

Abstract
Control over cellular delivery of different functionalities and their synchronized activation is a challenging task. We report several RNA and RNA/DNA-based nanoparticles designed to conditionally activate the RNA interference in various human cells. These nanoparticles allow precise control over their formulation, stability in blood serum, and activation of multiple functionalities. Importantly, interferon and pro-inflammatory cytokine activation assays indicate the significantly lower responses for DNA nanoparticles compared to the RNA counterparts, suggesting greater potential of these molecules for therapeutic use.
Keywords: RNA and DNA nanotechnology, RNA and DNA nanoparticles, RNAi, FRET, RNA−DNA hybrids reassociation
RNA interference (RNAi) is a natural cellular post-transcriptional gene regulation process that utilizes small double-stranded RNAs to trigger guided gene silencing.1 By introducing synthetic RNA duplexes called small-interfering RNAs (siRNAs),2 we can harness the RNAi machinery for therapeutic gene control and the treatment of various diseases.3−6 RNA nanotechnology offers an advantage of precise control over the composition and stoichiometry of the delivered cargo.5,7−15 RNA molecules can be programmed7,8,16−23 to form a wide variety of compact and stable artificial three-dimensional nanostructures (called RNA nanoparticles) suitable for the broad range of clinical and nanotechnological applications.5,9,11,14,24−31 Recently, we developed a strategy based on RNA–DNA hybrids that can be generally used for triggering activation of different functionalities in vitro and in vivo.(32,33) The key idea is to split the functional entity (e.g., Dicer Substrate RNAs or DS RNAs, RNA aptamers, FRET pair of dyes) into two RNA–DNA hybrids. The resulting inactive hybrids are decorated with complementary ssDNA toeholds which will interact and trigger the reassociation process when both of the cognate hybrids are present in close proximity. The reassociation releases the split functionalities and restores their original function. Utilizing this novel approach involving RNA–DNA hybrids7,8,32,33 and our expertise in RNA nanotechnology,8,17 we have developed RNA–DNA and DNA–RNA hybrid nanocubes consisting of either RNA or DNA cores (composed of six strands), respectively, with six attached RNA–DNA hybrid duplexes. After addition of six cognate hybrids to the hybrid nanoparticles, the complementary toeholds in each duplex initiate reassociation of the DNA strands, displacing the RNA strands. This regulated displacement from a DNA partner induces the reassociation of the RNA duplexes, which are further processed by the human Dicer enzyme, thus activating RNAi.
Results and Discussion
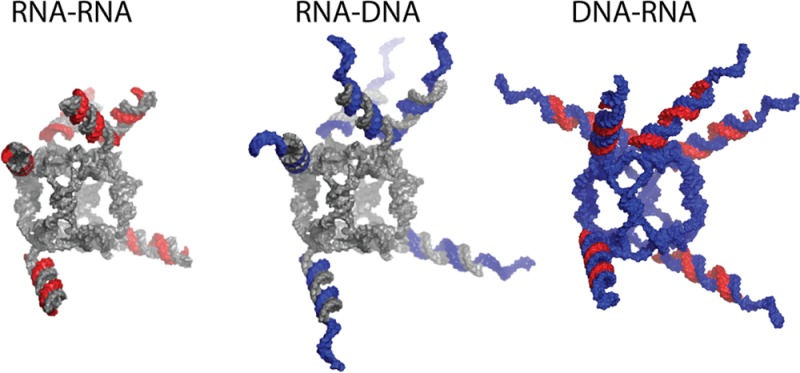
Overall, three different types of RNA–RNA, RNA–DNA and DNA–RNA functional nanoparticles (schematically shown in Supporting Information Figure S1) were designed and tested in this work. All three-dimensional models of the functional nanoparticles presented in this work were generated as detailed in Methods. The experimental results are presented and discussed below.
RNA Nanocubes
We first used previously characterized six-stranded RNA nanocubes17 with three single-stranded uracils at the corner16 as scaffolds for the controlled delivery of multiple siRNAs. Through 3′-side extensions of individual scaffold strands, RNA nanocubes were functionalized with six Dicer Substrate RNAs or DS RNAs34 (Figure 1a). DS RNAs are needed to promote the intracellular release of siRNAs through dicing (Supporting Information Figure S2a).24 To study the potential use of RNA scaffolds for simultaneous delivery of multiple siRNAs, nanocubes functionalized with six fluorescently labeled DS RNAs were transfected using Lipofectamine 2000 (L2K) into human breast cancer cells (Figure 1b, Supporting Information Figure S1), visualized by confocal fluorescence microscopy and analyzed by fluorescence-activated cell sorting (FACS). The results reveal significant intracellular uptake of the functionalized nanocubes through endocytosis (Figure 1c). Intracellular release of siRNAs activating RNAi (Figure 1d and Supporting Information Figure S3) was assessed in experiments with human breast cancer cells stably expressing enhanced green fluorescent protein (eGFP). MDA-MB-231/eGFP cells were transfected with nanocubes carrying six DS RNAs against eGFP34 and the same individual siRNAs at 6-fold higher concentrations. After 3 days, the levels of eGFP expression were analyzed with fluorescence microscopy and FACS. Extensive levels of silencing in eGFP production were observed and the silencing in GFP production remained significant even on the 12th day post-transfection (Figure 1d).
Figure 1.

Activation of RNAi with RNA nanocubes 3′-side functionalized with six Dicer substrate RNAs. (a) Schematics of nanocube formation and release of siRNAs through dicing. (b) Cellular uptake of fluorescently labeled cubes (10 nM) and duplexes (60 nM) was assessed with confocal microscopy and analyzed by flow cytometry. (c) Localization of fluorescently labeled functional nanocubes (10 nM) with commonly used markers for endosomal compartments EEA1 and Rab7. (d) GFP knockdown assays for human breast cancer cells (MDA-MB-231/GFP) which stably express GFP. At 3, 5, 6, 7, 12, and 14 days after the transfection of cells with nanocubes, eGFP expression was statistically analyzed with flow cytometry experiments. As the control, siRNA duplexes against eGFP were used for all time points. gMFI corresponds to the geometric mean fluorescence intensity. Error bars denote SEM. The images were taken 3 days post-transfection.
Functional RNA Nanocubes against HIV-1
To demonstrate the generality and the feasibility of the approach in using functionalized nanocubes as potential therapeutic moieties, several different HIV-1 genes were targeted with six different DS RNAs35 carried by the RNA nanocubes (Figure 2). 293T cells were used to produce HIV-1 pseudotyped with VSV-g in the presence of increasing amounts of functional nanocubes or a mixture of the six individual DS RNAs. Equal amounts of virus-containing supeRNAtant were used to infect HeLa cells, and the percentage of infected cells was detected using FACS analysis. The results shown in Figure 2 confirm that functional RNA nanocubes have comparable negative effect on HIV-1 production to the mixture of six DS RNAs alone. Western blot analysis using α-HIV-Ig confirms that both the functional RNA nanocubes and the mixture of DS RNAs reduce the level of p55 (Gag polyprotein) and p24 in the virus producing cells (Supporting Information Figure S4). Functional nanocubes (at ≤5 nM) designed to target HIV-1 significantly reduce the levels of viral RNA in infected cells and subsequently the expression of Gag, while nonfunctionalized cubes have no such effect. This approach can be used as a part of a combinatorial RNAi (co-RNAi) strategy for highly effective simultaneous suppression of multiple viral genes preventing the possibility of mutation-assisted escape from RNAi.36,37
Figure 2.
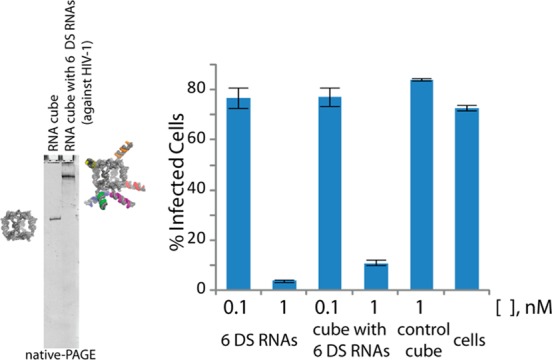
Assembly of RNA nanocubes functionalized with six different Dicer substrate RNAs against HIV-1. Virus produced from cells transfected with a mixture of siRNAs or an RNA–RNA nanocube targeting HIV-1 RNA was assessed for infectivity. Error bars denote SD; N = 3.
RNA–DNA Nanocubes
To test the idea of simultaneous delivery and activation of multiple split functionalities (e.g., RNAi and FRET) that can be triggered in a controlled manner, the same RNA nanocubes were decorated with RNA–DNA hybrids containing DNAs fluorescently labeled with Alexa 546 that are complementary to the antisense strands of the DS RNAs (Figure 3a). The DNAs were designed to have ssDNA toeholds oriented outward from the nanocubes. The inward orientation of toeholds was also tested but the assembly yields of the nanoparticles were very low (data not shown). The assemblies were confirmed by native-PAGE and DLS experiments, shown in Figure 3b. These nanoparticles do not activate the RNAi pathway in cells due to the inability of Dicer to process the RNA–DNA hybrids (Supporting Information Figure S2a). Cognate RNA–DNA hybrids were designed to carry the sense RNA strands of DS RNAs hybridized to DNAs fluorescently labeled with Alexa 488. The in vitro reassociation of RNA–DNA nanoparticles with cognate hybrids was monitored using FRET (Figure 3c). These experiments demonstrated DNA duplex formation that positioned the pair of Alexa dyes within their Förster radius, thus, activating FRET. To confirm the release of active DS RNAs on the RNA nanocube scaffolds that accompanied the formation of duplex DNA, silencing experiments were carried out using eGFP expressing cells (Figure 3d). The cells were transfected with RNA–DNA nanocubes alone, and the corresponding cognate free RNA–DNA duplexes alone, or cotransfected with both using individually prepared complexes with L2K. It is evident that eGFP silencing occurs only when RNA–DNA nanocubes and the cognate RNA–DNA hybrids are simultaneously delivered into the cells.
Figure 3.

Activation of split functionalities during reassociation of RNA nanocubes 3′-side decorated with six RNA–DNA (in blue) hybrids (carrying six Dicer substrate RNA antisense) with six cognate hybrids (carrying Dicer substrate RNA senses). (a) Schematics of reassociation and activation of FRET and RNAi. (b) Assembled RNA cubes were analyzed by total SYBR Gold staining native PAGE and DLS experiments. (c) FRET time traces during reassociation of fluorescently labeled cubes and hybrids labeled with Alexa 488 and Alexa 546. (d) GFP knockdown was quantified through flow cytometry. Please note that the individual hybrids and RNA nanocubes decorated with hybrids cause no decrease in eGFP production.
DNA–RNA Nanocubes
As an alternative approach for simultaneous activation of multiple split functions in cells, we used DNA nanocubes as scaffolds (Figure 4a). These scaffolds were decorated with six hybrids carrying antisense strands of DS RNAs. The correct formation of DNA–RNA hybrid nanocubes was verified by native PAGE (Figure 4b). The reassociation of the RNA parts of the hybrids resulted in the release of functional DS RNAs and activated FRET (Figure 4a). The effective activation of FRET was confirmed in vitro (Figure 4c and Supporting Information Figure S5) and in the cultured cells (Figure 4d). The release of DS RNA was observed through gene silencing experiments (Figure 4e, Supporting Information Figure S5). The DNA–RNA hybrid cubes containing sense strands of DS RNAs with the corresponding cognate RNA–DNA heteroduplexes carrying the antisense RNA strands were also extensively tested (Supporting Information Figure S6).
Figure 4.

Activation of different split functionalities during reassociation of DNA nanocubes (in blue) 3′-side decorated with six RNA–DNA hybrids (carrying six DS RNA antisenses, in red) with six cognate hybrids (carrying DS RNA senses). (a) Schematics of reassociation and activation of FRET and RNAi. (b) The formation of DNA cubes was confirmed by total SYBR Gold staining native PAGE and DLS experiments. (c) FRET time traces during reassociation of fluorescently labeled cubes and hybrids labeled with Alexa 546 and Alexa 488. (d) FRET experiments: cells were cotransfected with cubes and cognate hybrids labeled with Alexa 546 and Alexa 488 and images were taken on the next day. (e) GFP knockdown was measured via flow cytometry. Please note that the individual hybrids and DNA nanocubes decorated with hybrids cause no decrease in eGFP expression. Image numbers in (d) correspond to differential interference contrast (DIC) images (1), Alexa 488 emission (2), Alexa 546 emission (3), bleed-through corrected FRET image (4), 3D chart representation of zoomed fragment indicated by a white box of bleed-through corrected FRET image with the white dot indicating the correspondence (5).
Activation of Interferons by Functional Nanoparticles
Since secretion of inflammatory cytokines and type I interferons by the immune cells in response to traditional nucleic acid based therapeutics (siRNA, antisence oligonucleotides, etc.) is a common dose-limiting toxicity that restricts the therapeutic benefits of these compounds38−41 and has recently been reported for RNA-based nanoparticles,27 we also examined the type I IFN and cytokine responses to RNA–RNA, RNA–DNA and DNA–RNA nanoparticles in both primary human peripheral blood mononuclear cells (PBMC) and in a monocyte–macrophage reporter cell line (Figure 5 and Supporting Information Figure S7a). While all constructs resulted in elevation in IFNβ levels above baseline, only RNA-containing cubes were very potent inducers of type I IFN in human PBMC (Figure 5a). The potency of RNA-containing cubes was comparable to or even stronger than that of the assay positive control ODN2216 specifically designed to produce high levels of type I interferon.42
Figure 5.
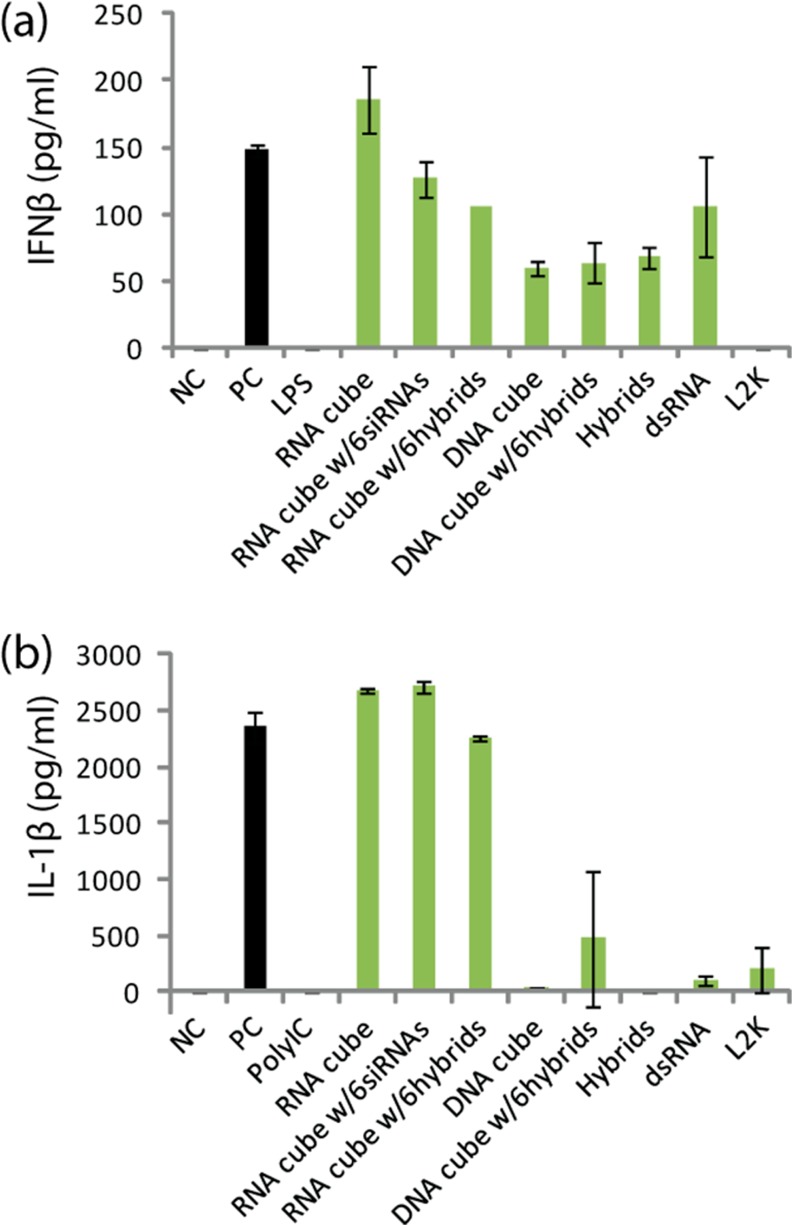
Induction of type I interferon and pro-inflammatory cytokine in human peripheral blood mononuclear cells in vitro. Human PBMC from 2 healthy donor volunteers was incubated with control and test samples for 24 h. Cell culture supernatants were analyzed by conventional ELISA to detect IFNb (a) or IL-1b (b). Error bars denote SD; N = 2 donors. Variability of response within each individual donor was low (% CV < 20). NC is negative control (PBS); PC is positive control in (a) 5 μg/mL of ODN2216, in (b) 20 ng/mL of ultrapure K12 Escherichia coli LPS. Lipofectamine 2000 (L2K) at the same concentration as used to deliver constructs and 5 μg/mL polyIC were used as additional controls.
This data was further supported in the model of the monocyte–macrophage reporter cell line in which RNA-containing cubes were potent stimulators of type I IFN, while DNA-containing cubes were relatively inert toward the type I IFN response. Importantly, triggering of type I IFN was not related to any inherent toxicity of the nanocubes based on cell viability assays (Supporting Information Figure S7b). RNA-induced stimulation of IFN activity required the adaptor protein MAVS, suggesting signaling occurred through the RNA sensing pathways involving RIG-I and MDA5. Although DNA-based cubes failed to trigger measurable type I IFN responses, as expected, long dsDNA (93 bps) were able to induce a type I IFN response via cGAS, a DNA-binding receptor in the type I IFN signaling pathway.43 These data suggest that the large DNA cubes used in this work might not be recognized by cGAS. Furthermore, mixing RNA or DNA cubes containing RNA–DNA hybrids with cognate RNA–DNA hybrids carrying DNAs complementary to the DNAs of nanocubes did not result in measurable DNA-dependent type I IFN-induction. These results are not unexpected since reassociation results in only 39 bps DNA duplexes, and IFN induction is directly proportional to the number of base pairs of the dsDNA.44 In agreement, we have shown previously that reassociation of RNA–DNA hybrids yielding short dsDNA molecules are less efficient at stimulating type I IFN compared to those releasing long dsDNA molecules.32
Induction of pro-inflammatory cytokine IL-1β, recognized as a marker of pyrogenicity and an innate immune response triggered by TLR-mediated NFkB activation,45−47 was also detected in PBMC cultures only in response to RNA-containing cubes (Figure 5b), suggesting that in addition to the intracellular RIG/MDA5 pathway, these constructs trigger activation of inflammatory pathways through endosomal TLRs (TLR3, TLR7 and/or TLR9).
Together, these data indicate that DNA–RNA nanocubes might be less immunogenic than RNA–RNA and RNA–DNA nanocubes, and thus might be better suited for therapeutic purposes to reduce the potential side effects of cytokine release. RNA-based nanocubes, on the other hand, may become possible candidates for conditionally controlled immunostimulation enhancement analogous to the work of Khisamutdinov et al.(27)
Conclusion
In conclusion, we have developed three different nanoparticle-based strategies for conditional activation of RNAi. We show how RNA and DNA nanoscaffolds can be functionalized with multiple DS RNAs (e.g., targeting six different parts of HIV) as well as RNA–DNA hybrids. The RNA–DNA hybrids cannot be diced and, thus, are not active. Cognate hybrids reassociate through ssDNA toeholds interactions and release either the functional RNA nanoparticles or siRNAs. Several different cell culture experiments demonstrate FRET and RNAi activation by conditional triggering of split functionalities in cells. The use of RNA nanocubes as scaffolds for multiple DS RNAs provides a simple and precisely programmable mechanism to control composition. However, the application of DNA nanoscaffolds carrying multiple split functionalities appears to have some additional advantages summarized in Table 1. These nanocubes are more resistant to human blood serum nucleases and have lower immunogenicity compared to their RNA-based counterparts. The intracellular fluorescent tracking of hybrid reassociation in the nanocubes is more practical, because fluorescent dyes and any other additional functionality attached to the DNAs will not interfere with RNAi, mediated by the DS RNA released from the nanoparticles. Furthermore, besides being the carriers for RNAi activators, nanocubes can easily be modified with various ligands such as aptamers48,49 for targeted delivery (Supporting Information Figure S8). An inherent utility of this novel technique will be to exploit the multiple existing three-dimensional shapes formed by the DNA molecules50−58 and RNA–DNA hybrid structures.59−61
Table 1. Relative Differences for RNA–RNA, RNA–DNA, and DNA–RNA Nanoparticles Described in This Work.
| functional nanoparticle | processed by Dicer | stable in human serum | activates type I IFNs | activates pro-inflammatory cytokine | activates RNAi |
|---|---|---|---|---|---|
| RNA–RNA nanocube | YES | NO | YES | YES | YES |
| RNA–DNA nanocube | NO | NO | YES | YES | NOa |
| DNA–RNA nanocube | NO | YES | NO | NO | NOa |
Cognate RNA–DNA hybrids are required for activation of RNAi during intracellular reassociation.
Methods
3D Modeling of Functional RNA and DNA Cube Assemblies
All three-dimensional models of the functional nanoparticles presented in this work were generated using custom-written PyMOL scripts (http://www.pymol.org/). In the case of the RNA cube scaffolds, the previously generated and tested RNA cube model16 was mated with separately generated multiple alteRNAtive models of a helix and siRNA arm junctions generated with the aid of RNA2D3D, MC-Sym, and RNAComposer.62−64 Overlapping fragments were best fit together (minimum RMSD), then any structural duplicates were removed, and the final models were converted into PDB files and imaged in PyMOL. The DNA cube models were built using RNA cube helices as guides to lay out separately generated B-form DNA helices bridged in the corners with single-stranded (B-form) DNA fragments. Overlapping backbones were best fit and ultimately fused into individual chains (with duplicate fragments removed). The junction models were best-mated to the cubic DNA scaffold and separately generated A-form RNA–DNA (hybrid), and B-form DNA–DNA helical arms were best-fit, depending on the modeled cube.
Functional RNA and DNA Cube Assemblies and Native PAGE
The designing principles and production of RNA strands entering the composition of nanocubes functionalized with six siRNAs is comprehensively described elsewhere.24 The full list of RNA and DNA sequences used is available in Supporting Information. RNA molecules were purchased from Integrated DNA Technologies, Inc. or prepared by transcription of PCR amplified DNA templates; synthetic DNA molecules coding for the sequence of the designed RNA were purchased and amplified by PCR using primers containing the T7 RNA polymerase promoter. RNA molecules were prepared enzymatically by in vitro transcription using T7 RNA polymerase.65 Transcription was performed in 50 mM Tris-HCl, pH 7.5; 2 mM spermidine; 1 mM DTT; 5 mM MgCl2; 0.5 mM MnCl2; 1 mM NTPs; 0.1 μM of DNA template; and T7 RNA polymerase (Promega). RNA, RNA–DNA and DNA–RNA nanocubes were assembled as detailed in Afonin et al.(17,24) Cognate RNA–DNA hybrids were assembled as described in Afonin et al.(33) For the visualization of assembled nanoparticles, native-PAGE was used.66,67 Typically, assembly experiments were analyzed at 10 °C on 7% (29:1) native polyacrylamide gels in the presence of 89 mM Tris-borate, pH 8.3, and 2 mM Mg(OAc)2. A Hitachi FMBIO II Multi-View Imager was used to visualize SYBR Gold stained RNA-, RNA–DNA- and DNA–RNA-based nanoparticles.
Dynamic Light Scattering (DLS) Experiments
For DLS experiments, 400 μL of sample solutions containing assembled nanoparticles (500 nM final) was used. The samples were measured at 25 °C with a Zetasizer nano (Malvern Instruments Ltd.) equipped with a 633 nm laser.68 Readings with polydispersity index (PDI) below 0.5 were used.
Recombinant Human Dicer Assay
For dicing experiments, purchased DS RNA sense strand molecules were 5′-end radiolabeled using T4 Polynucleotide Kinase. Nanocubes with either six DS RNAs or six RNA–DNA hybrids were prepared as described above (3 μM final). For dicing, samples were incubated for 3 h at 37 °C with recombinant human turbo dicer enzyme kit (Genlantis) prior to analysis on 2 mM Mg(OAc)2 native 7% PAGE (described above). The electrophoresis was performed for 1.5 h at 15 W at 10 °C followed by the exposure to a phosphorimaging screen and analysis using phosphorimaging instrument (Storm).
Human Blood Serum Degradation Studies
Aliquots of freshly drawn human whole blood serum (blood was allowed to coagulate, then spun down and supeRNAtant was collected) were used. Different radiolabeled constructs (3 μM final) were incubated with 5% (v/v) human blood serum at 37 °C for various time periods. Prior to immediate loading on native-PAGE, degradation time courses were quenched on dry ice and loaded on the gel in reverse order. The electrophoresis was performed as described above.
Fluorescence Studies
Reassociation of RNA–DNA and DNA–RNA nanocubes with cognate RNA–DNA hybrids in vitro was tracked with FRET measurements using a FluoroMax3 (Jobin-Yvon, Horiba). All fluorescently labeled sequences are presented in Supporting Information (Fluorescently labeled molecules). For all the experiments, the excitation wavelength was set at 460 nm and the excitation and emission slit widths were set at 2 nm. Alexa 488 labeled constructs were first incubated for 2 min at 37 °C and then Alexa 546 labeled constructs were added. Upon excitation at 460 nm, the emissions at 520 and 570 nm were recorded simultaneously every 30 s to follow the process of reassociation through FRET measurements. This was done with naked constructs and constructs individually preincubated with L2K in the amounts relevant for the transfection conditions (see below).
Transfection of Human Breast Cancer Cells
Human breast cancer cell line MDA-MB-231 (with or without eGFP) was grown in D-MEM media (Gibco BRL) supplemented with 10% FBS and penicillin-streptomycin in a 5% CO2 incubator. All transfections in this project were performed using Lipofectamine 2000 (L2K) purchased from Invitrogen. 10× or 50× solutions of nanoparticles were individually preincubated at room temperature with L2K. RNA–DNA and DNA–RNA cubes were cotransfected with their cognate RNA–DNA hybrids (at 6-fold higher concentrations). To avoid reassociation in media, RNA–DNA or DNA–RNA NPs and cognate hybrids were preincubated with L2K separately. In vitro fluorescent experiments show no reassociation of these complexes in solution (Supporting Information Figures S4a and S5d); thus, the reassociation occurs only in cells. Prior to each transfection, the cell media was swapped with OPTI-MEM mixed with prepared 50× of NP/L2K complexes and/or Hybrid/L2K complexes to the final concentration of 1×. The cells were incubated for 4 h followed by the media change (D-MEM, 10% FBS, 1% pen–strep).
Endosomal Colocalization Studies
To confirm the endosomal location of endocytosed fluorescently labeled functional RNA NP in cells, costaining experiments with endosomal markers (EEA1 and Rab7) were performed.33 Cells were transfected with RNA NPs labeled with six Alexa 546 dyes. On the next day, transfected cells were fixed with 4% paraformaldehyde for 20 min at room temperature and handled at this temperature thereafter. Samples were washed three times with PBS and then permeabilized with 0.2% Triton X-100 for 20 min. Upon washing three times with PBS, samples were blocked for 1 h with 1% BSA and then exposed to primary antibodies against the early endosome associated protein EEA1 (Cell signaling) or against the late endosome marker Rab 7 (Cell signaling). Upon washing three times with PBS, the samples were stained with a secondary Alexa 488 antibody (Molecular Probes). As the comparison, fluorescently labeled DS RNAs were used at 6-fold higher concentrations.
Microscopy
Reassociation of RNA–DNA hybrids in cells was assessed through FRET.33 All measurements were performed using a LSM 710 confocal microscope (Carl Zeiss) with a 63×, 1.4 NA magnification lens. All images were taken with a pinhole adjusted to 1 airy unit. Fluorescently labeled hybrid NPs and cognate hybrids were individually preincubated with L2K and cotransfected into cells. On the next day, the samples were fixed by incubation in 4% paraformaldehyde for 20 min at room temperature. Images of the cells were then taken to assess the appearance of FRET within the sample. For Alexa 488 imaging, the 488 nm line of an argon laser was used as excitation and the emission was collected between 493 and 557 nm. For Alexa 546 imaging, a DPSS 561 laser was used for excitation and emission was collected between 566 and 680 nm. In order to evaluate the sensitized emission through FRET, images were taken exciting the sample with the 488 nm line and collecting emission between 566 and 680 nm. Because of spectral overlap, the FRET signal is contaminated by donor emission into the acceptor channel and by the excitation of acceptor molecules by the donor excitation wavelength. This bleed through was assessed through measurements performed with samples transfected with individual dyes and mathematically removed from the images of FRET.
Flow Cytometry Experiments
For analysis with flow cytometry experiments, the MDA-MB-231 (with or without eGFP) cells grown in 12-well plates (1.0 × 105 cells per well) were lifted with cell dissociation buffer, and the level of nanoparticle uptake or expression of eGFP was determined by fluorescence-activated cell sorting (FACS) analysis on a FACScalibur flow cytometer (BD Bioscience).68 At least 20 000 events were collected and analyzed using the CellQuest software. For statistical analysis, the geometric mean fluorescence intensity (gMFI) and standard error of the mean (SEM) were calculated and plotted.
Virus Production
293T cells were transfected with HIV-RFP and VSV-g in the presence of increasing amounts of DS RNA mixture and functionialized RNA nanocubes. As a control the nonfunctionalized RNA nanocubes and pcDNA (to compensate for the amounts of transfected DS RNAs, nanocubes and functional RNA nanocubes) were used. At 48 h post-transfection, culture supeRNAtants were collected and filtered with 0.45 μm pore-size sterile filters.
Infectivity Assay
The day before the infection, 2.5 × 104 HeLa cells/well were plated in 24-well tissue culture plates. On the day of the infection, the culture media (DMEM, 10%FBS) was replaced with new culture media containing Polybrene at the final concentration of 5 μg/mL. Different dilutions of virus-containing supeRNAtant were added to the plates before incubation at 37 °C for 48 h. After this incubation, the cells were trypsinized and resuspended in PBS containing 2% FBS. The percentages of infected cells were measured by fluorescence-activated cell sorter (FACS) analysis.
Western Blot Analysis
RIPA buffer (SIGMA) was used to lyse transfected 293T cells. Cell lysates were mixed with 2× Laemmli sample loading buffer (BioRad), boiled, and loaded on native-PAGE gels. Subsequently, proteins were transferred to a PVDF membrane by electroblotting. Then the membrane was incubated for 1 h at room temperature in blocking buffer (5% nonfat dry milk in PBS). The blocked blot was exposed to the HIV-Ig or tubulin antibody in blocking buffer with constant mixing. After extensive washing, bound antibodies were detected by chemiluminescence using horseradish peroxidase-conjugated species-specific secondary antibodies as described by the manufacturer (GE Healthcare).
Reporter-Cell Line for Analysis of Interferon Activation
Type I IFN activity was measured using THP-1 reporter cells which express secreted alkaline phosphatase (SEAP) in response to type I IFN (Invivogen). THP-1 cells were depleted for cGAS or MAVS by siRNA to assess whether IFN induction was dependent on DNA- or RNA-sensing, respectively. THP-1 cells were cultivated in RPMI 1640 with 10% FBS, 10 mM HEPES, 1 mM pyruvate, penicillin–streptomycin, and normocin (100 μg/mL). THP-1 cells were transfected with control or SMART pool siRNAs (Thermo) at a final concentration of 50 nM with RNAiMax (Invitrogen). After 24 h, cells were differentiated with 50 ng/mL phorbol 12-myristate 13-acetate (PMA)(Sigma) for 24 h. Cells were transferred to a 96-well dish and incubated for an additional 24 h in media lacking PMA prior to transfection of nanocubes. Nucleic acids were transfected using Lipofectamine 2000 reagent according to the manufacturer’s protocol (Invitrogen) at a final concentration of 10 nM. Culture supeRNAtants were harvested 24 h post transfection and assayed for alkaline phosphatase activity by incubating with the QUANTI-BLUE substrate (Invivogen) and absorbance was measured at 625 nm using a spectrophotometer. The viability of the THP-1 cells was assessed with the CellTiter-Blue assay (Promega, Madison, WI) following manufacturer’s instructions. Briefly, the CellTiter-Blue reagent was added to the wells at a 1 to 6 ratio and the fluorescence was recorded (560 ex/590 em) upon 1–2 h incubation at 37 °C.
Primary Human Peripheral Blood Mononuclear Cell (PBMC) Culture for Analysis of Interferon and Cytokine Secretion
Healthy volunteers’ blood was collected under FNLCR Protocol OH9-C-N046. Blood was drawn into tubes containing Li-heparin as anticoagulant and processed within 2 h after collection. PBMC were isolated using Ficoll-Paque Plus (GE Healthcare Biosciences, Pittsburgh, PA) according to the manufacturer’s protocol. One million cells in the total final volume of 1 mL of complete RPMI was incubated in the presence of negative control (PBS), positive control (20 ng/mL ultrapure K12 E. coli LPS for IL-1β assay or 5 μg/mL ODN2216 for IFNβ assay) or test compounds in L2K at a final concentration of 0.5 μM. L2K mixed with cell culture grade endotoxin free water was analyzed as a vehicle control. After 24 h incubation at 37 °C, cell culture supernatants were collected and analyzed for the presence of IFNβ and IL-1β using commercial ELISA kits from PBL Assay Science (Piscataway, NJ) and R&D Systems (Carlsbad, CA), respectively.
Acknowledgments
We thank Eckart Bindewald for helpful discussion. This publication was funded in part with federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under Contract HHSN261200800001E. This research was additionally supported in part by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Supporting Information Available
All sequences used in this project and supplementary data are included. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fire A.; Xu S.; Montgomery M. K.; Kostas S. A.; Driver S. E.; Mello C. C. Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis Elegans. Nature 1998, 391, 806–811. [DOI] [PubMed] [Google Scholar]
- Elbashir S. M.; Lendeckel W.; Tuschl T. RNA Interference Is Mediated by 21- and 22-Nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Xie J. Progress on RNAi-Based Molecular Medicines. Int. J. Nanomed. 2012, 7, 3971–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot C. V.; Calin G. A.; Coleman R. L.; Lopez-Berestein G.; Sood A. K. RNA Interference in the Clinic: Challenges and Future Directions. Nat. Rev. Cancer 2011, 11, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P. The Emerging Field of RNA Nanotechnology. Nat. Nanotechnol. 2010, 5, 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramsen J. B.; Kjems J. Development of Therapeutic-Grade Small Interfering RNAs by Chemical Engineering. Front. Genet. 2012, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Viard M.; Koyfman A. Y.; Martins A. N.; Kasprzak W. K.; Panigaj M.; Desai R.; Santhanam A.; Grabow W. W.; Jaeger L.; et al. Multifunctional RNA Nanoparticles. Nano Lett. 2014, 14, 5662–5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Kasprzak W. K.; Bindewald E.; Kireeva M.; Viard M.; Kashlev M.; Shapiro B. A. In Silico Design and Enzymatic Synthesis of Functional RNA Nanoparticles. Acc. Chem. Res. 2014, 47, 1731–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla G. C.; Haque F.; Tor Y.; Wilhelmsson L. M.; Toulme J. J.; Isambert H.; Guo P.; Rossi J. J.; Tenenbaum S. A.; Shapiro B. A. A Boost for the Emerging Field of RNA Nanotechnology. ACS Nano 2011, 5, 3405–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Lindsay B.; Shapiro B. A. Engineered RNA Nanodesigns for Applications in RNA Nanotechnology. RNA Nanotechnol. 2013, 1, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y.; Pi F.; Sharma A.; Rajabi M.; Haque F.; Shu D.; Leggas M.; Evers B. M.; Guo P. Stable RNA Nanoparticles as Potential New Generation Drugs for Cancer Therapy. Adv. Drug Delivery Rev. 2014, 66, 74–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaled A.; Guo S.; Li F.; Guo P. Controllable Self-Assembly of Nanoparticles for Specific Delivery of Multiple Therapeutic Molecules to Cancer Cells Using RNA Nanotechnology. Nano Lett. 2005, 5, 1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y.; Haque F.; Shu D.; Li W.; Zhu Z.; Kotb M.; Lyubchenko Y.; Guo P. Fabrication of 14 Different RNA Nanoparticles for Specific Tumor Targeting without Accumulation in Normal Organs. RNA 2013, 19, 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H.; Kobayashi T.; Kabata R.; Endo K.; Iwasa T.; Yoshimura S. H.; Takeyasu K.; Inoue T.; Saito H. Synthetic RNA-Protein Complex Shaped Like an Equilateral Triangle. Nat. Nanotechnol. 2011, 6, 116–120. [DOI] [PubMed] [Google Scholar]
- Osada E.; Suzuki Y.; Hidaka K.; Ohno H.; Sugiyama H.; Endo M.; Saito H. Engineering RNA-Protein Complexes with Different Shapes for Imaging and Therapeutic Applications. ACS Nano 2014, 8, 8130–8140. [DOI] [PubMed] [Google Scholar]
- Afonin K. A.; Kasprzak W.; Bindewald E.; Puppala P. S.; Diehl A. R.; Hall K. T.; Kim T. J.; Zimmermann M. T.; Jernigan R. L.; Jaeger L.; et al. Computational and Experimental Characterization of RNA Cubic Nanoscaffolds. Methods 2014, 67, 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Bindewald E.; Yaghoubian A. J.; Voss N.; Jacovetty E.; Shapiro B. A.; Jaeger L. In Vitro Assembly of Cubic RNA-Based Scaffolds Designed in Silico. Nat. Nanotechnol. 2010, 5, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabow W. W.; Zakrevsky P.; Afonin K. A.; Chworos A.; Shapiro B. A.; Jaeger L. Self-Assembling RNA Nanorings Based on RNAi/Ii Inverse Kissing Complexes. Nano Lett. 2011, 11, 878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chworos A.; Severcan I.; Koyfman A. Y.; Weinkam P.; Oroudjev E.; Hansma H. G.; Jaeger L. Building Programmable Jigsaw Puzzles with RNA. Science 2004, 306, 2068–2072. [DOI] [PubMed] [Google Scholar]
- Jaeger L.; Chworos A. The Architectonics of Programmable RNA and DNA Nanostructures. Curr. Opin. Struct. Biol. 2006, 16, 531–543. [DOI] [PubMed] [Google Scholar]
- Guo P.; Zhang C.; Chen C.; Garver K.; Trottier M. Inter-RNA Interaction of Phage Phi29 PRNA to Form a Hexameric Complex for Viral DNA Transportation. Mol. Cell 1998, 2, 149–155. [DOI] [PubMed] [Google Scholar]
- Geary C.; Rothemund P. W.; Andersen E. S. RNA Nanostructures. A Single-Stranded Architecture for Cotranscriptional Folding of RNA Nanostructures. Science 2014, 345, 799–804. [DOI] [PubMed] [Google Scholar]
- Grabow W. W.; Jaeger L. RNA Self-Assembly and RNA Nanotechnology. Acc. Chem. Res. 2014, 47, 1871–1880. [DOI] [PubMed] [Google Scholar]
- Afonin K. A.; Grabow W. W.; Walker F. M.; Bindewald E.; Dobrovolskaia M. A.; Shapiro B. A.; Jaeger L. Design and Self-Assembly of SiRNA-Functionalized RNA Nanoparticles for Use in Automated Nanomedicine. Nat. Protocol 2011, 6, 2022–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binzel D. W.; Khisamutdinov E. F.; Guo P. Addition to Entropy-Driven One-Step Formation of Phi29 PRNA 3wj from Three RNA Fragments. Biochemistry 2014, 53, 3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binzel D. W.; Khisamutdinov E. F.; Guo P. Entropy-Driven One-Step Formation of Phi29 PRNA 3wj from Three RNA Fragments. Biochemistry 2014, 53, 2221–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khisamutdinov E. F.; Li H.; Jasinski D. L.; Chen J.; Fu J.; Guo P. Enhancing Immunomodulation on Innate Immunity by Shape Transition among RNA Triangle, Square and Pentagon Nanovehicles. Nucleic Acids Res. 2014, 42, 9996–10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khisamutdinov E. F.; Jasinski D. L.; Guo P. RNA as a Boiling-Resistant Anionic Polymer Material to Build Robust Structures with Defined Shape and Stoichiometry. ACS Nano 2014, 8, 4771–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu D.; Khisamutdinov E. F.; Zhang L.; Guo P. Programmable Folding of Fusion RNA in Vivo and in Vitro Driven by PRNA 3wj Motif of Phi29 DNA Packaging Motor. Nucleic Acids Res. 2014, 42, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibrov S. M.; McLean J.; Parsons J.; Hermann T. Self-Assembling RNA Square. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6405–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao C.; Li X.; Tian C.; Jiang W.; Wang G.; Mao C. Construction of RNA Nanocages by Re-Engineering the Packaging RNA of Phi29 Bacteriophage. Nat. Commun. 2014, 5, 3890. [DOI] [PubMed] [Google Scholar]
- Afonin K. A.; Desai R.; Viard M.; Kireeva M. L.; Bindewald E.; Case C. L.; Maciag A. E.; Kasprzak W. K.; Kim T.; Sappe A.; et al. Co-Transcriptional Production of RNA–DNA Hybrids for Simultaneous Release of Multiple Split Functionalities. Nucleic Acids Res. 2014, 42, 2085–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Viard M.; Martins A. N.; Lockett S. J.; Maciag A. E.; Freed E. O.; Heldman E.; Jaeger L.; Blumenthal R.; Shapiro B. A. Activation of Different Split Functionalities on Re-Association of RNA–DNA Hybrids. Nat. Nanotechnol. 2013, 8, 296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose S. D.; Kim D. H.; Amarzguioui M.; Heidel J. D.; Collingwood M. A.; Davis M. E.; Rossi J. J.; Behlke M. A. Functional Polarity Is Introduced by Dicer Processing of Short Substrate RNAs. Nucleic Acids Res. 2005, 33, 4140–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low J. T.; Knoepfel S. A.; Watts J. M.; ter Brake O.; Berkhout B.; Weeks K. M. Shape-Directed Discovery of Potent ShRNA Inhibitors of Hiv-1. Mol. Ther 2012, 20, 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D.; Kay M. A. Combinatorial RNAi: A Winning Strategy for the Race against Evolving Targets?. Mol. Ther 2007, 15, 878–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B.; Sanders R. W. Molecular Strategies to Design an Escape-Proof Antiviral Therapy. Antiviral Res. 2011, 92, 7–14. [DOI] [PubMed] [Google Scholar]
- Holmlund J. T.; Monia B. P.; Kwoh T. J.; Dorr F. A. Toward Antisense Oligonucleotide Therapy for Cancer: Isis Compounds in Clinical Development. Curr. Opin. Mol. Ther. 1999, 1, 372–385. [PubMed] [Google Scholar]
- Advani R.; Lum B. L.; Fisher G. A.; Halsey J.; Geary R. S.; Holmlund J. T.; Kwoh T. J.; Dorr F. A.; Sikic B. I. A Phase I Trial of Aprinocarsen (Isis 3521/Ly900003), an Antisense Inhibitor of Protein Kinase C-Alpha Administered as a 24-h Weekly Infusion Schedule in Patients with Advanced Cancer. Invest. New Drugs 2005, 23, 467–477. [DOI] [PubMed] [Google Scholar]
- Yu D.; Zhu F. G.; Bhagat L.; Wang H.; Kandimalla E. R.; Zhang R.; Agrawal S. Potent Cpg Oligonucleotides Containing Phosphodiester Linkages: In Vitro and in Vivo Immunostimulatory Properties. Biochem. Biophys. Res. Commun. 2002, 297, 83–90. [DOI] [PubMed] [Google Scholar]
- Ishii K. J.; Akira S. Tlr Ignores Methylated RNA?. Immunity 2005, 23, 111–113. [DOI] [PubMed] [Google Scholar]
- Krug A.; Rothenfusser S.; Hornung V.; Jahrsdorfer B.; Blackwell S.; Ballas Z. K.; Endres S.; Krieg A. M.; Hartmann G. Identification of Cpg Oligonucleotide Sequences with High Induction of Ifn-Alpha/Beta in Plasmacytoid Dendritic Cells. Eur. J. Immunol. 2001, 31, 2154–2163. [DOI] [PubMed] [Google Scholar]
- Sun L.; Wu J.; Du F.; Chen X.; Chen Z. J. Cyclic Gmp-Amp Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson D. B.; Medzhitov R. Recognition of Cytosolic DNA Activates an Irf3-Dependent Innate Immune Response. Immunity 2006, 24, 93–103. [DOI] [PubMed] [Google Scholar]
- Brencicova E.; Diebold S. S. Nucleic Acids and Endosomal Pattern Recognition: How to Tell Friend from Foe?. Front. Cell Infect. Microbiol. 2013, 3, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby R. E.; Webb L. M.; Mackenzie K. J.; Li Y.; Leitch A.; Reijns M. A.; Lundie R. J.; Revuelta A.; Davidson D. J.; Diebold S.; et al. RNA:DNA Hybrids Are a Novel Molecular Pattern Sensed by Tlr9. EMBO J. 2014, 33, 542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Chen Z. J. Innate Immune Sensing and Signaling of Cytosolic Nucleic Acids. Annu. Rev. Immunol. 2014, 32, 461–488. [DOI] [PubMed] [Google Scholar]
- Shangguan D.; Li Y.; Tang Z.; Cao Z. C.; Chen H. W.; Mallikaratchy P.; Sefah K.; Yang C. J.; Tan W. Aptamers Evolved from Live Cells as Effective Molecular Probes for Cancer Study. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 11838–11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangguan D.; Tang Z.; Mallikaratchy P.; Xiao Z.; Tan W. Optimization and Modifications of Aptamers Selected from Live Cancer Cell Lines. ChemBioChem 2007, 8, 603–606. [DOI] [PubMed] [Google Scholar]
- Chen J. H.; Seeman N. C. Synthesis from DNA of a Molecule with the Connectivity of a Cube. Nature 1991, 350, 631–633. [DOI] [PubMed] [Google Scholar]
- Lee H.; Lytton-Jean A. K.; Chen Y.; Love K. T.; Park A. I.; Karagiannis E. D.; Sehgal A.; Querbes W.; Zurenko C. S.; Jayaraman M.; et al. Molecularly Self-Assembled Nucleic Acid Nanoparticles for Targeted in Vivo SiRNA Delivery. Nat. Nanotechnol. 2012, 7, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen E. S.; Dong M.; Nielsen M. M.; Jahn K.; Subramani R.; Mamdouh W.; Golas M. M.; Sander B.; Stark H.; Oliveira C. L.; et al. Self-Assembly of a Nanoscale DNA Box with a Controllable Lid. Nature 2009, 459, 73–76. [DOI] [PubMed] [Google Scholar]
- Kuzyk A.; Schreiber R.; Fan Z.; Pardatscher G.; Roller E. M.; Hogele A.; Simmel F. C.; Govorov A. O.; Liedl T. DNA-Based Self-Assembly of Chiral Plasmonic Nanostructures with Tailored Optical Response. Nature 2012, 483, 311–314. [DOI] [PubMed] [Google Scholar]
- Rothemund P. W. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Dietz H.; Liedl T.; Hogberg B.; Graf F.; Shih W. M. Self-Assembly of DNA into Nanoscale Three-Dimensional Shapes. Nature 2009, 459, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Ye T.; Su M.; Zhang C.; Ribbe A. E.; Jiang W.; Mao C. Hierarchical Self-Assembly of DNA into Symmetric Supramolecular Polyhedra. Nature 2008, 452, 198–201. [DOI] [PubMed] [Google Scholar]
- Goodman R. P.; Heilemann M.; Doose S.; Erben C. M.; Kapanidis A. N.; Turberfield A. J. Reconfigurable, Braced, Three-Dimensional DNA Nanostructures. Nat. Nanotechnol. 2008, 3, 93–96. [DOI] [PubMed] [Google Scholar]
- Seeman N. C. Nanomaterials Based on DNA. Annu. Rev. Biochem. 2010, 79, 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M.; Yamamoto S.; Tatsumi K.; Emura T.; Hidaka K.; Sugiyama H. RNA-Templated DNA Origami Structures. Chem. Commun. 2013, 49, 2879–2881. [DOI] [PubMed] [Google Scholar]
- Ko S. H.; Su M.; Zhang C.; Ribbe A. E.; Jiang W.; Mao C. Synergistic Self-Assembly of RNA and DNA Molecules. Nat. Chem. 2010, 2, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Ko S. H.; Tian C.; Hao C.; Mao C. RNA–DNA Hybrid Origami: Folding of a Long RNA Single Strand into Complex Nanostructures Using Short DNA Helper Strands. Chem. Commun. 2013, 49, 5462–5464. [DOI] [PubMed] [Google Scholar]
- Martinez H. M.; Maizel J. V. Jr.; Shapiro B. A. RNA2d3d: A Program for Generating, Viewing, and Comparing 3-Dimensional Models of RNA. J. Biomol. Struct. Dyn. 2008, 25, 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M.; Major F. The Mc-Fold and Mc-Sym Pipeline Infers RNA Structure from Sequence Data. Nature 2008, 452, 51–55. [DOI] [PubMed] [Google Scholar]
- Popenda M.; Szachniuk M.; Antczak M.; Purzycka K. J.; Lukasiak P.; Bartol N.; Blazewicz J.; Adamiak R. W. Automated 3d Structure Composition for Large RNAs. Nucleic Acids Res. 2012, 40, e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Kireeva M.; Grabow W. W.; Kashlev M.; Jaeger L.; Shapiro B. A. Co-Transcriptional Assembly of Chemically Modified RNA Nanoparticles Functionalized with SiRNAs. Nano Lett. 2012, 12, 5192–5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Cieply D. J.; Leontis N. B. Specific RNA Self-Assembly with Minimal Paranemic Motifs. J. Am. Chem. Soc. 2008, 130, 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Leontis N. B. Generating New Specific RNA Interaction Interfaces Using C-Loops. J. Am. Chem. Soc. 2006, 128, 16131–16137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.; Afonin K. A.; Viard M.; Koyfman A. Y.; Sparks S.; Heldman E.; Grinberg S.; Linder C.; Blumenthal R. P.; Shapiro B. A. In Silico, in Vitro, and in Vivo Studies Indicate the Potential Use of Bolaamphiphiles for Therapeutic SiRNAs Delivery. Mol. Ther. Nucleic Acids 2013, 2, e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.