-fit mechanism to bind and select the complementary nucleotide during DNA synthesis. DNA polymerase I enzymes belong to the A family of DNA polymerases and are shaped like a human hand, with a thumb subdomain that grasps the DNA, a palm subdomain that contains the active site, and a fingers subdomain that contains the substrate binding site.5,6 According to numerous crystal structures of enzymes in this class,4,7−9 DNA polymerase I enzymes transition from an empty enzyme–DNA binary complex with a wide active site cleft (the open conformation) to a clamped ternary complex (the closed conformation) when bound to a complementary incoming 2′-deoxynucleoside triphosphate (dNTP). This transition requires a large rotation of the fingers subdomain, specifically of the O helix, to clamp around the bound dNTP. Kermekchiev and colleagues3 suggested that the I707L mutation in KlenTaq1 could obstruct the transition between open and closed conformations because of its location near the mobile O helix, thereby slowing the enzyme.
We sought to determine the reason for the cold sensitivity of I707L KlenTaq1 by solving its crystal structure in the ternary and binary complexes. The atomic structure of the mutant enzyme might allow the visualization of the perturbation of the enzyme structure by the alternative placement of the branching methyl group at the side chain of residue 707. Surprisingly, the binary crystal structure of I707L KlenTaq1 revealed a large reorganization of the fingers subdomain and the DNA at low temperatures, resulting in blockage of the polymerase active site.
Materials and Methods
Materials
The wild-type and I707L mutant (Cs3C) KlenTaq1 DNA polymerase, which is an N-terminal deletion of 278 amino acids of Taq DNA polymerase, were produced and purified as previously described.3,10 Purified protein was dialyzed into buffer A (50 mM Tris-HCl, pH 7.5, 1 mM ethylenediaminetetraacetic acid, 6.5 mM 2-mercaptoethanol) and then bound to an equilibrated heparin agarose (Bio-Rad Laboratories, Hercules, CA) column to remove detergents for crystallography. The protein was eluted with buffer A plus 1.5 M NaCl and then desalted back into buffer A using diafiltration in an Amicon Ultra filter (EMD Millipore, Billerica, MA). Synthetic complementary oligonucleotides were designed for crystallization (16-mer Template, 5′-AAAGGGCGCCGTGGTC-3′; TemplateTTT, 5′-TTTGGGCGCCGTGGTC-3′; and 11-mer primer, 5′-GACCACGGCGC-3′), and solution kinetics assays (60-mers AAGTemplate, 5′-AAGCACTGTCCTTGACACGTTGATGGATTAGAGCAATCACACCCGAGACTGGCTATGCAC-3′; TTGTemplate, 5′- TTGCACTGTCCTTGACACGTTGATGGATTAGAGCAATCACACCCGAGACTGGCTATGCAC-3′; CCGTemplate, 5′- CCGCACTGTCCTTGACACGTTGATGGATTAGAGCAATCACACCCGAGACTGGCTATGCAC-3′; and 57-mer 6FAMPrimer, 5′-[6FAM] GTGCATAGCCAGTCTCGGGTGTGATTGCTCTAATCCATCAACGTGTCAAGGACAGTG-3′ with a 6-carboxyfluorescein (6FAM) fluorophore attached to the 5′-end) (Eurofins MWG/Operon, Huntsville, Alabama).
X-ray Crystallography of I707L KlenTaq1 DNA Polymerase
The I707L mutant KlenTaq1 DNA polymerase was crystallized with primer–template and substrate 2′,3′-dideoxycytidine triphophosphate (ddCTP) essentially as described for the wild-type KlenTaq1 DNA polymerase.4 Briefly, the protein was mixed with excess annealed 11-mer Primer and 16-mer Template or TemplateTTT, ddCTP, and magnesium chloride. The crystals formed in hanging drops over a well solution of 0.1 M HEPES, pH 7.5, 20 mM MnCl2, 0.1 M Na acetate, and 10% (w/v) PEG4000. To create the binary enzyme–DNA complex, the ddCTP was soaked out of single crystals in drops of mother liquor for over a week. Crystals were cryoprotected as previously described4 prior to being plunged into liquid nitrogen. Diffraction data sets for the binary and ternary complexes were collected at beamlines 22-ID and 22-BM, respectively, of the Advanced Photon Source, Argonne National Laboratory (Argonne, IL) (Table 1).
Table 1. Crystallographic Statisticsa.
| I707L–DNA–ddCTP | I707L–DNA(AAA) | I707L–DNA(TTT) | |
|---|---|---|---|
| space group | P3121 | P3121 | P3121 |
| cell dimensions a, b, c (Å) | 107.68, 107.68, 89.68; α = β = 90°, γ = 120° | 110.04, 110.04, 91.29; α = β = 90°, γ = 120° | 110.03, 110.03, 90.67; α = β = 90°, γ = 120° |
| resolution (Å) | 32.80–1.67 (1.70–1.67) | 47.65–2.20 (2.26–2.20) | 47.64–2.50 (2.56–2.50) |
| no. of reflections | 67 931 (2624) | 32 282 (2330) | 21 497 (1557) |
| Rwork/Rfree | 0.161/0.192 | 0.223/0.273 | 0.223/0.286 |
| I/σ(I) | 30.2 (2.78) | 12.0 (2.75) | 11.6 (2.56) |
| percent completeness | 97.3 (76.3) | 98.5 (98.6) | 96.4 (97.4) |
| redundancy | 3.6 (2.7) | 3.6 (3.7) | 4.6 (4.0) |
| Rsym | 0.047 (0.376) | 0.060 (0.681) | 0.120 (0.842) |
| atoms in asymmetric unit | 5441 | 4431 | 4723 |
| Average B-Factors | |||
| protein | 24.1 | 56.6 | 55.3 |
| DNA | 22.0 | 48.6 | 39.5 |
| waters | 35.7 | 46.3 | 34.0 |
| ligands/ions | 24.8 | 45.6 | 45.0 |
| Root-Mean-Square Deviation | |||
| bond lengths (Å) | 0.011 | 0.013 | 0.009 |
| bond angles (deg) | 1.6 | 1.7 | 1.4 |
| Ramachandran outliersb | 0.2% | 1.0% | 1.5% |
| Protein Data Bank code | 4N5S | 4N56 | 4XIU |
Data for the highest-resolution shell are in parentheses.
From Molprobity.32
Data sets were indexed, integrated, and scaled with HKL200011 for the ternary complex and XDS12 and phased by molecular replacement using Phaser.13 Model editing and refinement were performed using Coot14,15 and REFMAC5.16
Kinetics of Single Nucleotide Addition
The fluorescent pre-steady-state kinetics assay for nucleotide addition was performed essentially as previously described.17 Briefly, the fluorescent 6FAMPrimer was mixed with AAGTemplate, TTGTemplate, and CCGTemplate at equimolar concentrations in separate tubes and annealed by heating to 85 °C and slowly cooling to room temperature. To measure the nucleotide incorporation rate of deoxycytosine triphosphate (dCTP) opposite a 5′-XXG-3′ (where XX represents AA, CC, or TT) overhang on a 6-FAM tagged template and primer DNA complex in the I707L mutant and wild-type Klentaq1 polymerases, separate quench flow assays were performed. The assays were conducted by mixing the enzyme (4 μM)–DNA (0.2 μM) complex with equal volumes of 20 μM dCTP and quenching the reaction at four time points using four reaction volumes of a 95% formamide, 20 mM ethylenediaminetetraacetic acid solution. The enzymes, fluorescent DNA, and dCTP were all diluted from their original concentrations with reaction buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 10 mM MgCl2). All room-temperature reactions were executed manually on the benchtop. The primer lengths and amounts were measured by capillary electrophoresis with fluorescence detection (Duke University DNA Analysis Facility, Durham, NC), and the results were analyzed using PeakScanner software (Applied Biosystems) to determine the fraction of fluorescent primer extended. KaleidaGraph software (Synergy Software, Reading, PA) was used to fit plots of fraction primer extended versus time to the equation y = c(1 – e–kt).
Molecular Dynamics Simulation
The structure of KlenTaq1 DNA polymerase was obtained from the Protein Data Bank (PDB code: 4KTQ). In silico changes were made to modify residue 707 from an isoleucine to a leucine and to match the template sequence to that of the Template oligonucleotide. Explicit hydrogen atoms were added using the tleap module of AmberTools18 followed by the addition of Na+ counterions to neutralize the system. The system was solvated with a truncated octahedron unit cell with TIP3P water molecules19 using a 12.0 Å solvent buffer between the solute and the closest edge of the unit cell for a total atom count of 73 778 atoms. The Amber ff12SB force field18 was applied to the protein and DNA residues. The GPU-accelerated pmemd code19 of Amber 1218 was used to perform all minimizations and molecular dynamics (MD). The initial structure was minimized using a seven-step procedure involving 1000 steps of steepest descent minimization followed by 4000 steps of conjugate gradient minimization at each step. Positional restraints on all solute heavy atoms were initially set at 10.0 kcal/mol/Å2 and subsequently reduced at each step until the last minimization was performed without restraints. From the final minimized structure, five simulations were started using different initial random seeds to set the initial velocities. Because of the unknown complexity of the conformational change we were trying to simulate, we decided that using different initial velocities would increase our conformational sampling during MD20 and more quickly lead to the proper orientation of the I707L mutant. Each of the five simulations were separately heated linearly from 10 to 335 K over 2.0 ns while maintaining 10 kcal/mol/Å2 positional restraints on all heavy atoms of the protein and DNA. Once each simulation reached the desired 335 K, they each underwent a 3.5 ns equilibration process where the positional restraints were reduced in logical intervals until the final 500 ps of equilibration was performed without restraints. Unrestrained MD was performed on each simulation for 2.0 μs (a total of 10 μs) at constant pressure (1 atm) and temperature (335 K) maintained with a Langevin thermostat21 using periodic boundary conditions, saving the coordinates, velocities, and energies every 100 ps. The SHAKE algorithm22 was used to fix all covalent bond distances involving hydrogen, allowing a 2 fs time step for each simulation. All simulations were visualized using VMD v.1.9.1,23 and analysis was performed with the cpptraj module24 of AmberTools 13.
I707L KlenTaq1 Simulation
The final structure from the simulation of 4KTQ with the in silico I707L mutation was used to model in the missing residues of the fingers domain in the I707L mutant binary complex crystal structure. To accomplish this, the final MD structure from the 4KTQ-I707L simulation was aligned to the I707L mutant binary complex crystal structure, and the coordinates of the missing regions from simulated structure were saved and inserted into the crystal structure PDB. Next, we followed the same procedure for the 4KTQ-I707L to add hydrogens, neutralize, and solvate to a total of 81 212 atoms using AmberTools. After solvating, the same procedure (as stated for the 4KTQ-I707L simulation) was followed for performing minimization, heating, equilibration, and a total of 2.2 μs of unrestrained MD at 298 K using the GPU-accelerated pmemd code of Amber 12.
Results
To determine how the I707L mutation causes a cold-sensitive phenotype, we co-crystallized the mutant KlenTaq1 DNA polymerase with DNA and a substrate nucleotide using similar procedures as those for wild-type KlenTaq1.4 The incorporation of a 2′,3′-dideoxycytidine triphosphate (ddCTP) into the 3′-end of the 11 nucleotide primer by the polymerase prevents further elongation of the primer strand and traps the enzyme in a ternary (enzyme–DNA–dNTP) complex with excess ddCTP. To facilitate comparisons between wild-type KlenTaq1 and the I707L mutant, we used an identical DNA template and primer sequence as that in previous studies with wild-type KlenTaq1 that produced 3KTQ.pdb and 4KTQ.pdb, and our crystals, grown under the same conditions, were isomorphous to those crystals.4 A crystal diffracted to 1.67 Å resolution, and its crystal structure was solved by molecular replacement using 3KTQ.pdb (Table 1). The crystal structure of the I707L mutant polymerase ternary complex is nearly identical to the wild-type ternary structure, 3KTQ.pdb, except at higher resolution (Figure 1A). The improved resolution allowed us to model some external side chains missing in the wild-type KlenTaq1 structure.
While the mutant polymerase ternary complex has an identical backbone structure as that of the wild type, its side chain rotamers differ at two notable residues. Kermekchiev and colleagues3 noted that isoleucine 707 in the P helix was packed closely with nearby phenylalanine 749 in the Q helix and that a leucine placed in the most similar rotameric state as isoleucine 707 would result in a steric clash with F749. The crystal structure of the I707L mutant KlenTaq1 revealed that the leucine 707 side chain is not in a similar rotameric state as isoleucine 707. The leucine side chain vacates a space occupied by isoleucine’s γ1 and δ carbons, and that space is filled by a rotation of the phenylalanine 749 side chain (Figure 1B). Thus, the change in the location of a side chain methyl group from the γ to δ carbon via mutation changes the conformation of a single neighboring residue in the polymerase ternary complex.
The I707L mutant KlenTaq1 ternary complex reveals little about its low relative activity at low temperatures. The trapped intermediate structure contained a ddCTP paired opposite the template G along with two metal ions chelated by active site acidic residues and the triphosphate moiety of the ddCTP (Figure 1C). An anomalous difference density map showed strong density at one of the two divalent cation sites, suggesting that its identity was manganese(II) from the crystallization and cryoprotectant solutions. No major deviations were observed between the active sites of the mutant ternary complex and wild-type KlenTaq1 ternary complex in the closed conformation.
To gain additional insight into the cold sensitivity of the I707L mutant, we attempted to determine the structure of the binary polymerase–DNA complex. The substrate ddCTP was removed by soaking a ternary complex crystal in the mother liquor, as previously described for the wild-type KlenTaq1 binary “open” complex (4KTQ.pdb).4 The resultant crystal structure was solved to 2.2 Å using 4KTQ.pdb as a molecular replacement model (Table 1). As expected, most of the polymerase 3′-to-5′ exonuclease domain (inactive), thumb domain, and palm domain retain their structures (Figure 2A). The fingers subdomain and the DNA, however, underwent major rearrangements.
Figure 2.
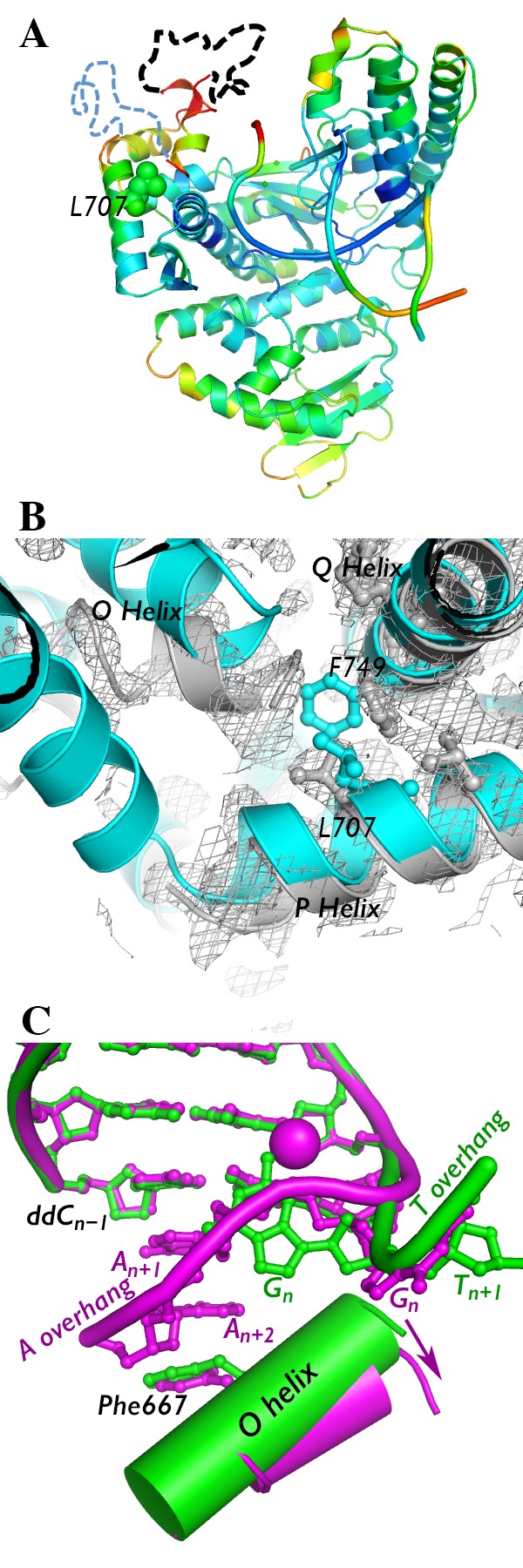
Crystal structure of I707L KlenTaq1–DNA binary complex. (A) The crystal structure of the I707L mutant binary complex is colored by B-factor (ranging from 30 (blue) to 100 (red)). Portions of the fingers subdomain that could not be modeled are denoted by dashed lines (black, residues 637–660; blue, residues 673–699). (B) The fingers subdomain was more difficult to model in the binary complex (gray) compared to the ternary complex (cyan). Gray mesh, 2Fo – Fc map of binary complex at 1.0σ. (C) Conformation of the DNA (colored ball and sticks) and O helix near the active site of I707L KlenTaq1 binary complex containing an AAA overhang (magenta) vs a TTT overhang (green). The DNA backbone is traced with a thick tube. The shift in the O helix angle is shown with an arrow. Nucleotides are numbered relative to the nucleotide to be copied (n). Manganese(II) ion bound to DNA is shown as a magenta ball.
The conservative mutation of isoleucine 707 to leucine resulted in a large conformation change in the polymerase. Unlike the I707L KlenTaq1 ternary complex, the leucine side chain adopts a rotamer pointing inward toward the protein interior in the binary complex, but phenylalanine 749 rotates away (Figure 2B). Phenylalanine 749 lies near the mobile O and O1 helices of the fingers subdomain. In this case, it also appears to function as a fulcrum for the fingers subdomain. Without the stability afforded by the presence of the dNTP substrate in the active site and phenylalanine 749, the fingers subdomain appears to be generally disordered in the I707L KlenTaq1 binary complex. While the electron density for most of the protein is easily interpretable, the electron density for the fingers, particularly portions distal from the protein, is weak and discontinuous (Figure 2B). Some parts could not be modeled (Figure 2B). The temperature factors (B-factors) for the fingers subdomain are much higher than the rest of the protein (Figure 2A). Together, these results suggest that, in the I707L mutant, the fingers subdomain either is destabilized relative to the rest of the protein or adopts multiple conformations.
In addition to alterations in the fingers, the DNA bound to the I707L mutant also adopts a dramatically different structure than two different wild-type binary complexes containing an 5′-AAAG overhang (4KTQ.pdb and 3SZ2.pdb).4,25 Structures of two A family DNA polymerase enzymes show that the template DNA is fed into the active site from a cleft made by the fingers and palm subdomains.7,8 In the I707L mutant binary complex, we observed strong electron density in the active site that appeared to be nucleotides with bases stacked. When modeled into the electron density, the template guanosine to be copied (Gn) is flipped out away from the active site while the previous two adenosines in the template 5′-overhang fit inside and stack neatly against the cytosine at the primer 3′-terminus (Figure 2C). This unusual DNA conformation coincides with the appearance of a manganese(II) ion near the flipped template guanosine (Figure 2C). The importance of this manganese ion is not clear. In the I707L mutant, the adenosines in the overhang are sandwiched between the primer terminus and phenylalanine 667 in the O helix. This residue plays a role in discrimination against ddNTPs in A family DNA polymerases.26−28 The active site cleft is occupied by the two adenosines in the overhang (An+1 and An+2) and is made larger by a shift in the O helix position away from the palm domain (Figure 2C). This is associated with the alternate rotameric state of phenylalanine 749 (Figure 2B).
In the I707L mutant binary complex, two adenosines blocked the active site by stacking against the primer terminus and phenylalanine 667. The adenine base is much better able to stack than pyrimidines,29 so we hypothesized that by changing the template overhang from adenosines to cytosines and thymidines we would decrease the free energy contribution from stacking and thereby unblock the I707L mutant. Increasing the free energy of the blocked state should tilt the equilibrium toward the unblocked state, thereby allowing the enzyme to match the template with the incoming dNTP. We tested this by measuring the activity of I707L and wild-type KlenTaq1 using a fluorescent primer strand annealed to a complementary template containing an AAG, CCG, or TTG 5′-overhang at room temperature. The addition of 10 μM dCTP to the reaction results in the incorporation of a single nucleotide to the fluorescent primer chain opposite the template G, which is detectable using a capillary electrophoresis sequencer. The wild-type KlenTaq1 showed no substantial change to the rate of nucleotide incorporation at room temperature for all three templates (Table 2), but the I707L mutant KlenTaq1 was substantially slowed by the AA overhang. These results support the hypothesis that base stacking by adenines aids in blocking the active site, but not permanently. In fact the I707L mutant appeared to be slightly faster than wild-type KlenTaq1 for templates with overhanging pyrimidines (Table 2), suggesting that blockage may occur only near pairs of adenines or purines more generally. We did not test a GG overhang in our single-hit assay because multiple deoxycytidines would be incorporated, creating multiple peaks in the electropherogram. These solution kinetics data are consistent with the hypothesis that aromatic stacking contributes to blockage in the I707L KlenTaq1 active site. No manganese was added to the reaction, suggesting that the unusual base stacking in the active site does not require the manganese ion bound near the template guanosine.
Table 2. First-Order Rate Constants of Nucleotide Incorporation by Wild-Type and I707L KlenTaq1 at 10 μM dCTP and Room Temperaturea.
| 5′-overhang | wild-type KlenTaq1 rate (s–1) | I707L KlenTaq1 rate (s–1) |
|---|---|---|
| AAG | 0.20 ± 0.05 | 0.061 ± 0.004 |
| CCG | 0.10 ± 0.02 | 0.21 ± 0.05 |
| TTG | 0.16 ± 0.02 | 0.20 ± 0.03 |
Errors were derived from curve fitting to four time points for each enzyme and substrate.
On the basis of these results, we hypothesized that pyrimidines in 5′-overhangs in the template would base stack poorly and not cause blockage in the active site of I707L KlenTaq1 like adenosines. To test this hypothesis, we co-crystallized I707L KlenTaq1 using a similar template with TTT instead of AAA in the 5′-overhang under the same conditions and solved its structure using molecular replacement (Table 1). The resulting TTT-overhang ternary crystal structure with I707L KlenTaq1 was indistinguishable from the AAA-overhang structure described earlier, including the rotamer change at F749 (results not shown). We then soaked out the ddCTP from the TTT-overhang ternary crystal to produce a binary complex. Instead of blocking the I707L KlenTaq1 active site, the TTT-overhang is located in a cleft between the fingers and palm domain (Figure 2C), as has been described previously in other A family DNA polymerase I enzymes. We observed no electron density in the active site cleft corresponding to stacked bases from the overhang. Conserved tyrosine 671 is stacked against the last template nucleotide in the duplex, and the overall structure, including that of the fingers subdomain, is nearly identical to that of the open, binary wild-type KlenTaq1 complex (4KTQ.pdb).
Our results indicate that the I707L mutant has a higher tendency than wild-type KlenTaq1 to adopt the blocked conformation in the presence of at least two adenosines in the 5′-overhang. We mutated isoleucine 707 of the open, wild-type KlenTaq1 binary complex (4KTQ.pdb) to leucine in silico and ran molecular dynamics (MD) simulations at a high temperature (62 °C) to see if I707L KlenTaq1 would transition to the blocked conformation. Of the five simulations starting from different random initial atomic velocities, one showed a substantial change in structure during the 2 μs simulation time. Although the template overhang does not swing in to block the active site in this simulation, the fingers subdomain moves substantially away from the rest of the polymerase to a position similar to the I707L mutant binary complex (Figure 3A,B). This conformational change opens the active site further and disrupts the last C:G base pair in the n – 1 position. The template G along with Gn–1 base stack with the last C in the primer strand and tyrosine 667 (Figure 3C). The quadruple stack of aromatic rings is similar to the stacking observed in the I707L mutant binary crystal structure, except for the n – 1 base pair and the identity of the bases (AA in the crystal structure and GG in the MD simulation). For comparison, we filled in the missing fingers subdomain of I707L KlenTaq1 binary complex from the crystal structure and simulated the molecular dynamics of I707L KlenTaq1 once trapped in the blocked conformation. After 2.2 μs of simulation time at 298 K, the mutant enzyme remained blocked with no major alterations in the structure (Figure 3D). The root-mean-square deviation of the final structure was 1.88 Å from the starting model.
Figure 3.

A comparison of the I707L KlenTaq1 DNA polymerase structures from MD simulations relative to crystal structures. (A) An overlay of the final MD structure (ice blue ribbons) with the I707L crystal structure (cyan ribbons). (B) An overlay of the final MD structure (ice blue ribbons) with the wild-type crystal structure (yellow ribbons). (C) A depiction of the active site from the simulated I707L mutant showing the four-residue pi-stacking interaction occurring between Phe667 on the O-helix of DNA polymerase, the base of Cn-1 on the DNA primer strand, and the bases of Gn–1 and Gn from the template strand. The residue numbers correspond to the original 4KTQ.pdb. (D) A comparison of the I707L mutant binary complex (cyan) with the final structure of a molecular dynamics simulation of the I707 mutant binary complex at 2.2 μs (pink).
Discussion
We employed X-ray crystallography to solve a biochemical mystery. How can a conservative mutation located over 20 Å away from the active site affect activity selectively at low temperature?
The I707L KlenTaq1 binary complex structure reveals a plausible explanation for its relatively low activity at low temperature. The conservative mutation of isoleucine 707 to leucine, while distant from the active site, occurred near a critical fulcrum for the mobile O helix of the fingers subdomain. The side chain methyl group, when bonded to the γ carbon (leucine) instead of the β carbon (isoleucine), allows its neighbor, phenylalanine 749, to adopt an alternate rotamer. The space vacated by the phenylalanine 749 side chain is filled by a movement of the O helix, the O1 helix, and the loop between the two helices. This chain reaction of small movements creates a larger active site cleft that can be occupied by two stacked adenosines from the template overhang, which block the incoming nucleotide and template base from accessing the active site. Pyrimidines, in contrast, base stack relatively poorly compared to adenosines and do not block the active site, according to the I707L mutant binary crystal structure containing the TTT-overhang (Figure 2C). If the free energy of the blocked state is not substantially lower than that of the unblocked state in the I707L mutant, then both states could be occupied at low and high temperatures. If, however, the transition state energy between the two states is substantial, then the enzyme might be transiently trapped in the blocked state at low temperatures, but less so high temperatures. This model is supported by the observed slow rate of dCTP incorporation by the I707L mutant at room temperature for a template with an AAG 5′-overhang (Table 2) and by the stability of the blocked I707L mutant complex in our room temperature molecular dynamics simulation (Figure 3D). The speed bumps presented by consecutive adenosines or possibly purines could slow the enzyme at low temperatures during PCR reaction assembly to the point that all bound primers produce only short extension products.
The blocked conformation may constitute an alternate conformational state for KlenTaq1. This conformation has been previously observed in a wild-type KlenTaq1 binary complex with 5′-AAAT, 5′-AAA-tetrahydrofuran, and a 5′-AAA-dNaM overhangs,25,30 showing the same two adenosines stacked against a primer ddC and F667. Although their crystallization conditions were different than ours and Li and colleagues,4 these crystals had nearly identical unit cell dimensions in the same space group. The appearance of the blocked conformation in multiple contexts and under multiple conditions suggests that the fingers subdomain of KlenTaq1 is more dynamic than previously thought. Our molecular dynamics simulations of I707L KlenTaq1 also indicate that the fingers subdomain is highly mobile. Although the template did not swing into the active site to block it during the 10 μs of total MD simulation time, the simulations do suggest that the blocked conformation is energetically accessible by I707L KlenTaq1. They also support our hypothesis that blockage is, in part, driven by base stacking in the polymerase active site. These structural studies point to the possibility that blockage of the polymerase active site is dependent on not only the enzyme sequence (as in the I707L mutation) but also the template sequence to be copied. To our knowledge, the blocked conformation has been observed only in KlenTaq1 and not other DNA polymerase I crystal structures.
In conjunction with Kermekchiev et al.,3 our results suggest a complex network of effects from polymerase conformation, primer–template annealing, thermodynamic states, and temperature produced the cold-sensitive phenotype from a conservative mutation distant from the polymerase active site. Wild-type KlenTaq1 is active at low and high temperatures, with a 74°/37° activity ratio between 2.5 and 4. During the PCR assembly process at room temperature, the primer and template are allowed to mix and anneal. Long (>15 nucleotide) primers match specifically with a designed target sequence, but they can also match to sequences that are complementary to shorter portions of the primer. Because an eight-nucleotide match with 50% GC content has a melting temperature at 25 °C,31 a substantial proportion of nonspecific, but similar, template sites are occupied by primers in the wrong places at room temperature. According to the KlenTaq1 crystal structure,4 only eight base pairs of DNA make contact with the polymerase. Thus, the polymerase cannot distinguish between primers paired to the wrong versus the right sequence at low temperature. When a polymerase that contains substantial activity at room temperature is added, the enzyme will extend the 3′-end of the primers bound to a semispecific site as well as to the target site, creating a population of unintended products mixed with the target product. If a primer at the wrong site is extended past another semispecific site on the opposite strand, then its product is subject to amplification by the polymerase. This produces unintended amplified products that can compete with the target sequence, resulting in less yield and spurious PCR products. The higher the activity at room temperature, the longer the unintended products will be, and the more likely they will serve as a template for the next amplification step.
An 8 nucleotide sequence is expected to randomly arise, on average, every 48 = 65 536 nucleotides. When combined with the fact that two strands of template DNA are involved, and any combination of semispecific sites for both the forward primer and the reverse primer (i.e., forward with forward, forward with reverse, reverse with forward, reverse with reverse) can cause mispriming, having two sites close to each other on opposite template strands is quite possible. Wild-type KlenTaq1 and polymerases with substantial activity at room temperature can then generate spurious PCR products from these sites.
The I707L mutant KlenTaq1 has a higher 74°/37° activity ratio between 4 and 7 and produces much higher yields of difficult-to-amplify target PCR products.3 We propose that, at low temperature, the I707L mutant is slowed each time it encounters pairs of adenosines (or possibly purines generally) due to the blockage of the active site by the template DNA. Our molecular dynamics simulation of in silico I707L mutant suggests that pairs of G’s can also stack in the active site like pairs of A’s (Figure 3C). While some primers still attach to nontarget sequences and are extended, this process is slow, resulting in shorter unintended products. These products are less likely to serve as templates for the next round of amplification; thus, fewer spurious PCR products are made. Once the temperature is raised to the annealing temperature for the primer, the primer is much more specific for the target sequence (i.e., few primers are bound to incorrect sites). At this point, the short, unintended products made at room temperature during reaction assembly become dead ends for the PCR. Because only the target sequence is initially copied into a long product, yields are much higher for the I707L mutant KlenTaq1 than that for wild-type KlenTaq1.
We note that, for CCG and TTG template overhangs, the I707L mutant polymerase appears to be substantially faster than wild-type KlenTaq1 (Table 2). This result suggests that, when unblocked, the I707L mutant can copy DNA at a faster rate. Kermekchiev and colleagues3 also observed higher activity at 74 °C for the I707L mutant compared to wild-type KlenTaq1. Together, these data suggest that, when unblocked, the I707L KlenTaq1 is faster than wild type. Our crystal structure of the I707L KlenTaq1 ternary complex showed no major difference to the wild-type ternary complex that could explain the mutant’s faster rate. When I707L KlenTaq1 encounters pairs of adenosines at high temperatures, the enzyme can still become temporarily blocked, but we hypothesize that high temperatures allow the enzyme to return back to the open conformation quickly. Our molecular dynamics simulations suggest that the fingers subdomain of I707L KlenTaq1 is quite labile at high temperature. The slower rate of DNA synthesis when encountering adenosines is compensated by the faster rates when not encountering adenosines, resulting in a higher overall activity at 74 °C.
These results not only explain a cold-sensitive phenotype for a mutant polymerase but also demonstrate large, long-range effects from a conservative mutation. In this specific case, the subtle change in location of a methyl group by one carbon atom with no change in atomic composition caused a chain reaction of effects that destabilized an entire polymerase subdomain and altered the DNA conformation. Because the change is located near a fulcrum for a critical mobile region of the polymerase, the mutation can affect the structure and function of the enzyme despite the large distance between the mutation and the enzyme active site.
Acknowledgments
W.M.B. and M.B.K. are for profit/loss providers of some of the subject technology for amplification and hot starts. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID (or 22-BM) beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. W-31-109-Eng-38. Beamtime was allocated through a General User Proposal. We were aided by the capable assistance of the SER-CAT staff, S. Langdon of the Duke DNA Analysis Facility, and M. Reyes of University of Richmond. This work also used the Extreme Science and Engineering Discovery Environment (XSEDE) allocation TG-MCB140003, which is supported by National Science Foundation grant OCI-1053575.
Glossary
Abbreviations
- KlenTaq1
Klenow fragment of DNA polymerase I from Thermus aquaticus
- F
phenylalanine
- I
isoleucine
- L
leucine
- PCR
polymerase chain reaction
- dNTP
2′-deoxynucleoside triphosphate
- ddNTP
2′,3′-dideoxycytidine triphophosphate
- 6FAM
6-carboxyfluorescein
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- PEG
poly(ethylene glycol)
- A
adenosine
- C
cytidine
- G
guanosine
- T
thymidine
- B-factor
temperature factor
- nm
nanometer
We thank University of Richmond School of Arts & Sciences, Thomas F. and Kate Miller Jeffress Memorial Trust, and SBIR grant R44 GM6081002A1 and 1R21HG006291 to W.M.B. from The National Institutes of Health for funding.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Saiki R. K.; Gelfand D. H.; Stoffel S.; Scharf S. J.; Higuchi R.; Horn G. T.; Mullis K. B.; Erlich H. A. (1988) Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239, 487–491. [DOI] [PubMed] [Google Scholar]
- Chou Q.; Russell M.; Birch D. E.; Raymond J.; Bloch W. (1992) Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplifications. Nucleic Acids Res. 20, 1717–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermekchiev M. B.; Tzekov A.; Barnes W. M. (2003) Cold-sensitive mutants of Taq DNA polymerase provide a hot start for PCR. Nucleic Acids Res. 31, 6139–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Korolev S.; Waksman G. (1998) Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 17, 7514–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese L. S.; Friedman J. M.; Steitz T. A. (1993) Crystal structures of the Klenow fragment of DNA polymerase I complexed with deoxynucleoside triphosphate and pyrophosphate. Biochemistry 32, 14095–14101. [DOI] [PubMed] [Google Scholar]
- Beese L. S.; Derbyshire V.; Steitz T. A. (1993) Structure of DNA polymerase I Klenow fragment bound to duplex DNA. Science 260, 352–355. [DOI] [PubMed] [Google Scholar]
- Doublie S.; Tabor S.; Long A. M.; Richardson C. C.; Ellenberger T. (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature 391, 251–258. [DOI] [PubMed] [Google Scholar]
- Johnson S. J.; Taylor J. S.; Beese L. S. (2003) Processive DNA synthesis observed in a polymerase crystal suggests a mechanism for the prevention of frameshift mutations. Proc. Natl. Acad. Sci. U.S.A. 100, 3895–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublie S.; Sawaya M. R.; Ellenberger T. (1999) An open and closed case for all polymerases. Structure 7, R31–5. [DOI] [PubMed] [Google Scholar]
- Barnes W. M. (1992) The fidelity of Taq polymerase catalyzing PCR is improved by an N-terminal deletion. Gene 112, 29–35. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P. C. K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E. Y.; Beese L. S. (2011) The structure of a high fidelity DNA polymerase bound to a mismatched nucleotide reveals an “ajar” intermediate conformation in the nucleotide selection mechanism. J. Biol. Chem. 286, 19758–19767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A., Darden T. A., Cheatham T. E. I., Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., Gotz A. W., Kolossvary I., Wong K. F., Paesani F., Vanicek J., Wolf R. M., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M.-J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Salomon-Ferrer R., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., and Kollman P. A. (2012) AMBER 12, University of California, San Francisco, CA.
- Jorgensen W. L.; Madura J. D. (1983) Quantum and statistical mechanical studies of liquids. 25. Solvation and conformation of methanol in water. J. Am. Chem. Soc. 105, 1407–1413. [Google Scholar]
- Caves L. S.; Evanseck J. D.; Karplus M. (1998) Locally accessible conformations of proteins: multiple molecular dynamics simulations of crambin. Protein Sci. 7, 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwanzig R. (1973) Nonlinear generalized Langevin equations. J. Stat. Phys. 9, 215–220. [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- Betz K.; Malyshev D. A.; Lavergne T.; Welte W.; Diederichs K.; Dwyer T. J.; Ordoukhanian P.; Romesberg F. E.; Marx A. (2012) KlenTaq polymerase replicates unnatural base pairs by inducing a Watson–Crick geometry. Nat. Chem. Biol. 8, 612–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor S.; Richardson C. C. (1995) A single residue in DNA polymerases of the Escherichia coli DNA polymerase I family is critical for distinguishing between deoxy- and dideoxyribonucleotides. Proc. Natl. Acad. Sci. U.S.A. 92, 6339–6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astatke M.; Grindley N. D.; Joyce C. M. (1998) How E. coli DNA polymerase I (Klenow fragment) distinguishes between deoxy- and dideoxynucleotides. J. Mol. Biol. 278, 147–165. [DOI] [PubMed] [Google Scholar]
- Wang W.; Wu E. Y.; Hellinga H. W.; Beese L. S. (2012) Structural factors that determine selectivity of a high-fidelity DNA polymerase for deoxy-, dideoxy-, and ribo-nucleotides. J. Biol. Chem. 287, 28215–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kool E. T. (2001) Hydrogen bonding, base stacking, and steric effects in dna replication. Annu. Rev. Biophys. Biomol. Struct. 30, 1–22. [DOI] [PubMed] [Google Scholar]
- Obeid S.; Welte W.; Diederichs K.; Marx A. (2012) Amino acid templating mechanisms in selection of nucleotides opposite abasic sites by a family a DNA polymerase. J. Biol. Chem. 287, 14099–14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibbe W. A. (2007) OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 35, W43–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B. III; Snoeyink J.; Richardson J. S.; Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]