

Abstract
NMDA receptors (NMDAR) are voltage- and glutamate-gated heteromeric ion channels found at excitatory neuronal synapses, the functions of which are to mediate the mechanisms of brain plasticity and, thereby, its higher order functions. In addition to Glu, the activation of these heteromeric receptors requires Gly or d-Ser as a coagonist. However, it is not fully known as to why coagonism is required for the opening of NMDAR ion channels. We show herein that the ligand binding domains (LBD) of the GluN1 and GluN2A subunits of the NMDAR heterodimerize only when both coagonists, Glu and Gly/d-Ser, bind to their respective sites on GluN2 and GluN1. In the agonist-free state, these domains form homomeric interactions, which are disrupted by binding of their respective agonists. Also, in a heteromer formed by the LBDs, GluN2A is more sensitized to bind Glu, while the affinity of Gly for GluN1 remains unchanged. We thus provide direct evidence to show that coagonism is necessary for heteromeric pairing of LBDs, which is an essential step in forming functional ion channels in NMDARs.
N-methyl-d-aspartate receptors (NMDAR) are heteromeric cation channels that are essential for neuronal synaptic plasticity mechanisms in the brain.1 They function as coincidence detectors of presynaptic and postsynaptic activities, leading to synapse strengthening.2 Because of the central role that NMDARs play in glutamatergic neuronal transmission, their dysregulation is associated with several pathophysiologies such as stroke, epilepsy, Parkinson’s disease, Alzheimer’s disease, schizhophrenia, and neuropathic pain.1,3−5 Functional NMDARs possess a tetrameric arrangement of subunits with a central ion channel pore surrounded by two of any of eight splice variants of GluN1 (a–h) and any of two separate gene products of GluN2 (A–D) subunits.6 The temporal and spatial nature of the subunit composition of the NMDAR confers considerable plasticity on the electrical properties of the resulting ion channel of this receptor. However, in all cases, activation of these channels requires l-Glu and Gly or d-Ser as coagonists,6,7 where they specifically bind to GluN2 and GluN1 subunits, respectively.7−9 Recent studies show that Gly acts on GluN1 at extrasynaptic sites, while d-Ser acts on GluN1 at synaptic sites.10 Although structures of the extracellular and transmembrane domains of the NMDAR have been recently reported, it is still not clear as to why coagonism is required for channel activation and how agonist binding leads to channel opening.11−14 Earlier studies had proposed that binding of agonists to the ligand binding domain (LBD) clefts lead to the closure of the cleft which, in-turn, opens the channel pore. However, later studies suggested that cleft closure step in itself does not explain the channel opening in NMDARs.15,16
Many of the ionotropic glutamate receptors (iGluRs) have been shown to form homomeric ion channels, whereas NMDARs are known to form only heteromeric channels.17 Obtaining functionally active LBDs of the heteromeric NMDARs has been challenging. However, we were successful in isolating function-intact LBDs of GluN1 and GluN2A subunits of the NMDAR for this current study. We present data analyzed from analytical ultracentrifugation (AUC), functional binding assays, and electrophysiological characteristics of NMDAR currents from cultured primary neurons, which provide explanations for the essentiality of coagonism in NMDAR channel activation. Specifically, binding of coagonists induces rearrangements at LBD regions by disrupting homomeric interactions and inducing heteromeric contacts. We also show that there is a clear functional advantage to coagonist binding in a heteromeric complex of GluN1 and GluN2A LBDs.
Experimental Procedures
Protein Expression and Purification
Rat cDNAs were constructed that encoded the extracellular LBD of GluN1, which encompassed the S1 region (Met394-Lys544), linked to its S2 domain (Arg663-Ser800), via a Gly-Thr linker. Similarly, the LBD of GluN2A was constructed by joining its S1 region (Pro401-Arg539) to S2 (Glc661-Asn802 also via a Gly-Thr linker. Each of the LBDs were expressed in Drosophila S2 cells as secreted proteins using a vector system with a Drosophila BiP secretory signal,18 as well as a C-terminal (His)8 tag for detection and affinity chromatography purification. The proteins were purified using a three-step protocol involving cation exchange chromatography with cellulose phosphate resin, followed by Ni-NTA affinity purification, and finally size exclusion chromatography on an Akta Superdex 200 column. All purifications were carried out in 30 mM Hepes/75 mM NaCl, pH 7.6.
AUC
Sedimentation velocity and sedimentation equilibrium analyses were performed in a Beckman Coulter Optima XL-I analytical ultracentrifuge. Sedimentation velocity studies were accomplished at 35 000 rpm at protein concentrations of 0.2–0.8 mg/mL in the presence or absence of ligands. Sedimentation equilibrium data were obtained at three different rotor speeds, viz., 17 000, 21 000, and 23 000 rpm, also at protein concentrations of 0.2–0.8 mg/mL. The detection method used was A280nm. The samples were run in two-sector centerpieces for velocity analyses and six-sector centerpieces for equilibrium analyses at a temperature of 20 °C. The buffer used was 30 mM Hepes/75 mM NaCl, pH 7.6, with or without 400–500 μM l-Glu or Gly/d-Ser. Equilibrium scans were collected every 6 h until the last two scans overlapped completely. The partial specific volumes of the proteins, as well as solvent viscosities and densities, were calculated using Sednterp. The partial specific volumes of proteins were calculated from the respective sequences. The velocity data were analyzed by SEDFIT19 and plotted for both continuous sedimentation coefficient distributions c(s) or molar mass distributions, c(M). SEDFIT uses estimates of partial specific volume and frictional ratio of the macromolecules to calculate the diffusion coefficients to fit for c(s) or c(M) distributions approximated based on Lamm equation solutions as described.19,20 Equilibrium data were analyzed using SEDPHAT analysis software by fitting to the species analysis model.
Equilibrium Ligand Binding Assays
The purified LBDs of GluN1 and GluN2A were assessed for binding of ligands, Gly and l-Glu, respectively. [3H]-Gly (American Radiolabeled Chemicals, St. Louis, MO) and [3H]-l-Glu (PerkinElmer, Waltham, MA) were utilized for the assays. Rapid Equilibrium Dialysis (RED) Devices (Thermo Fisher Scientific, Waltham, MA) with a molecular weight cutoff of 8 kDa were used to equilibrate the purified LBDs with the radiolabeled ligands. A range of radioligand concentrations, from 0.5–75 μM, was used for the assays. Apart from the radioligands, each reaction mixture had 4–6 μM purified protein in 50 mM Tris/50 mM NaCl, pH 7.8. The reaction mixtures were incubated on ice for 30–60 min before transferring to the sample chamber of the RED devices. The sample (100 μL) and a corresponding volume of 300 μL of buffer were added to the sample and buffer chambers, respectively. The samples were equilibrated at room temperature with gentle shaking for 6–7 h. Nonspecific binding was excluded by assaying a control reaction with 1000× molar excess of unlabeled ligand for each concentration point used in each set. After equilibration, equal volumes of samples were counted from both the sample and buffer chambers in a LS6500 Beckman Scintillation Counter. The counts from the buffer chamber correspond to the free ([F]) ligand concentration, while the counts from sample chamber correspond to bound ([B]) + [F] ligand concentrations. The bound ligand concentrations were plotted against the initial concentrations. The data were fitted by nonlinear regression to a one-site binding model using GraphPad Prism (La Jolla, CA) software to obtain the affinity constant, KD from the following equation:
Primary Neuron Culture
Cortical neurons were dissociated with 1 mg/mL papain from embryos of GluN2B–/– mice and plated on 35 mm tissue culture-treated dishes coated with poly-l-Lys. Neurobasal medium (Invitrogen, Carlsbad, CA), Supplemented with 2% B27 Supplement (Invitrogen)/1% l-Glu were used for the cell cultures maintained at 37 °C in a humidified atmosphere with 5% CO2.
Electrophysiology
Whole-cell patch clamp electrophysiology recordings of GluN2B–/– neurons of day-in vitro (DIV) 13–20 were performed at room temperature. For recordings, neurons were bathe in an extracellular solution composed of 140 mM NaCl/3 mM KCl/2 mM CaCl2/10 mM HEPES/1 μM tetrodotoxin (TTX)/20 mM dextrose, pH 7.35. Borosilicate glass recording pipettes with a resistance of 2–4 MΩ, were constructed using a Flaming/Brown Micropipette Puller, model P-97 (Sutter Instrument Company, Novato, CA). Pipettes were backfilled with an intracellular solution of 140 mM CsF/2 mM CaCl2/10 mM EGTA/10 mM HEPES/2 mM tetraethylammonium chloride/4 mM Na2ATP, pH 7.35. The test solutions were applied using a nine-barrel Rapid Solution Changer, RSC-200 (BioLogic, Claix, Fr). An extracellular solution containing 100 μM l-Glu/10 μM Gly or d-Ser/1 μM TTX/0.5 μM strychnine was applied to elicit NMDA induced currents. An Axopatch-200B amplifier (Molecular Devices, Sunnyvale, CA) was used to record the whole-cell current, which was low-pass filtered at 5 kHz by a built-in eight-pole Bessel filter, digitized at 1 kHz sampling frequency using a Digidata 1322A digitizer (Molecular Devices). The cells were voltage-clamped at −70 mV, pH 7.35. pCLAMP-8 software (Molecular Devices) was used to acquire data. Data were analyzed utilizing Clampfit and Prism Graphpad. Peak current amplitudes, as well as the area integral of the peak current, were quantified separately. Statistical significance for the differences between preincubations and controls were tested by paired t-test analyses.
Results and Discussion
Agonist Binding Disrupts Homodimers of GluN1 or GluN2A LBDs
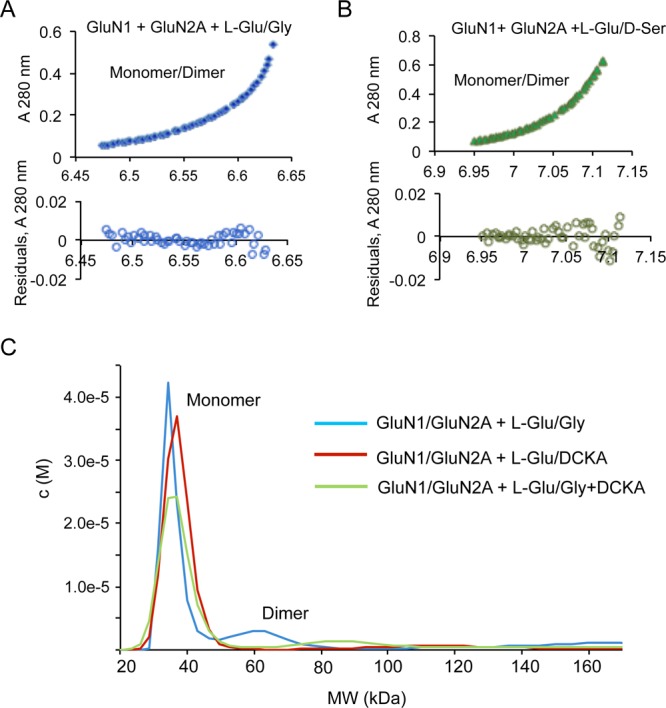
It is known that LBDs of many iGluR subtypes are able to self-associate, although self-association of LBDs of the NMDAR has not been as clear.21 We performed sedimentation velocity and sedimentation equilibrium studies on purified recombinant LBDs of GluN1 and GluN2A subunits of the NMDAR (Figure 1A, inset). Theoretical monomer masses used for sedimentation equilibrium data fitting were 36.5 kDa for GluN1 and 34.9 kDa for GluN2A and the homodimer molar masses used was two times the corresponding monomer molar masses. Our results show that LBDs of GluN1 or GluN2A can self-associate to form homomers. Both GluN1 and GluN2A LBDs showed the presence of dimeric forms in addition to monomeric forms (Figure 1A,B). However, addition of l-Glu led to a substantial increase in the monomeric fraction of GluN2A (Figure 1A), as was also the case with the addition of Gly to GluN1 (Figure 1C, D). In either case, the addition of agonists resulted in dimeric forms of each LBD dissociating into monomeric forms. This shows that agonist binding disrupts homomers of both GluN1 and GluN2A. There have been conflicting reports regarding the self-associating potential of the LBDs of the NMDAR.22,23 However, our results show that in the absence of ligands these domains self-associate. This finding assumes significance because most of the structural data available have utilized ligand bound forms of the receptor, which potentially may not reveal the nature of the homomeric interactions.
Figure 1.

Self-association of LBDs is disrupted by agonist binding. (A) c(M) distribution overlay after sedimentation velocity analysis of a primarily dimeric sample of GluN2A to highlight the increase in monomer population in the presence of l-Glu. Inset, SDS-PAGE showing purified protein preparations of GluN1-LBD and GluN2A-LBD respectively. (B) Sedimentation equilibrium analysis of GluN1 analyzed using SEDPHAT and fitted with the Species Analysis model for a combination of corresponding GluN1 monomeric and dimeric molecular weights. The bottom panel shows the residuals of the fitted data. (C) Sedimentation equilibrium data of GluN1 + 400 μM Gly were analyzed with SEDPHAT and fitted for GluN1 monomer size using the Species Analysis model. (D) Sedimentation velocity analysis of the GluN1-LBD analyzed using SEDFIT and showing c(M) distribution of molecular masses corresponding to monomeric and dimeric species in the presence or absence of Gly.
Coagonism Is a Requirement for Heteromeric Association of GluN1 and GluN2 LBDs
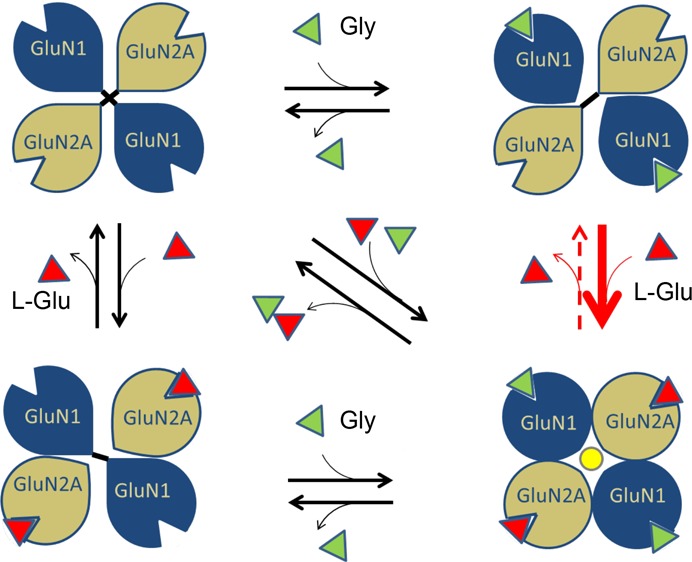
In order to study the effects of agonists on the hetero-oligomerization of the LBDs of GluN1 and GluN2, we performed both sedimentation velocity and sedimentation equilibrium analyses of an equimolar mixture of GluN1 and GluN2A and evaluated the changes of the dimeric population in the presence or absence of l-Glu and Gly/d-Ser. The results from sedimentation velocity analyses are shown in Figure 2A–D. A combined average molecular weight of 36 kDa for the monomer and a corresponding dimer size of 72 kDa were used to fit the GluN1/GluN2A heterodimer molar mass for sedimentation equilibrium analyses. We found that the dimeric fraction is substantially increased only when both the coagonists are present (Figure 2A,B). Similar results were obtained when Gly was replaced by d-Ser (Figure 2C,D). Sedimentation equilibrium analyses led to the same conclusions (Figure 3A,B). These data show that binding of both the agonists to their respective LBDs is necessary to drive heteromerization. Thus, the two-coagonist requirement of NMDAR is likely a mechanism to initiate and stabilize the heteromeric state of NMDAR during its activation. We also demonstrate that agonist-driven heteromerization is prevented by 5,7-dichlorokynurenic acid (DCKA), a selective Gly binding site inhibitor of NMDAR (Figure 3C). This finding reveals that the mechanism of DCKA inhibition involves interference with GluN1-GluN2 interaction at the LBD region of NMDAR. In addition, this also shows that heteromerization of NMDAR subunits is a feature that could be targeted by antagonists.
Figure 2.

Heteromerization of GluN1 and GluN2A LBDs requires both coagonists. (A) Sedimentation velocity data analyzed using SEDFIT and showing the c(M) distribution of molecular masses corresponding to monomeric and dimeric species for the combination of GluN1 + GluN2A in the presence or absence of coagonists. (B) Bar diagrams showing the ratio of percent fraction of dimer/monomer species as in (A), quantified by the peak integration method in SEDFIT. The binding of coagonists leads to a higher ratio than that in the absence of ligands, l-Glu alone or Gly alone. * denotes p = 0.02, 0.03, and 0.02 respectively obtained from unpaired t-test analyses from two independent experimental sets. (C) c(M) distribution overlay to highlight the increase in dimer population in the presence of l-Glu/Gly or l-Glu/d-Ser combinations, but not in the presence of d-Ser alone. (D) Bar diagrams showing the ratio percent of dimer/monomer species as in (C), quantified by the peak integration method in SEDFIT. ** denotes p = 0.003 obtained from unpaired t-test analyses.
Figure 3.
Heterodimerization of GluN1 and GluN2 requires both coagonists. (A, B) SEDPHAT analysis of GluN1/GluN2A in the presence of both l-Glu and Gly (A) or l-Glu and d-Ser (B) fitted to monomer and dimer molar masses using a combined average molecular weight of GluN1 and GluN2A of 36 000. (C) c(M) distribution overlay showing attenuation of dimerization in the presence of DCKA compared to that in the presence of l-Glu/Gly.
Glutamate Affinity to GluN2A Is Enhanced by Gly-bound GluN1, but not GluN1 Alone
In order to ascertain whether a functional effect is associated with the inter-LBD interactions of GluN1 and GluN2A, we performed equilibrium binding assays using radiolabeled Gly and l-Glu with the purified preparations of GluN1 and GluN2A in microdialysis devices. The binding dissociation constants (KD) for Gly and l-Glu for the LBDs of GluN1 and GluN2A, respectively, were measured with a combination of both proteins (Figure 4A–D, Table 1). Remarkably, the experiments showed that while the presence of GluN1 did not affect the binding affinity of l-Glu to GluN2A, the addition of Gly in the reaction significantly enhanced the affinity of l-Glu toward GluN2A with an ∼3 fold reduction in KD value and a correspondingly higher Bmax (Figure 4C, Figure 5A,B, Table 1). The increase in Bmax suggests that a shift of the binding equilibrium occurs toward a stable ligand bound state for GluN2A, with a slower ligand dissociation rate. This implies that the Gly-bound GluN1 enters into a modulatory interaction with GluN2A. We did not observe a similar enhancement in binding of Gly to GluN1 in the presence of l-Glu bound to GluN2A (Figure 4D, Figure 5C,D, Table 1). These results clearly show that the cooperative enhancement observed for l-Glu binding to GluN2A subunits in the intact heteromeric receptor is primarily driven by inter-LBD interactions, while any allosteric enhancement of Gly binding is not through the LBD.24−27 This finding also highlights the fact that coagonists are necessary for functional modulation in addition to channel activation of NMDARs.
Figure 4.

Binding of coagonists to NMDAR subunits. (A) 3H-l-Glu binding profile of GluN2A-LBD measured by equilibrium dialysis. (B) 3H-Gly binding profile of the GluN1-LBD. (C) Combination assay using both GluN1-LBD and GluN2A-LBD for 3H-l-Glu binding in comparison with GluN2A alone. (D) Combination assay using both GluN1-LBD and GluN2A-LBD for [3H]-Gly binding in comparison with GluN1 alone.
Table 1.
| KD (μM) | Bmax (pmol) | |
|---|---|---|
| GluN1-LBD + GluN2A-LBD: [3H]-l-glutamate | 7.8 ± 0.3 | 2.5 ± 0.3 |
| GluN1-LBD + GluN2A-LBD + glycine: [3H]-l-glutamate | 2.8 ± 0.7 | 6.4 ± 0.2 |
| GluN1-LBD + GluN2A-LBD: [3H]-glycine | 7.8 ± 2 | 7.4 ± 0.8 |
| GluN1-LBD + GluN2A-LBD + l-Glu: [3H]-glycine | 7.6 ± 2.5 | 7.7 ± 0.2 |
Figure 5.

Binding of l-Glu to GluN2A is enhanced by Gly binding to GluN1. (A) [3H]-l-Glu equilibrium binding assay using equilibrium dialysis performed for the combination of GluN2A and GluN1 in the presence or absence of Gly. (B) Averaged Bmax values from (A) are represented as bar graphs. (C) Combination assays using both GluN1 and GluN2A for [3H]-Gly binding in the presence or absence of l-Glu. (D) Averaged Bmax values from (C) represented as bar graphs. No significant differences were observed for [3H]-Gly binding in the presence or absence of l-Glu. The KD and Bmax values obtained from the assays are summarized in Table 1. *** denotes p = 0.0002 obtained from unpaired t test analysis, n = 3.
Pretreatment of Either Coagonist Prepares the Receptor for Faster Channel Opening
If an agonist-driven rearrangement event occurs for the channel activation, we assumed that it would be directly observed on the channel currents. Therefore, we measured l-Glu induced whole cell currents with either Gly or d-Ser as coagonists on GluN2B–/– derived mouse cortical neurons in culture, and evaluated the effects of preincubation of any of the three coagonists before fully stimulating channel opening with both the agonists. We found a significant increase in overall conductance in all three cases (Figure 6A–C; quantifications are presented in Figure 7A–F). Enhanced conductance after Gly or d-Ser preincubations were mostly displayed by a significant rise in peak amplitudes, while the increased current after l-Glu preincubation was mostly contributed by a slower desensitization step. The data from electrophysiological recordings support our sedimentation equilibrium and sedimentation velocity data, which demonstrated that binding of one of the two coagonists to the LBD of its respective subunit could disrupt a homomeric interaction, which would be the diagonally placed subunit in a 1–2–1–2 tetrameric arrangement.13,14,28 A coagonist preincubation achieves this first step so that when the second agonist binds, the channel opening event is faster with a higher peak current than when both the agonists are applied together.
Figure 6.
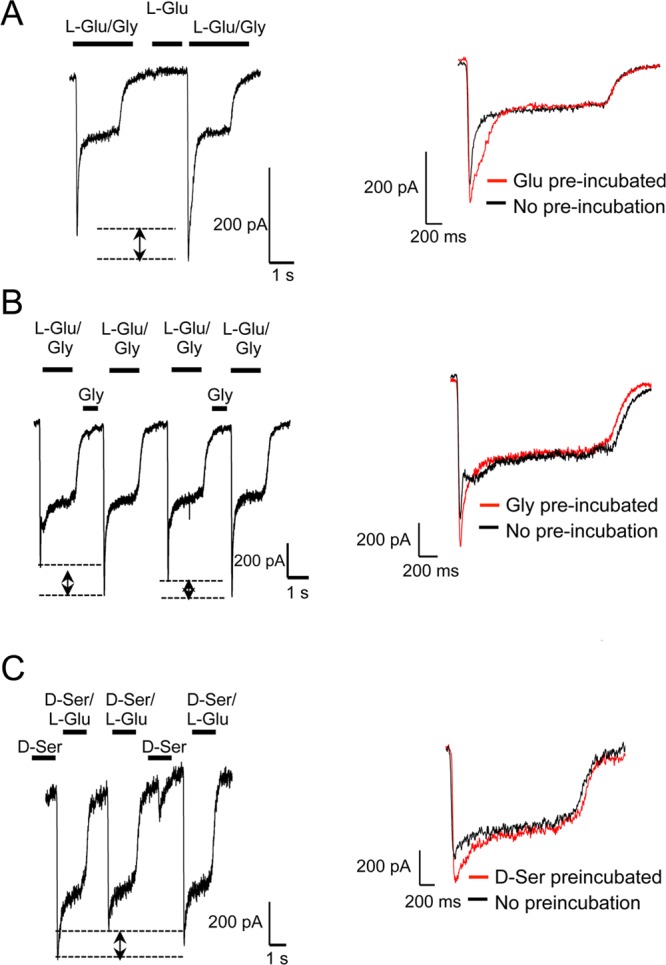
Enhanced NMDAR channel conductance follows coagonist preincubation. (A) Whole cell recordings of l-Glu/Gly induced currents before and after preincubation with l-Glu. (B) Traces for l-Glu/Gly currents before and after Gly preincubation. (C) Traces for l-Glu/d-Ser currents before and after d-Ser preincubation. Each trace from (A–C) represents at least three recordings. Each inset shows overlay of traces with (red) or without (black) preincubation.
Figure 7.

Comparison of NMDA-induced current peak amplitudes, as well as the area under the current peak between the control and coagonist preincubated samples. (A, B) Gly preincubation, (C, D) d-Ser preincubation, (E, F) l-Glu preincubation. p values obtained from paired t-tests in each case are shown.
Agonist-Driven LBD Reorganization for NMDAR Channel Activation
Previous studies on amino-terminal truncated mutants of NMDARs have shown that LBDs form the essential region that is sufficient to provide the agonist-gated channel activation property.29,30 Structural studies revealed that the LBDs adopted a clamshell cleft-like structure with an upper and a lower lobe that close upon agonist binding.31 It was suggested that the cleft closure would pull the linker between this domain and the ion channel pore to result in channel opening. However, later studies with partial agonists that also lead to similar degree of cleft closure, but reduced channel activation, suggested the possible existence of additional mechanisms for complete channel activation.32 In recent studies on LBDs in other iGluRs, such as kainate receptors, it had been observed that these domains undergo major quaternary structural rearrangements upon activation, compared to other regions of the receptor.33 The mechanical coupling between the LBD and M3 helices of NMDARs, as reported recently, also shows that any realignment at the LBD region can directly affect the M3 helices.34 This indicates that agonist-induced pairings of GluN1 and GluN2 LBDs, which we report here, could act in concert with M3-S2 linker stretching to lead to the final open state of the ion channel pore. On the basis of these observations, we propose a model highlighting the realignment of GluN1/GluN2A LBDs of the NMDAR upon binding of coagonists (Figure 8). In a GluN1–2–1–2 tetrameric arrangement of the NMDAR, homomeric interactions between the diagonally placed subunits could attract the LBDs inward, thereby blocking the ion channel pore. Simultaneous binding of coagonists disrupts the diagonal homomeric contacts and instead promotes heteromeric contacts between adjacent GluN1 and GluN2 LBDs leading to subunit realignments resulting in channel pore opening. The GluN2 subunit in this quaternary arrangement has a greater probability to retain the l-Glu agonist in its cleft and hence would be more resistant to variations in the neurotransmitter concentrations.
Figure 8.

Proposed mechanism of coagonist regulation in NMDAR activation. A model showing the possible mechanism of partial agonism for NMDAR activation based on homomeric and heteromeric interactions in the LBDs. In the absence of agonists the diagonally placed LBDs will tend to form homomeric interactions resulting in the closing of the ion channel pore. Binding of one of the agonists can disrupt the homomeric interaction of that subunit. Binding of the second coagonist will initiate heteromeric interactions. The agonist induced heteromeric interactions between LBDs causes realignment that could transduce down leading to channel opening. Inability of a single coagonist to induce domain realignments maintains the channel in the closed state. The red arrow in bold indicates that binding of l-Glu is favored by a Gly bound GluN1 as per our data. The red dotted arrow indicates that the dissociation rate of l-Glu is reduced when Gly remains bound to GluN1.
In conclusion, this study provides a plausible explanation of a previously unknown mechanism of agonist-induced heteromeric pairing and subsequent functional modulation of NMDAR-LBDs and thereby provides critical insights to answer the question as to why coagonism is required for NMDAR activation.
Acknowledgments
The authors thank Dr. Victoria. A. Ploplis, Ms. Deborah Donahue, and Dr. Mary. F. Prorok for help or suggestions during the course of this study.
The authors declare no competing financial interest.
Supported by Grant HL019982 from the NIH.
Funding Statement
National Institutes of Health, United States
References
- Paoletti P.; Bellone C.; Zhou Q. (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Dan Y.; Poo M. M. (2004) Spike timing-dependent plasticity of neural circuits. Neuron 44, 23–30. [DOI] [PubMed] [Google Scholar]
- Lai T. W.; Shyu W. C.; Wang Y. T. (2011) Stroke intervention pathways: NMDA receptors and beyond. Trends Mol. Med. 17, 266–275. [DOI] [PubMed] [Google Scholar]
- Ahmed I.; Bose S. K.; Pavese N.; Ramlackhansingh A.; Turkheimer F.; Hotton G.; Hammers A.; Brooks D. J. (2011) Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain 134, 979–986. [DOI] [PubMed] [Google Scholar]
- Ghasemi M.; Schachter S. C. (2011) The NMDA receptor complex as a therapeutic target in epilepsy: a review. Epilepsy Behav. 22, 617–640. [DOI] [PubMed] [Google Scholar]
- Traynelis S. F.; Wollmuth L. P.; McBain C. J.; Menniti F. S.; Vance K. M.; Ogden K. K.; Hansen K. B.; Yuan H.; Myers S. J.; Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner N. W.; Dingledine R. (1988) Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 241, 835–837. [DOI] [PubMed] [Google Scholar]
- Johnson J. W.; Ascher P. (1987) Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325, 529–531. [DOI] [PubMed] [Google Scholar]
- Mothet J. P.; Parent A. T.; Wolosker H.; Brady R. O.; Linden D. J.; Ferris C. D.; Rogawski M. A.; Snyder S. H. (2000) D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. U.S.A. 97, 4926–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papouin T.; Ladepeche L.; Ruel J.; Sacchi S.; Labasque M.; Hanini M.; Groc L.; Pollegioni L.; Mothet J. P.; Oliet S. H. (2012) Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 150, 633–646. [DOI] [PubMed] [Google Scholar]
- Kazi R.; Gan Q.; Talukder I.; Markowitz M.; Salussolia C. L.; Wollmuth L. P. (2013) Asynchronous movements prior to pore opening in NMDA receptors. J. Neurosci. 33, 12052–12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersen A.; Tajima N.; Fernandez-Cuervo G.; Garnier-Amblard E. C.; Furukawa H. (2014) Structural insights into competitive antagonism in NMDA receptors. Neuron 81, 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E.; Furukawa H. (2014) Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 344, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. H.; Lu W.; Michel J. C.; Goehring A.; Du J.; Song X.; Gouaux E. (2014) NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 511, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussius C. L.; Popescu G. K. (2010) NMDA receptors with locked glutamate-binding clefts open with high efficacy. J. Neurosci. 30, 12474–12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borschel W. F.; Murthy S. E.; Kasperek E. M.; Popescu G. K. (2011) NMDA receptor activation requires remodelling of intersubunit contacts within ligand-binding heterodimers. Nat. Commun. 2, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J.; Mayer M. L. (2013) Functional insights from glutamate receptor ion channel structures. Annu. Rev. Physiol. 75, 313–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaki T.; Figuera M.; Ploplis V. A.; Castellino F. J. (2003) Rapid selection of Drosophila S2 cells with the puromycin-resistant gene. BioTechniques 35, 482–486. [DOI] [PubMed] [Google Scholar]
- Schuck P.; Perugini M. A.; Gonzales N. R.; Howlett G. J.; Schubert D. (2002) Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 82, 1096–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veran J.; Kumar J.; Pinheiro P. S.; Athane A.; Mayer M. L.; Perrais D.; Mulle C. (2012) Zinc potentiates GluK3 glutamate receptor function by stabilizing the ligand binding domain dimer interface. Neuron 76, 565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H.; Singh S. K.; Mancusso R.; Gouaux E. (2005) Subunit arrangement and function in NMDA receptors. Nature 438, 185–192. [DOI] [PubMed] [Google Scholar]
- Ivanovic A.; Reilander H.; Laube B.; Kuhse J. (1998) Expression and initial characterization of a soluble glycine binding domain of the N-Methyl-D-aspartate receptor NR1 subunit. J. Biol. Chem. 273, 19933–19937. [DOI] [PubMed] [Google Scholar]
- Fadda E.; Danysz W.; Wroblewski J. T.; Costa E. (1988) Glycine and D-serine increase the affinity of N-methyl-D-aspartate sensitive glutamate binding sites in rat brain synaptic membranes. Neuropharmacology 27, 1183–1185. [DOI] [PubMed] [Google Scholar]
- Grimwood S.; Wilde G. J.; Foster A. C. (1993) Interactions between the glutamate and glycine recognition sites of the N-methyl-D-aspartate receptor from rat brain, as revealed from radioligand binding studies. J. Neurochem. 60, 1729–1738. [DOI] [PubMed] [Google Scholar]
- Danysz W.; Parsons C. G. (1998) Glycine and N-methyl-D-aspartate receptors: physiological significance and possible therapeutic applications. Pharmacol. Rev. 50, 597–664. [PubMed] [Google Scholar]
- Regalado M. P.; Villarroel A.; Lerma J. (2001) Intersubunit cooperativity in the NMDA receptor. Neuron 32, 1085–1096. [DOI] [PubMed] [Google Scholar]
- Riou M.; Stroebel D.; Edwardson J. M.; Paoletti P. (2012) An alternating GluN1–2-1–2 subunit arrangement in mature NMDA receptors. PloS One 7, e35134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madry C.; Mesic I.; Betz H.; Laube B. (2007) The N-terminal domains of both NR1 and NR2 subunits determine allosteric Zn2+ inhibition and glycine affinity of N-methyl-D-aspartate receptors. Mol. Pharmacol. 72, 1535–1544. [DOI] [PubMed] [Google Scholar]
- Mony L.; Zhu S.; Carvalho S.; Paoletti P. (2011) Molecular basis of positive allosteric modulation of GluN2B NMDA receptors by polyamines. EMBO J. 30, 3134–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H.; Singh S. K.; Mancusso R.; Gouaux E. (2005) Subunit arrangement and function in NMDA receptors. Nature 438, 185–192. [DOI] [PubMed] [Google Scholar]
- Kussius C. L.; Popescu A. M.; Popescu G. K. (2010) Agonist-specific gating of NMDA receptors. Channels 4, 78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauder D. M.; Kuybeda O.; Zhang J.; Klymko K.; Bartesaghi A.; Borgnia M. J.; Mayer M. L.; Subramaniam S. (2013) Glutamate receptor desensitization is mediated by changes in quaternary structure of the ligand binding domain. Proc. Natl. Acad. Sci. U.S.A. 110, 5921–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazi R.; Dai J.; Sweeney C.; Zhou H. X.; Wollmuth L. P. (2014) Mechanical coupling maintains the fidelity of NMDA receptor-mediated currents. Nat. Neurosci. 17, 914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]