

Abstract

To explore the feasibility of developing ligands targeted to the atypical C1 domains of protein kinase C ζ and ι, we have prepared diacylglycerol lactones substituted with hydrophilic groups on their side chains, which potentially could interact with the arginine residues that distinguish the atypical C1 domains of PKCζ and PKCι from typical C1 domains, and we have measured their binding to mutated versions of the C1b domain of PKCδ that incorporate one or more of these arginine residues. The most selective of the diacylglycerol lactones showed only a 10-fold reduction in binding affinity with the triple arginine mutant (N7R/S10R/L20R) compared to the wild-type, whereas phorbol 12,13-dibutyrate showed a 6000-fold loss of affinity. Molecular modeling confirms that these ligands are indeed able to interact with the arginine residues. Our results show that dramatic changes in selectivity can be obtained through appropriate substitution of diacylglycerol lactones.
Introduction
The C1 domain has emerged as a critical structural motif functioning as a recognition module for the lipophilic second messenger sn-1,2-diacylglycerol (DAG).1−3 Initially identified as the regulatory site on the classic and novel isoforms of protein kinase C (PKC) for DAG and their ultrapotent analogues the phorbol esters, C1 domains serve a similar function while coupled to distinct effector domains on six other families of proteins.4−6 These families comprise isoforms of protein kinase D, chimaerin, RasGRP, unc13, MRCK, and DAG kinase. DAG, the endogenous ligand for C1 domains, plays a central role in cellular signaling. It is generated through breakdown of phosphoinositol 4,5-bisphosphate by phospholipase Cβ, activated by G-protein-coupled receptors, as well as by phospholipase Cγ, activated by receptor tyrosine kinases.7 It is also generated indirectly through the activity of phospholipase D on phosphatidylcholine.8 Given the critical role played by the many pathways signaling through DAG, it is not surprising that PKC and the other family members have been identified as attractive therapeutic targets for cancer, Alzheimer’s disease, and a variety of other conditions.9,10 Drugs in clinical trials targeting the C1 domain of PKC include bryostatin 1 and PEP005 (ingenol 3-angelate).11−13
The structure of the complex between phorbol ester and the C1b domain of PKCδ was solved by X-ray crystallography.14 The ligand binds to the C1 domain by inserting into a hydrophilic cleft in an otherwise hydrophobic surface. The bound ligand promotes membrane insertion of the C1 domain both by capping the hydrophilic cleft and by contributing additional hydrophobicity provided by its acyl side chains. The membrane lipids, in turn, provide further stabilization of the complex. Functional consequences of the membrane insertion of the C1 domain for its protein host include translocation of the protein to membranes, bringing it into proximity with substrates or interacting partners, and conformational change. In the cases of both β2-chimaerin15 and PKCβII,16 X-ray crystallography reveals that the C1 domain in its unliganded state is involved in intramolecular interactions which are lost when the C1 domain shifts to the membrane, providing a further mechanistic component for the conformational change.
Analysis of the C1 domains in PKC family members indicated that whereas the C1 domains of the so-called classical PKC isoforms α, βI, βII, and γ and those of the so-called novel PKC isoforms δ, ε, η, and θ were DAG-responsive, those of the atypical PKC isoforms ζ and ι/λ failed to recognize DAG. Such DAG-unresponsive C1 domains have been termed “atypical”. Structural analysis indicates, moreover, that the atypical C1 domains can be further subdivided.
The atypical C1 domains of PKCζ17 and Vav118 retain a conformation of the binding cleft very similar to that of the typical C1 domains but possess residues that interfere with formation of the C1 domain–ligand–phospholipid complex (Figure 1). In the case of PKCζ/ι, we have identified four arginine residues along the rim of the the binding cleft of the C1 domain that are primarily responsible for its failure to bind phorbol ester.17 Replacement of these four arginine residues with the residues in the corresponding positions in the typical C1b domain of PKCδ confers high affinity phorbol ester binding on this atypical C1 domain, as evidenced by its membrane translocation in response to PMA.17 Conversely, replacement of the corresponding residues on the typical C1b domain of PKCδ with these arginines led to loss of phorbol ester binding activity.17 In the case of Vav1, we similarly identified five residues responsible for the loss of binding activity.19 Four residues increase the hydrophilicity along the rim of the binding cleft, disfavoring membrane insertion of the C1 domain. The fifth confers hydrophobicity in what is a hydrophilic collar in typical C1 domains that stabilizes interaction of the membrane-inserted C1 domain with lipid head groups. Once again, substitution of these five identified residues from Vav1 into the corresponding positions in the C1b domain of PKCδ abolished ligand binding activity and replacement of these five residues in the atypical C1 domain of Vav1 with the corresponding residues in the C1b domain of PKCδ generated potent binding activity (quintuple mutant of Vav1 C1 domain, Kd for PDBu = 1.05 ± 0.14 nM; PKCδ C1b domain, Kd = 0.193 ± 0.005 nM).19
Figure 1.

Amino acid sequence alignment of the C1b domain of PKCδ, the C1 domains of PKCζ, PKCι, and hVav1. The residues in PKCζ/ι largely responsible for the lack of phorbol ester binding are shown in red.17 The residues in Vav1 largely responsible for the lack of phorbol ester binding are shown in blue.18
The second class of atypical C1 domains differs conceptually from the above in that they involve deletions of residues in the loops constituting the DAG/phorbol ester binding cleft. Examples include the atypical C1 domains of Raf,20 of KSR (kinase suppressor of Ras),21 and of Nore1.22 In each instance, these deletions disrupt the structure of the binding cleft and destroy binding.
The atypical PKC isoforms ζ and ι play critical roles in cellular signaling. Unique among PKC isoforms, PKCι is the only isoform for which knockout is embryonic lethal.23 PKCι functions as an oncogene in cancer in multiple tissue types including lung and colon.24,25 PKCζ plays a critical role in NFκB signaling and in IL-4 response, and the role of PKCι in cell polarity has been highlighted.26
In PKCζ, the role of the atypical C1 domain, like that of the typical C1 domains of the novel and classical PKCs, has been suggested to be regulatory, acting as an allosteric inhibitor of enzyme activity by binding to the PIF (PDK-1 interacting fragment) pocket in the small lobe of the kinase domain, stabilizing interactions of the inhibited kinase.27 Ligand binding to the PKCζ C1 domain might therefore be stimulatory, by relieving this inhibition. A possible precedent is provided by the λ-interacting protein (LIP), which binds to the C1 domain of PKCλ/ι and activates the enzyme.28 On the other hand, a prominent feature of the atypical PKCs is their coupling to other proteins mediated through their N-terminal PB1 (Phox Bem 1) domain.29 Ligand binding to the PKCζ C1 domain might lead to functional inhibition if it caused the mislocalization or sequestration of the PKC. A possible precedent is provided by the par-4 gene product, which binds to the C1 domain and leads to inhibition.30
Diacylglycerol lactones have proven to provide a powerful scaffold for the design of novel, potent ligands targeting C1 domains.31 They combine relative structural simplicity with potencies approaching those of the phorbol esters for optimized structures. Manipulation of the side chains using combinatorial chemistry has yielded compounds with dramatic differences in functional consequences at the whole cell level, plausibly reflecting the diversity for interactions afforded by the C1 domain–ligand–membrane ternary complex for different cellular membrane subdomains.32 Since the atypical C1 domains of PKCζ/ι retain a binding cleft conformation similar to that of the typical C1 domains, we postulated that DAG lactones appropriately substituted to interact with the positively charged residues that distinguish the C1 domains of PKCζ/ι from the typical C1 domains might provide ligands for these atypical C1 domains. We describe here a first step for exploring this strategy. We prepared a series of DAG lactones incorporating polar side chains that might interact with one or more of the critical arginine residues that distinguish the PKCζ/ι C1 domains. We compared their binding to the typical C1b domain of PKCδ and to mutated versions of this C1 domain incorporating one or more of these arginine residues. Up to a 600-fold gain in relative affinity was obtained, and computer modeling predicted that ligand–arginine interactions should be occurring. Our findings provide encouragement that our approach is promising.
Results
Diacylglycerol lactones 1–13 (Table 1) were prepared and characterized as described in Scheme 1.
Table 1. Structures of DAG-lactones 1–13 including log P Values.
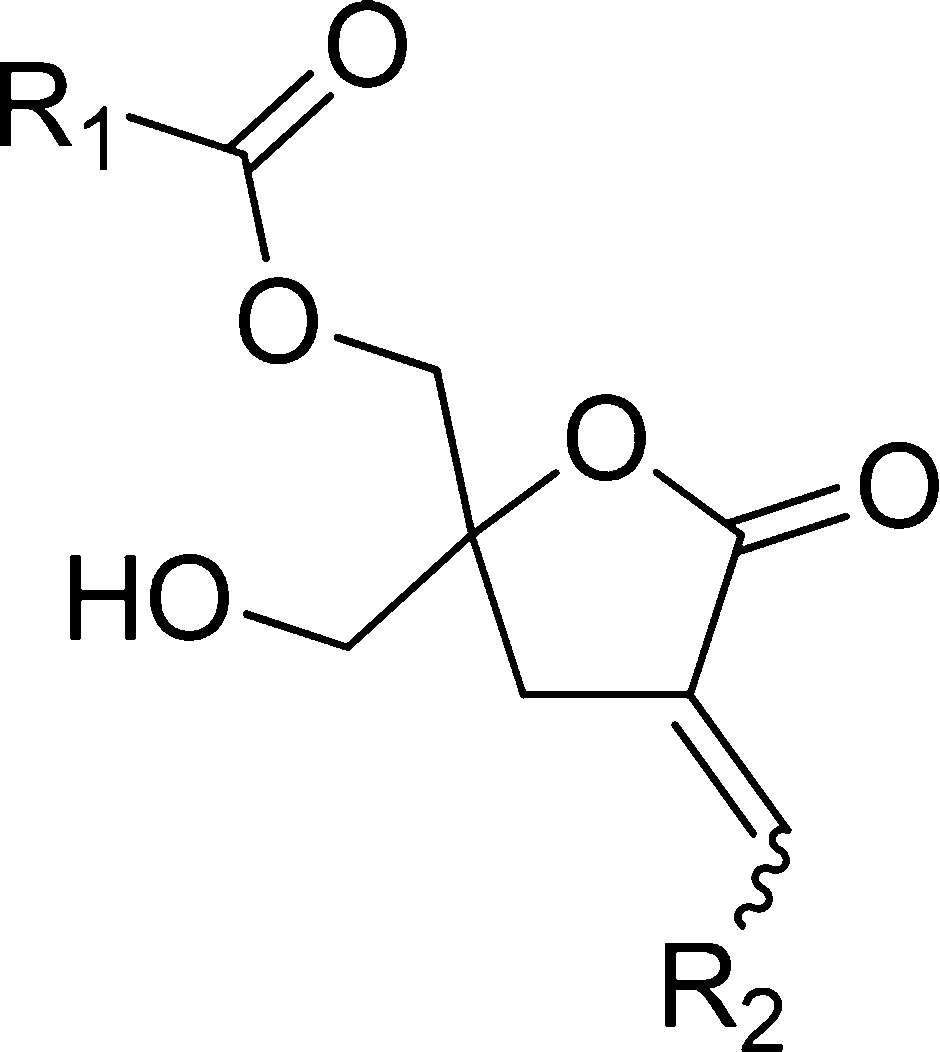
| R1 | R2 | log P a | |
|---|---|---|---|
| 1-E | [(CH3)2CH]2CHCH2- | o-C6H4OH | 3.60 |
| 2-E | [(CH3)2CH]2CHCH2- | m-C6H4OH | 3.60 |
| 3-E | [(CH3)2CH]2CHCH2- | p-C6H4OH | 3.60 |
| 4-E | [(CH3)2CH]2CHCH2- | m-C6H4CO2H | 3.54 |
| 5-E | [(CH3)2CH]2CHCH2- | p-C6H4CO2H | 3.54 |
| 5-Z | [(CH3)2CH]2CHCH2- | p-C6H4CO2H | 3.54 |
| 6-E | (CH3)3C- | m-C6H4CO2H | 2.58 |
| 7-E | (CH3)3C- | p-C6H4CO2H | 2.58 |
| 8-E | [(CH3)2CHCH2]2CHCH2- | m-C6H4CO2H | 4.38 |
| 9-E | [(CH3)2CHCH2]2CHCH2- | p-C6H4CO2H | 4.38 |
| 10-E | [(CH3)2CHCH2]2CHCH2- | o-C6H4CH[CO2C(CH3)3]2 | 6.24 |
| 11-E | [(CH3)2CHCH2]2CHCH2- | m-C6H4CH[CO2C(CH3)3]2 | 6.24 |
| 12-E | [(CH3)2CHCH2]2CHCH2- | p-C6H4CH[CO2C(CH3)3]2 | 6.24 |
| 13-E | (HOCH2)2(CH3)C- | -CH2CH[CH2CH(CH3)2]2 | 3.20 |
The log P values were calculated using ChemBioDraw Ultra, version 12.0.2.
Scheme 1.
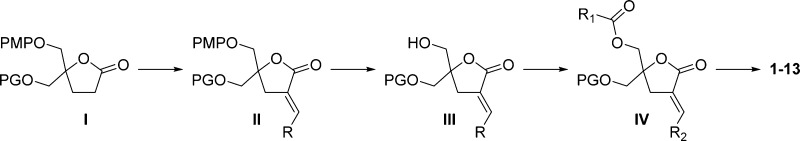
The syntheses of DAG-lactone analogues 1–13 were completed utilizing a well-established methodology developed in our laboratory.33−35 From commercially available starting reagents the doubly protected lactone I [PMP = p-methoxyphenyl and PG = either benzyl (Bn) or tert-butyldiphenylsilyl (TBDPS)] was synthesized and reacted with different aldehydes (RCHO) to form the corresponding aldol intermediates, which then underwent elimination of the resultant β-hydroxylactone to give the corresponding olefin (II). In some cases, this generated mixtures of E- and Z-isomers. Consistent with previously synthesized DAG-lactones, the E/Z geometry around the double bond was assigned by 1H NMR: the vinyl proton for the E-isomers displayed a characteristic multiplet that was farther downfield than that of the corresponding Z-isomers. After separation of the geometric E/Z-isomers and removal of the PMP group with ceric ammonium nitrate (CAN), the compounds were individually converted to the corresponding DAG-lactones with different R′ acyl groups by conventional methods. The target compounds (1–13) were obtained by deprotection of intermediate IV. When the protecting group was the benzyl ether (PG = Bn), debenzylation was performed with BCl3, (CH2Cl2, −78 °C), whereas in the case of the silyl ether (PG = TBDPS), deprotection was accomplished with tetra-n-butylammonium fluoride (TBAF) under standard conditions. The decision to prepare racemic DAG-lactones was mainly practical, as these compounds are easier to synthesize and we have found the differences in binding affinity between the enantiomers to be minimal.36
Our initial objective was to examine the influence of various hydrophilic substitutions of the DAG-lactone acyl side chains on binding affinities to variants of the PKCδ C1b domain substituted with one of the four critical arginine residues found in PKCζ/ι. All of the single arginine substitutions led to only a modest decrease in PDBu binding (Table 2, first row), permitting the measurement of DAG-lactone binding affinities by competition for [3H]PDBu binding. Table 3 shows the ratio of the Kd or Ki values for mutant binding to wild-type binding, giving an indication of the relative change in binding affinity due to the arginine mutation(s). Thus, in the first row of Table 3 it can be seen that (as described previously17) binding of [3H]PDBu to the N7R and P11R mutants was about 5-fold weaker than to the wild-type δC1B domain, binding to L20R was about 35-fold weaker, and binding to S10R was essentially the same. A value of less than 1 in Table 3, as seen for some of the DAG-lactones, indicates that binding to the mutant was stronger than to the wild-type domain.
Table 2. Binding Affinities of Ligands to the Wild-Type C1b Domain of PKCδ and to Mutated Versions Incorporating One or More of the Residues of PKCζ/ι Responsible for Its Loss of Ligand Binding Activity.
|
Ki, nMa |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | Kd, nM,a wtδC1B | N7R | S10R | P11R | L20R | N7R/P11R | N7R/L20R | N7R/S10R/P11R | N7R/S10R/L20R |
| PDBuc | 0.23 | 1.18 ± 0.07 | 0.32 ± 0.10 | 1.19 ± 0.19 | 8.0 ± 1.8 | 33.8 ± 1.8 | 480 ± 44 | 19.6 ± 2.3 | 1360 ± 290 |
| 1-E | 2.95 ± 0.17 | 3.29 ± 0.37 | 2.33 ± 0.34 | 10.4 ± 0.76 | 14.0 ± 0.99 | 37 ± 10 | 239 ± 39 | 63 ± 19 | 661 ± 35 |
| 2-E | 2.46 ± 0.67 | 2.48 ± 0.44 | 2.08 ± 0.27 | 2.32 ± 0.65 | 19.4 ± 8.0 | NTb | NTb | NTb | NTb |
| 3-E | 2.95 ± 0.29 | 1.99 ± 0.18 | 1.18 ± 0.11 | 1.48 + 0.35 | 9.4 ± 2.1 | NTb | NTb | NTb | NTb |
| 4-E | 2102 ± 531 | 640 ± 110 | 840 ± 280 | 421 ± 44 | 2820 ± 500 | 5190 ± 930 | 45900 ± 9200 | NTb | NTb |
| 5-E | 560 ± 99 | 787 ± 79 | 813 ± 98 | 710 ± 150 | 6900 ± 1600 | 3800 ± 440 | 117000 ± 63000 | NTb | NTb |
| 5-Z | 865 ± 99 | 473 ± 75 | 496 ± 60 | 940 ± 150 | 2810 ± 470 | 2810 ± 240 | 51900 ± 1900 | NTb | NTb |
| 6-E | 1206 ± 208 | 1737 ± 99 | 1765 ± 440 | 3360 ± 380 | 5500 ± 330 | 15970 ± 470 | 105000 ± 31000 | NTb | NTb |
| 7-E | 1070 ± 87 | 880 ± 110 | 667 ± 51 | 998 ± 19 | 6140 ± 490 | 3700 ± 1400 | 83400 ± 5200 | NTb | NTb |
| 8-E | 300 ± 45 | 281 ± 25 | 468+ 96 | 100 ± 5 | 1600 ± 120 | NTb | NTb | NTb | NTb |
| 9-E | 96 ± 18 | 120 ± 12 | 94 ± 9 | 120 ± 12 | 739 ± 91 | NTb | NTb | NTb | NTb |
| 10-E | 1.0 ± 0.02 | 0.9 ± 0.1 | 1.5 ± 0.12 | 1.7 ± 0.6 | 17.3 ± 0.4 | NTb | NTb | NTb | NTb |
| 11-E | 0.5 ± 0.04 | 0.6 ± 0.09 | 0.6 ± 0.06 | 1.0 ± 0.25 | 8.8 ± 0.5 | NTb | NTb | NTb | NTb |
| 12-E | 0.6 ± 0.08 | 0.7 ± 0.08 | 0.6 ± 0.12 | 1.2 ± 0.4 | 10.5 ± 0.5 | NTb | NTb | NTb | NTb |
| 13-E | 777 ± 59 | 806 ± 20 | 1120 ± 140 | 2030 ± 140 | 810 ± 120 | 23800 ± 2500 | 7640 ± 510 | 47000 ± 5300 | 7830 ± 890 |
Values (in nanomolar concentrations) represent the mean ± SEM of at least triplicate determinations.
NT, not tested.
Values for the binding of [3H]PDBu to the various mutants are from Pu et al.17
Table 3. Relative Binding Affinities of Ligands to the Wild-Type C1b Domain of PKCδ and to Mutated Versions Incorporating One or More of the Residues of PKCζ/ι Responsible for Its Loss of Ligand Binding Activity.
|
Kd or Ki relative
to that for PDBu binding to the wtδC1ba |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | Kd, nM,a wtδC1B | N7R | S10R | P11R | L20R | N7R/P11R | N7R/L20R | N7R/S10R/P11R | N7R/S10R/L20R |
| PDBu | (0.23) | 5.13 | 1.39 | 5.17 | 34.7 | 147 | 2090 | 85.2 | 5900 |
| 1-E | (2.95) | 1.12 | 0.79 | 3.53 | 4.75 | 12.5 | 81.0 | 21.3 | 224 |
| 2-E | (2.46) | 1.01 | 0.85 | 0.94 | 7.87 | NTb | NTb | NTb | NTb |
| 3-E | (2.95) | 0.67 | 0.40 | 0.50 | 3.20 | NTb | NTb | NTb | NTb |
| 4-E | (2102) | 0.30 | 0.40 | 0.20 | 1.34 | 2.47 | 21.8 | NTb | NTb |
| 5-E | (560) | 1.41 | 1.45 | 1.27 | 12.4 | 6.79 | 209 | NTb | NTb |
| 5-Z | (865) | 0.55 | 0.57 | 1.09 | 3.25 | 3.25 | 60.0 | NTb | NTb |
| 6-E | (1206) | 1.44 | 1.46 | 2.79 | 4.57 | 13.2 | 87.0 | NTb | NTb |
| 7-E | (1070) | 0.82 | 0.62 | 0.93 | 5.74 | 3.49 | 78.0 | NTb | NTb |
| 8-E | (300) | 0.94 | 1.56 | 0.33 | 5.33 | NTb | NTb | NTb | NTb |
| 9-E | (96) | 1.25 | 0.98 | 1.25 | 7.70 | NTb | NTb | NTb | NTb |
| 10-E | (1) | 0.90 | 1.50 | 1.70 | 17.3 | NTb | NTb | NTb | NTb |
| 11-E | (0.5) | 1.20 | 1.20 | 2.00 | 17.6 | NTb | NTb | NTb | NTb |
| 12-E | (0.6) | 1.17 | 1.00 | 2.00 | 17.5 | NTb | NTb | NTb | NTb |
| 13-E | (777) | 1.04 | 1.44 | 2.61 | 1.04 | 30.7 | 9.83 | 60.5 | 10.1 |
Data are derived from Table 2.
NT, not tested.
The hydrophilic DAG-lactones fell into two classes. The R2 phenol (1–3) and aryl diester (10–12) derivatives retained good potency on the wild-type C1b domain. They also showed good retention of activity on the single variants N7R, S10R, and P11R and in some cases even improved potency over the wild-type domain (Table 2). The R2 phenol compounds (1–3) also averaged only a 5-fold decrease in binding affinity to L20R versus the wild-type C1 domain, compared to a 35-fold reduction in binding with [3H]PDBu. Compound 3 showed as much as a 10-fold improvement in selectivity for L20R, the mutant with the greatest effect on [3H]PDBu binding (Table 3). There was little improvement in the selectivity of the aryl diester class (10–12) for L20R, and this series was not further characterized.
The second class of DAG-lactones, the R2 benzoic acid derivatives 4–9 and the R1 methylpropane diol derivative 13, showed large reductions in affinity for the wild-type C1b domain, ranging from 96- to 2100-fold (Table 2). In the case of the benzoic acid derivatives, this was no doubt due in large part to the formal charge on these molecules and the solvation energy penalty for membrane interaction. Accordingly, the reduction in affinity was generally inversely proportional to the size and hydrophobicity of the R1 side chain; i.e., compounds 8 and 9 with the largest branched acyl groups had the highest affinities. This correlation of Ki with log P is believed to be a function of nonspecific membrane binding.37 Interestingly, however, the activity of this second class of DAG-lactones toward the single mutants did not decrease substantially relative to the wild-type domain (Table 3). Particularly strikingly, compounds 4 and 13 both bound almost as strongly to the L20R mutant as to the wild-type domain, in contrast to the 35-fold decrease in potency of [3H]PDBu for L20R. Compounds 4 and 8 also bound the P11R mutant 5 times and 3 times more strongly, respectively, than they bound the wild-type domain.
Discussion and Conclusions
We had observed previously that the decrease in affinity of [3H]PDBu for variant C1b domains incorporating more than one arginine substitution was greater than the product of the decreases induced by the individual substitutions.17 We next examined how the hydrophilic substituted DAG-lactones interacted with the N7R/P11R and N7R/L20R double mutants as well as with the triple mutants derived from these double mutants by addition of the S10R. As previously described, the S10R mutation had only a modest effect in combination, consistent with its modest effect when alone.17 For the N7R/L20R double C1b domain mutant, PDBu bound with a 2090-fold decrease in affinity. In dramatic contrast, 13 showed only a 9.8-fold loss in affinity, corresponding to a 212-fold increase in selectivity. The triple mutant N7R/S10R/L20R showed a further modest decrease in affinity for PDBu, with a total reduction in affinity of 5900-fold; compound 13 displayed no further loss of affinity for an increase in selectivity relative to PDBu of 585-fold. For the N7R/P11R double mutant, which displayed a 147-fold loss in PDBu binding affinity, compound 4 showed only a 2.5-fold loss in affinity, corresponding to a 59-fold increase in selectivity, whereas compound 13 was much less tolerant, with only a 4.8-fold increase in selectivity.
Modeling provides some insight into the interactions of the compounds with the various mutants. We docked the DAG-lactones 4 and 13 into modeled structures of the C1 domain mutants and explored the conformational space available to the arginine residues and hydrophilic R1 and R2 lactone side chains to identify low-energy salt-bridged or hydrogen-bonded complex structures.
DAG-lactone 4, a meta-substituted R2 benzoate, is shown in Figure 2. As seen in Figure 2A and in Table 3, the meta-isomer seems to be favorable for strong interactions with arginine at position 11. The carboxylate group is positioned to form strong hydrogen bonds with one of the Nη atoms and the Nε atom in the arginine side chain (R11), as well as with the backbone amide nitrogen. In Figure 2A compound 4 is shown simultaneously binding the arginine at position 7 (R7). We found that arginine at this position can bind any of the DAG-lactones, regardless of their R1 and R2 side chains, through interactions with the carbonyl oxygen of the ester adjacent to R1 along with the protein backbone carbonyl at either tyrosine Y8 (as shown) or L24. Figure 2B shows the docked complex of compound 4 with the L20R mutant. Here the charged carboxylate group on the ligand and the guanidinium group on the arginine (R20) are pulled down from the binding site, toward the location in the membrane-bound complex of the more hydrated interfacial region. The high selectivity of compound 4 to the L20R mutant may be an example of the previously observed nonadditivity of mutant ligand-binding behavior,17 as neither the other meta-benzoate compounds (6 and 8) nor the other compounds with the same R1 acyl group (1–3, 5) are as selective.
Figure 2.

Modeled complexes of DAG-lactone 4 with the N7R/P11R (A) and L20R (B) mutant C1 domains. The arginine residues are colored magenta. The ligand is colored green, and hydrogen bond interactions are indicated with dashed orange lines.
DAG-lactone 13 differs from the other compounds under discussion here in that its polar side chain is in the R1 position rather than the R2. We have shown previously that there is a strong solvation penalty for placing polar side chains at R1, because in binding the C1 domain, this polar group would be driven into the hydrophobic part of the membrane. Polar side chains at R2 are located in the membrane interfacial region and thus can remain hydrated.33 However, DAG-lactones are capable of adopting an alternative binding mode, albeit with somewhat weaker hydrogen bonding, in which the positions of the side chains are essentially flipped.38 This suggests that DAG-lactone 13 may be binding the C1 domain arginine mutants with a mixture of the two binding modes, one with worse solvation but better hydrogen bonding and one vice versa. Figure 3A shows the docked structure of compound 13, in the “standard” DAG-lactone binding mode, interacting with arginine at position 7 (R7). The arginine hydrogen-bonds to the hydroxyl groups in the diol but also, as with compound 4, to the ester carbonyl and the protein backbone. Figure 3B shows compound 13 in its alternative “reversed” orientation, with the diol side chain interacting with arginine at position 20 (R20). With the DAG-lactone in this orientation, the arginine can also hydrogen-bond weakly to the ether oxygen in the lactone ring. Compound 13 does not form particularly strong interactions with arginine at position 11 in either of its binding modes (not shown), which may explain its reduced selectivity for the P11R and the N7R/P11R mutants. However, this compound could bind the N7R/L20R double mutant with a mixture of its two binding modes which would add a favorable amount of configurational entropy to the binding free energy.
Figure 3.

Modeled complexes of DAG-lactone 13 with the N7R (A) and L20R (B) mutant C1 domains. The arginine residues are colored magenta. The ligand is colored green, and hydrogen bond interactions are indicated with dashed orange lines.
The core conclusion is that indeed marked improvements in selectivity are achievable for DAG-lactones incorporating moieties that may interact with arginines corresponding to those found in the atypical C1 domains of PKCζ/ι. Among compounds 1–3, the ortho derivative was clearly the least selective; for the other pairs of meta versus para there were in general relatively small differences, depending on the specific pair of compounds. Although the series of compounds examined is not extensive, it is noteworthy that compounds 4 and 13 showed different relative recognition for N7R/P11R versus N7R/L20R. The high selectivity of compound 13 in particular suggests that further exploration of this class of DAG-lactones may be of interest and that the exploitation of the DAG-lactones’ ability to bind in multiple binding orientations could be used to develop compounds targeted to multiple mutations simultaneously.
Experimental Section
General Procedures
All chemical reagents were commercially available. Melting points were determined on an MPA 100 OptiMelt automated melting point system (Stanford Research Systems, USA) or a MelTemp II apparatus, (Laboratory Devices, USA) and are uncorrected. Column chromatography was performed on a Teledyne Isco CombiFlash Companion instrument under gradient elution conditions with RediSep disposable flash columns. Analytical TLC was performed on Analtech Uniplates silica gel GF. All of the intermediates described in this section were oils purified by column chromatography and confirmed by TLC to be a single spot. These compounds were used directly for the ensuing step. 1H and 13C NMR spectra were recorded on a Varian Unity Inova instrument at 400 and 100 MHz, respectively. Spectra are referenced to the solvent in which they were run (7.24 ppm for CDCl3). Positive-ion fast atom bombardment mass spectra (FABMS) were obtained on a VG 7070E-HF double-focusing mass spectrometer operated at an accelerating voltage of 6 kV under the control of a MASPEC-II data system for Windows (Mass Spectrometry Services, Ltd.). Either glycerol or 3-nitrobenzyl alcohol was used as the sample matrix, and ionization was effected by a beam of xenon atoms generated in a saddle-field ion gun at 8.0 ± 0.5 kV. Nominal mass spectra were obtained at a resolution of 1200, and matrix-derived ions were background-subtracted during data system processing. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA.
Aldol Condensation Followed by Olefination (II)
Under argon, a solution of I (prepared as described previously34,35) (1 equiv) in THF (5 mL/mmol) at −78 °C was treated dropwise with [(CH3)3Si]2N-Li (LiHMDS, 1.5 equiv) and stirred at the same temperature for 1 h. A solution of ArCHO (1.5 equiv dissolved in THF) was then added dropwise, and the mixture was stirred at −78 °C until determined complete by TLC analysis. The reaction mixture was then quenched with a saturated aqueous solution of NH4Cl and allowed to warm to room temperature. The layers were separated, and the aqueous layer was extracted with Et2O (3×). The combined organics were washed with H2O (2×) and brine (1×), dried (MgSO4), and concentrated in vacuo to give the condensation product as a mixture of diastereomers, which was then directly dissolved in a solution of Et3N (4 equiv) in CH2Cl2 (10 mL/mmol), cooled to 0 °C, treated dropwise with CH3SO2Cl (MsCl, 2 equiv), and then stirred at room temperature for 1 h. The reaction mixture was then cooled again to 0 °C and treated dropwise with 1,8-diazabicyclo[5.4.0]non-5-ene (DBU, 5 equiv). When the addition of DBU was completed, the reaction mixture was allowed to reach room temperature overnight. The volatiles were removed in vacuo, and the residue was treated with EtOAc followed by 1 N HCl. The layers were separated, and the aqueous layer was extracted with EtOAc (1×). The combined organics were washed with H2O (2×) and brine (1×), dried (MgSO4), and concentrated in vacuo. Purification by silica gel flash column chromatography (gradient EtOAc/hexanes) gave II in 52–91% yield.
PMP Deprotection (III)
Ceric ammonium nitrate (CAN, 3 equiv) was added to a stirring solution of II (1 equiv) in acetonitrile (8 mL/mmol) and water (2 mL/mmol) at 0 °C. The reaction was monitored by TLC, quenched upon completion with a saturated aqueous Na2S2O3 solution, and warmed to room temperature. The resulting aqueous solution was extracted with CH2Cl2 (2×), dried (MgSO4), and concentrated in vacuo. Purification by silica gel flash column chromatography gave III in 29–97% yield.
Acylation (IV)
A solution of III (1 equiv) in CH2Cl2 (12 mL/mmol) was treated with Et3N (3 equiv) and dimethylaminopyridine (DMAP, 2.5 equiv) and reacted with the corresponding acid chloride (RCOCl, 1.2–1.5 equiv). The reaction was stirred at room temperature and monitored by TLC. Upon completion, the reaction was concentrated in vacuo and purified by silica gel flash column chromatography to give IV in 42–100% yield.
Benzyl Deprotection (1–9, 13) (Table 4)
Table 4. Protecting Groups for Compound IV in Scheme 1.
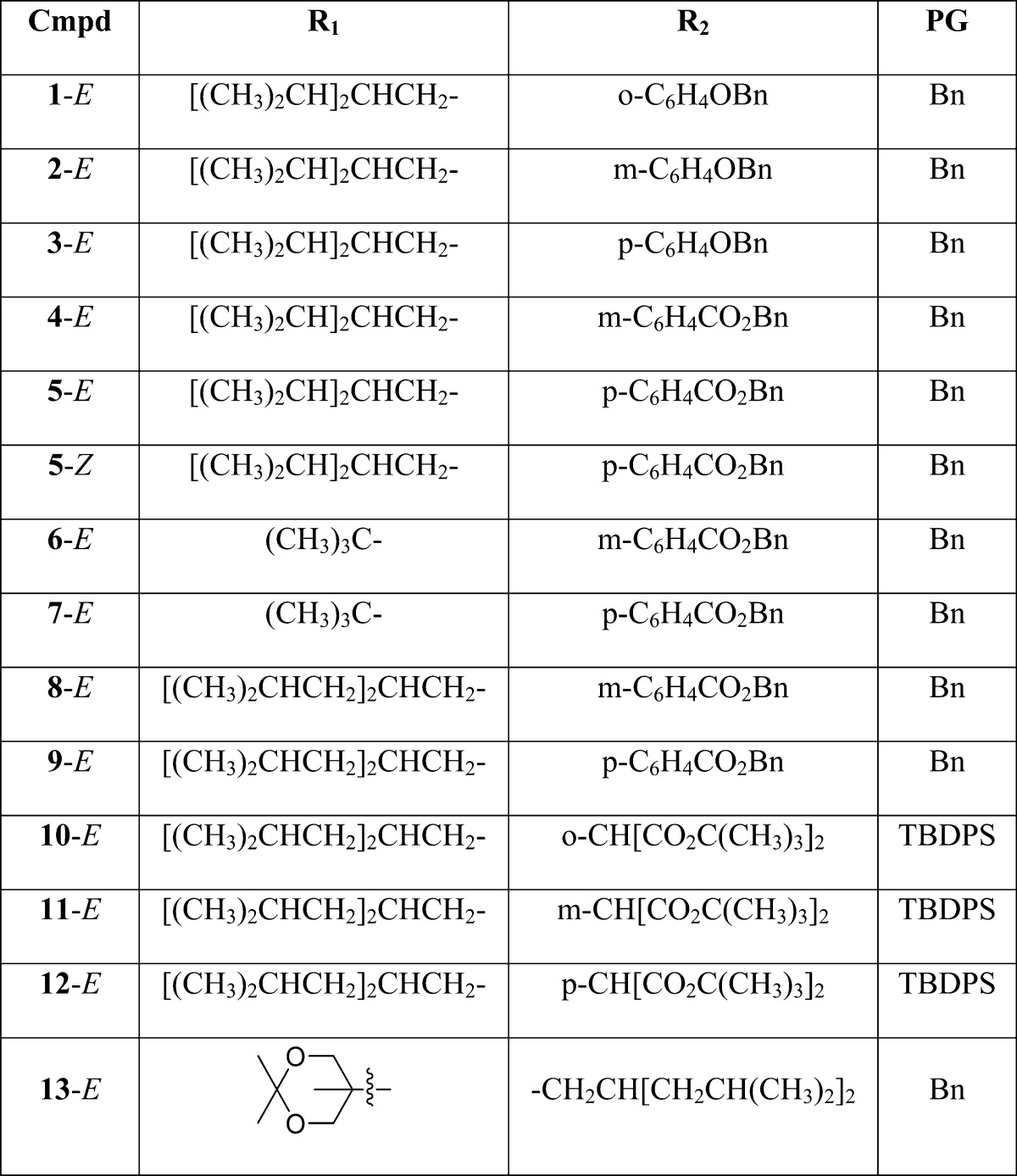
BCl3 (3 equiv) was added slowly to a stirring solution of IV (1 equiv) in CH2Cl2 (20 mL/mmol) at −78 °C. The reaction was monitored by TLC and quenched upon completion by MeOH. Immediate concentration in vacuo and purification by silica gel column chromatography (gradient elution) gave 1–9 (19–82% yield) and 13 (88% yield).
TBDPS Deprotection (10–12)
TBAF (2 equiv) was added slowly to a stirring solution of IV (1 equiv) in THF (20 mL/mmol) at room temperature. The reaction was monitored by TLC and directly concentrated in vacuo upon completion. Purification by silica gel column chromatography (gradient elution) gave 10–12 in 27–53% yield.
1-E: R1 = [(CH3)2CH]2CHCH2-; R2 = o-C6H4OH
This compound has been previously published.39
2-E: R1 = [(CH3)2CH]2CHCH2-; R2 = m-C6H4OH
This compound has been previously published.39
3-E: R1 = [(CH3)2CH]2CHCH2-; R2 = p-C6H4OH
This compound has been previously published.39
4-E: R1 = [(CH3)2CH]2CHCH2-; R2 = m-C6H4CO2H
1H NMR (400 MHz, CDCl3) δ 0.73, 0.76, 0.82, 0.83 (4 × d, J = 6.8 Hz, 12 H, CH2CH(CH(CH3)2)2), 1.53 (pentet, J ≈ 5.8 Hz, 1 H, CH2CH(CH(CH3)2)2), 1.69 (septuplet, J ≈ 6.2 Hz, 2 H, CH2CH(CH(CH3)2)2), 2.18 (dd, J = 5.8, 1.0 Hz, 2 H, CH2CH(CH(CH3)2)2), 3.08 (dd, J = 17.9, 2.8 Hz, 1 H, H-3a), 3.33 (dd, J = 17.9, 3.0 Hz, 1 H, H-3b), 3.81 (AB q, J = 12.2 Hz, 2 H, CCH2OH), 4.29 (AB q, J = 12.0 Hz, 2 H, CCH2OC(O)CH2CH(CH(CH3)2)2), 7.55 (t, J = 7.8 Hz, 1H, Ar), 7.60 (t, J = 2.8 Hz, 1H, C=CHC6H4COOH), 7.71 (d, J = 7.9 Hz, 1H, Ar), 8.11 (dm, 1 H, J = 7.8 Hz, Ar), 8.22 (br s, 1 H, Ar); 13C NMR (100 MHz, CDCl3) δ 174.83, 171.14, 170.55, 135.87, 135.30, 134.94, 131.49, 131.22, 130.28, 129.44, 126.12, 83.93, 65.65, 64.85, 47.08, 32.94, 32.24, 29.47, 21.42, 21.40, 18.84, 18.77; FAB-MS (m/z, relative intensity) 417 (M – H, 100). Anal. (C23H30O7) calcd C 66.01, H 7.73; found C 65.85, H 7.22.
5-E: R1 = [(CH3)2CH]2CHCH2-; R2 = p-C6H4CO2H
1H NMR (400 MHz, CDCl3) δ 0.74, 0.77, 0.84, 0.85 (4 × d, J = 6.8 Hz, 12 H, CH2CH(CH(CH3)2)2, 1.54 (pentet, J ≈ 5.8 Hz, 1 H, CH2CH(CH(CH3)2)2), 1.70 (septuplet, J ≈ 6.4 Hz, 2 H, CH2CH(CH(CH3)2)2), 2.18 (dd, J = 5.8, 1.3 Hz, 2 H, CH2CH(CH(CH3)2)2), 3.07 (dd, J = 18.0, 2.8 Hz, 1 H, H-3a), 3.29 (dd, J = 18.0, 2.8 Hz, 1 H, H-3b), 3.79 (AB q, J = 12.2 Hz, 2 H, CCH2OH), 4.29 (AB q, J = 12.0 Hz, 2 H, CCH2OC(O)CH2CH(CH(CH3)2)2), 7.59 (d, J = 8.3 Hz, 2 H, Ar), 7.60 (t, J = 2.8 (t, J = 2.8 Hz, 1H, C=CHC6H4COOH), 8.15 (d, J = 8.3 Hz, 2 H, Ar); 13C NMR (100 MHz, CD3OD) δ 175.74, 172.92, 164.91, 140.12, 135.56, 131.21, 131.10, 129.61, 113.95, 85.79, 67.14, 65.19, 33.80, 33.25, 30.58, 21.68, 19.07, 19.00; FAB-MS (m/z, relative intensity) 417 (M – H, 100). Anal. (C23H30O7·0.1H2O) calcd C 65.73, H 7.24; found C 65.56, H 7.28.
5-Z: R1 = [(CH3)2CH]2CHCH2-; R2 = p-C6H4CO2H
1H NMR (400 MHz, CDCl3) δ 0.76, 0.77, 0.86, 0.87 (4 × d, J = 6.7 Hz, 12 H, CH2CH(CH(CH3)2)2, 1.53 (pentet, J ≈ 5.7 Hz, 1 H, CH2CH(CH(CH3)2)2), 1.70 (septuplet, J ≈ 6.5 Hz, 2 H, CH2CH(CH(CH3)2)2), 2.12 (d, J = 5.7 Hz, 2 H, CH2CH(CH(CH3)2)2), 3.68 (s, 2 H, H-3a,b), 3.79 (AB s, 2 H, CCH2OH), 4.31 (AB q, J = 12 Hz, 2 H, CCH2OC(O)CH2CH(CH(CH3)2)2), 6.91 (s, 1 H, C=CHC6H4COOH), 7.33 (d, J = 8.0 Hz, 2 H, Ar), 8.02 (d, J = 8.0 Hz, 2 H, Ar); 13C NMR (100 MHz, CDCl3) δ 174.70, 172.20, 171.02, 148.06, 143.17, 135.77, 130.85, 129.15, 87.93, 63.60, 63.29, 47.02, 32.81, 32.00, 29.49, 29.45, 21.44, 18.85, 18.77; FAB-MS (m/z, relative intensity) 417 (M – H, 99), 229 (100). Anal. (C23H30O7) calcd C 66.01, H 7.73; found C 65.84, H 7.19.
6-E: R1 = (CH3)3C-; R2 = m-C6H4CO2H
1H NMR (400 MHz, CD3OD) δ 1.06 (s, 9 H, C(CH3)3), 3.13 (dd, J = 18.0, 2.8 Hz, 1 H, H-3a), 2.14 (dd, J = 18.0, 3.0 Hz, 1 H, H-3b), 3.73 (AB q, J = 12.1 Hz, 2 H, CCH2OH), 4.26 (AB q, J = 11.9 Hz, 2 H, CCH2OC(O)C(CH3)3), 7.49 (t, J = 2.9 Hz, 1H, C=CHC6H4COOH), 7.55 (t, J = 7.8 Hz, 1 H, Ar), 7.76 (d, J = 7.8 Hz, 1 H, Ar), 8.03 (d, J = 7.8 Hz, 1 H, Ar), 8.18 (s, 1 H, Ar); 13C NMR (100 MHz, CD3OD) δ 179.17, 173.10, 168.98, 136.23, 135.86, 135.78, 135.41, 132.76, 131.85, 131.76, 130.28, 128.48, 85.92, 67.23, 65.08, 39.89, 33.07, 27.37, 27.33; FAB-MS (m/z, relative intensity) 361 (M – H, 100). Anal. (C19H22O7·0.2H2O) calcd C 62.35, H 6.17; found C 62.35, H 6.12.
7-E: R1 = (CH3)3C-; R2 = p-C6H4CO2H
Mp = 220 °C; 1H NMR (400 MHz, CD3OD) δ 1.12 (s, 9 H, C(CH3)3), 3.20 (dd, J = 18.3, 2.9 Hz, 1 H, H-3a), 3.36–3.27 (overlapped with solvent, 1 H, H-3b), 3.78 (AB q, J = 12.1 Hz, 2 H, CCH2OH), 4.31(AB q, J = 11.9 Hz, 2 H, CCH2OC(O)C(CH3)3), 7.56 (t, J = 3.0 Hz, 1H, C=CHC6H4COOH), 7.71 (d, J = 8.4 Hz, 2H, Ar), 8.13 (d, J = 8.4 Hz, 2H, Ar); FAB-MS (m/z, relative intensity) 361 (M – H, 100). Anal. (C19H22O7·0.1H2O) calcd C 62.35, H 6.17; found C 62.26, H 6.25.
8-E: R1 = [(CH3)2CHCH2]2CHCH2-; R2 = m-C6H4CO2H
1H NMR (400 MHz, CDCl3) δ 0.78–0.82 (m, 12 H, CH2CH[CH2CH(CH3)2]2), 0.97–1.15 (m, 4 H, CH2CH[CH2CH(CH3)2]2), 1.54 (septuplet, 2 H, J ≈ 6.4 Hz, CH2CH[CH2CH(CH3)2]2), 1.88 (pentet, J ≈ 6.7 Hz, 1 H, CH2CH[CH2CH(CH3)2]2), 2.21 (d, J = 6.4 Hz, 2 H, CH2CH[CH2CH(CH3)2]2), 3.07 (dd, J = 17.8, 2.4 Hz, 1 H, H-3a), 3.35 (dd, J = 17.9, 2.4 Hz, 1H, H-3b), 3.81 (AB q, J = 12.2 Hz, 2 H, CCH2OH), 4.29 (AB q, J = 12.0 Hz, 2 H, CCH2OC(O)CH2CH(CH(CH3)2)2), 7.52 (t, J = 7.8 Hz, 1 H, Ar), 7.57 (br s, 1 H, C=CHC6H4COOH), 7.69 (d, J = 7.8 Hz, 1 H, Ar), 8.08 (d, J = 7.8 Hz, 1 H, Ar), 8.20 (s, 1 H, Ar); 13C NMR (100 MHz, CDCl3) δ 173.40, 171.27, 170.62, 135.86, 135.18, 134.87, 131.45, 131.28, 130.35, 129.37, 126.06, 83.95, 65.45, 64.77, 44.11, 39.42, 32.16, 30.74, 25.22, 22.94, 22.66, 22.63; FAB-MS (m/z, relative intensity) 445 (M – H, 100). Anal. (C25H34O7·0.3H2O) calcd C 66.44, H 7.72; found C 66.31, H 7.55.
9-E: R1 = [(CH3)2CHCH2]2CHCH2-; R2 = p-C6H4CO2H
1H NMR (400 MHz, CD3OD) δ 0.78–0.83 (m, 12 H, CH2CH[CH2CH(CH3)2]2), 1.01–1.10 (m, 4 H, CH2CH[CH2CH(CH3)2]2)), 1.55 (septuplet of doublets, J ≈ 6.7, 2.7 Hz, 2H, CH2CH[CH2CH(CH3)2]2), 1.88 (pentet, J ≈ 6.6 Hz, 1 H, CH2CH[CH2CH(CH3)2]2), 2.18 (dd, J = 6.5, 0.9 Hz, 2 H, CH2CH[CH2CH(CH3)2]2), 3.24 (AB quartet of doublets, J = 18.3, 2.8 Hz, 2 H, H-3a,b), 3.75 (AB q, J = 12.0 Hz, 2 H, CCH2OH), 4.31 (d, J = 2.8 Hz, 2 H, CCH2OC(O)CH2CH(CH(CH3)2)2), 7.53 (t, J = 2.8 Hz, 1H, C=CHC6H4COOH), 7.69 (d, J = 8.4 Hz, 2 H, Ar), 8.10 (d, J = 8.4 Hz, 2 H, Ar); 13C NMR (100 MHz, CD3OD) δ 172.85, 171.38, 167.57, 138.64, 134.13, 129.77, 129.64, 128.04, 84.21, 65.40, 63.75, 43.80, 38.74, 31.76, 30.44, 24.81, 21.84, 21.47, 21.45; FAB-MS (m/z, relative intensity) 445 (M – H, 100). Anal. (C25H34O7·0.2H2O) calcd C 66.71, H 7.70; found C 66.64, H 7.61
10-E: R1 = [(CH3)2CHCH2]2CHCH2-; R2 = o-C6H4CH[CO2C(CH3)3]2H
1H NMR (400 MHz, CDCl3) δ 0.83–0.86 (m, 12 H, CH2CH[CH2CH(CH3)2]2), 0.10–1.16 (m, 4 H, CH2CH[CH2CH(CH3)2]2), 1.46 and 1.47 (s, 18 H, CH[CO2C(CH3)3]2), 1.59 (septuplet, J ≈ 6 Hz, 2 H, CH2CH[CH2CH(CH3)2]2), 1.93 (pentet, J ≈ 6.7 Hz, 1 H, CH2CH[CH2CH(CH3)2]2), 2.24 (d, J = 6.5 Hz, 2 H, CH2CH[CH2CH(CH3)2]2), 2.89 (dd, J = 17.7, 2.8 Hz, 1 H, H-3a), 3.04 (dd, J = 17.7, 2.9 Hz, 1 H, H-3b), 3.67 (AB q, J = 12.1 Hz, 2 H, CCH2OH), 4.22 (AB q, J = 11.9 Hz, 2 H, CCH2OC(O)CH2CH(CH(CH3)2)2), 4.75 (s, 1 H, CH[CO2C(CH3)3]2), 7.37 (m, 2 H, Ar), 7.55 (d, J = 7.5 Hz, 2 H, Ar), 7.74 (t, J = 2.8 Hz, 1 H, C=CHC6H4CH[CO2C(CH3)3]2); 13C NMR (100 MHz, CDCl3) δ 173.17, 170.13, 167.15, 166.91, 134.72, 134.13, 133.30, 129.80, 129.52, 128.15, 128.02, 127.70, 83.26, 82.67, 82.53, 64.93, 64.40, 56.42, 44.00, 43.97, 39.20, 31.61, 30.58, 27.83, 27.81, 25.09, 25.07, 22.83, 22.78, 22.56, 22.52; FAB-MS (m/z, relative intensity) 655 (M + K+, 25), 57 (100). Anal. (C35H52O9) calcd C 68.16, H 8.50; found C 67.96, H 8.53.
11-E: R1 = [(CH3)2CHCH2]2CHCH2-; R2 = m-C6H4CH[CO2C(CH3)3]2H
1H NMR (400 MHz, CDCl3) δ 0.78, 0.80, 0.82 (overlapping doublets, J = 6.3 Hz, 12 H, CH2CH[CH2CH(CH3)2]2), 0.98–1.13 (m, 4 H, 2 × CH2CH[CH2CH(CH3)2]2), 1.45 (s, 18 H, CH[CO2C(CH3)3]2), 1.55 (septuplet, J ≈ 6.7 Hz, 2 H, CH2CH[CH2CH(CH3)2]2), 1.88 (pentet, J ≈ 6.7 Hz, 1 H, CH2CH[CH2CH(CH3)2]2), 2.20 (d, J = 6.5 Hz, 2 H, CH2CH[CH2CH(CH3)2]2), 2.89 (t, J = 6.5 Hz, 1 H, OH), 3.01 (dd, J = 17.7, 2.8 Hz, 1 H, H-3a), 3.22 (dd, J = 17.7, 2.9 Hz, 1 H, H-3b), 3.71 (AB quartet of doublets, J = 12.1, 6.4 Hz, 2 H, CCH2OH), 4.24 (AB q, J = 11.9 Hz, 2 H, CCH2OC(O)CH2CH(CH(CH3)2)2), 4.45 (s, 1 H, CH[CO2C(CH3)3]2), 7.38–7.46 (m, 3 H, Ar), 7.48 (br s, 1 H, Ar), 7.52 (t, J = 2.8 Hz, 1 H, C=CHC6H4CH[CO2C(CH3)3]2); 13C NMR (100 MHz, CDCl3) δ 173.13, 171.04, 166.95, 136.80, 134.45, 134.28, 131.28, 130.96, 129.27, 128.89, 124.42, 83.28, 82.30, 82.28, 65.27, 64.60, 59.74, 43.92, 39.18, 32.06, 30.48, 27.79, 25.00, 22.74, 22.46, 22.45; FAB-MS (m/z, relative intensity) 617 (MH+, <1), 57 (100). Anal. (C35H52O9) calcd C 68.16, H 8.50; found C 68.10, H 8.47.
12-E: R1 = -CCH3(CH2OH)2; R2 = p-C6H4CH[CO2C(CH3)3]2H
1H NMR (400 MHz, CDCl3) δ 0.80–0.84 (m, 12 H, CH2CH[CH2CH(CH3)2]2), 0.10–1.15 (m, 4 H, 2 × CH2CH[CH2CH(CH3)2]2), 1.46 (s, 18 H, CH[CO2C(CH3)3]2), 1.57 (septuplet, J ≈ 6.7 Hz, 2 H, CH2CH[CH2CH(CH3)2]2), 1.90 (pentet, J ≈ 6.7 Hz, 1 H, CH2CH[CH2CH(CH3)2]2), 2.22 (d, J = 6.5 Hz, 2 H, CH2CH[CH2CH(CH3)2]2), 3.03 (dd, J = 17.7, 2.8 Hz, 1 H, H-3a), 3.21 (dd, J = 17.7, 2.9 Hz, 1 H, H-3b), 3.73 (AB q, J = 12.1 Hz, 2 H, CCH2OH), 4.26 (AB q, J = 11.9 Hz, 2 H, CCH2OC(O)CH2CH(CH(CH3)2)2), 4.46 (s, 1 H, CH[CO2C(CH3)3]2), 7.47 (two doublets, J = 8.8 Hz, 4 H, Ar), 7.55 (t, J = 2.8 Hz, 1 H, C=CHC6H4CH[CO2C(CH3)3]2); 13C NMR (100 MHz, CDCl3) δ 173.25, 170.99, 166.90, 136.83, 135.51, 133.91, 130.09, 129.97, 124.19, 83.20, 82.40, 65.22, 64.77, 59.87, 44.00, 39.27, 32.21, 30.58, 27.85, 25.09, 22.82, 22.81, 22.54, 22.52; FAB-MS (m/z, relative intensity) 617 (MH+, 1), 57 (100). Anal. (C35H52O9·0.6H2O) calcd C 66.98, H 8.54; found C 66.74, H 8.32.
13-E: R1 = (HOCH2)2(CH3)C-; R2 = -CH2CH[CH2CH(CH3)2]2
1H NMR (400 MHz, CDCl3) δ 0.86 (d, J = 6.6 Hz, 12 H, CH2CH[CH2CH(CH3)2]2), 0.99–1.18 (m, 7 H, 2 × CH2CH[CH2CH(CH3)2]2 and C(CH2OH)2CH3), 1.61 (septuplet, J ≈ 6.9 Hz, 2 H, CH2CH[CH2CH(CH3)2]2), 1.70 (pentet, J ≈ 6.9 Hz, 1 H, CH2CH[CH2CH(CH3)2]2), 2.13 (dd, J = 7.2, 6.0 Hz, 2 H, CH2CH[CH2CH(CH3)2]2), 2.63 (dd, J = 17.3, 2.4 Hz, 1 H, H-3a), 2.83 (dm, J = 17. 3 Hz, 1 H, H-3b), 3.33 and 3.41 (br s, 2 H, C(CH2OH)2CH3), 3.60 (br s, 1 H, CCH2OH), 3.64–3.77 (m, 4 H, C(CH2OH)2CH3), 3.85 (t, J = 11.3 Hz, 2 H, CCH2OH), 4.21 (dd, J = 12.0, 2.6 Hz, 1 H, CCHHOC(O)C(CH2OH)2CH3), 4.40 (dd, J = 11.9, 2.9 Hz, 1 H, CCHHOC(O)C(CH2OH)2CH3), 6.80 (m, 1 H, C=CH); 13C NMR (100 MHz, CDCl3) δ 175.16, 170.01, 141.70, 126.24, 83.11, 67.71, 67.67, 65.83, 64.43, 49.64, 43.82, 34.86, 32.75, 30.39, 25.18, 25.17, 22.92, 22.62, 22.60, 17.05; FAB-MS (m/z, relative intensity) 415 (MH+, 95), 299 (100). Anal. (C22H38O7) calcd C 63.74, H 9.24; found C 63.71, H 9.25.
Modeling
Structures for the single arginine mutants of the PKCδ C1b domain were generated as described previously.17 Briefly, the selected residue in the crystal structure14 was mutated to arginine, then subjected to a conformational search of its four χ angles to identify a small set of low-energy conformers. Structures for the double, triple, and quadruple mutants were built using the lowest-energy conformer found for each single arginine side chain.
Docking of the DAG-lactones was performed using the software program GOLD 5.0.40 The binding site was defined by atoms within a 10.0 Å sphere around the Nε atom of the C1 domain residue Gln 27 (Gln 257 in full-length PKCδ). Ligand flexibility flags included internal hydrogen bond detection and ring corner flipping, and the default torsion angle distributions were used. The GoldScore scoring function was used with the default parameter file. The genetic algorithm settings were automatically optimized according to ligand flexibility with the search efficiency set to 100%.
After docking, the structures of the DAG-lactone–mutant PKCδ complexes underwent conformational searching using systematic torsional sampling in MacroModel41,42 to identify low-energy salt-bridged conformers for the charged ligand and protein side chains. The searches used 10 000 steps of sampling on the rotatable bonds in the DAG-lactone R1 and R2 side chains and in the side chains of residues 7–13, 20–25, and 27 in the mutated C1 domain. Minimization of each conformer found was done using the OPLS 2005 force field with octanol implicit solvent. All atoms in the DAG-lactone and residues 7–13, 20–25, and 27 in the binding site were free to move during minimization, and the rest of the protein was held fixed.
Biology
[20-3H]Phorbol 12,13-dibutyrate (PDBu) was obtained from PerkinElmer, Waltham, MA. PDBu was from LC Laboratories (Woburn, MA). Phosphatidyl-l-serine was from Avanti Polar Lipids (Alabaster, AL). We have previously described the preparation of the GST-fusion construct with the PKCδ C1b domain, together with the construction, production, and purification of the variants incorporating the single amino acid substitutions N7R, S10R, P11R, L20R, along with those incorporating the multiple substitutions N7R/P11R, N7R/L20R, N7R/S10R/P11R, and N7R, S10R, L20R.17 Binding affinities of the various compounds to the wild-type and mutated GST-C1b domain constructs were determined in competitive binding assays with [20-3H]PDBu as described previously43,44 in the presence of 100 μg/mL phosphatidylserine.
Acknowledgments
This work was supported in part through the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute (Project Z1A BC 005270) and in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under Contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Glossary
Abbreviations Used
- PDBu
phorbol 12,13-dibutyrate
- PKC
protein kinase C
- DAG
sn-1,2-diacylglycerol
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Hurley J. H.; Newton A. C.; Parker P. J.; Blumberg P. M.; Nishizuka Y. Taxonomy and function of C1 protein kinase C homology domains. Protein Sci. 1997, 6, 477–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahelin R. V. Lipid binding domains: more than simple lipid effectors. J. Lipid Res. 2009, 50, S299–S304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colon-Gonzalez F.; Kazanietz M. G. C1 domains exposed: from diacylglycerol binding to protein interactions. Biochim. Biophys. Acta 2006, 1761, 827–837. [DOI] [PubMed] [Google Scholar]
- Kazanietz M. G. Targeting protein kinase C and “non-kinase” phorbol ester receptors: emerging concepts and therapeutic implications. Biochem. Biophys. Acta 2005, 1754, 296–304. [DOI] [PubMed] [Google Scholar]
- Yang C.; Kazanietz M. G. Divergence and complexities in DAG signaling: looking beyond PKC. Trends Pharmacol. Sci. 2003, 24, 602–608. [DOI] [PubMed] [Google Scholar]
- Brose N.; Rosenmund C. Move over protein kinase C, you’ve got company: alternative cellular effectors of diacylglycerol and phorbol esters. J. Cell Sci. 2002, 115, 4399–4411. [DOI] [PubMed] [Google Scholar]
- Singer W. D.; Brown H. A.; Sternweis P. C. Regulation of eukaryotic phosphatidylinositol-specific phospholipase C and phospholipase D. Annu. Rev. Biochem. 1997, 66, 475–509. [DOI] [PubMed] [Google Scholar]
- Becker K. P.; Hannun Y. A. Protein kinase C and phospholipase D: intimate interactions in intracellular signaling. Cell. Mol. Life Sci. 2005, 62, 1448–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griner E. M.; Kazanietz M. G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [DOI] [PubMed] [Google Scholar]
- Reyland M. E. Protein kinase C isoforms: multi-functional regulators of cell life and death. Front. Biosci. 2009, 14, 2386–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G.; Swanson N. Clinical findings using ingenol mebutate gel to treat actinic keratoses. J. Am. Acad. Dermatol. 2013, 68, S39–48. [DOI] [PubMed] [Google Scholar]
- Ruan B. F.; Zhu H. L. The chemistry and biology of the bryostatins: potential PKC inhibitors in clinical development. Curr. Med. Chem. 2012, 19, 2652–2664. [DOI] [PubMed] [Google Scholar]
- Blumberg P. M.; Kedei N.; Lewin N. E.; Yang D.; Czifra G.; Pu Y.; Peach M. L.; Marquez V. E. Wealth of opportunity—the C1 domain as a target for drug development. Curr. Drug Targets 2008, 9, 641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Kazanietz M. G.; Blumberg P. M.; Hurley J. H. Crystal structure of the cys2 activator-binding domain of protein kinase C delta in complex with phorbol ester. Cell 1995, 81, 917–924. [DOI] [PubMed] [Google Scholar]
- Canagarajah B.; Leskow F. C.; Ho J. Y.; Mischak H.; Saidi L. F.; Kazanietz M. G.; Hurley J. H. Structural mechanism for lipid activation of the Rac-specific GAP, beta2-chimaerin. Cell 2004, 119, 407–418. [DOI] [PubMed] [Google Scholar]
- Leonard T. A.; Rozycki B.; Saidi L. F.; Hummer G.; Hurley J. H. Crystal structure and allosteric activation of protein kinase C βII. Cell 2011, 144, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu Y.; Peach M. L.; Garfield S. H.; Wincovitch S.; Marquez V. E.; Blumberg P. M. Effects on ligand interaction and membrane translocation of the positively charged arginine residues situated along the C1 domain binding cleft in the atypical protein kinase C isoforms. J. Biol. Chem. 2006, 281, 33773–33788. [DOI] [PubMed] [Google Scholar]
- Geczy T.; Peach M. L.; El Kazzouli S.; Sigano D. M.; Kang J. H.; Valle C. J.; Selezneva J.; Woo W.; Kedei N.; Lewin N. E.; Garfield S. H.; Lim L.; Mannan P.; Marquez V. E.; Blumberg P. M. Molecular basis for failure of “atypical” C1 domain of Vav1 to bind diacylglycerol/phorbol ester. J. Biol. Chem. 2012, 287, 13137–13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapley J.; Tybulewicz V. L.; Rittinger K. Crucial structural role for the PH and C1 domains of the Vav1 exchange factor. EMBO Rep. 2008, 9, 655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott H. R.; Carpenter J. W.; Zhong S.; Ghosh S.; Bell R. M.; Campbell S. L. The solution structure of the Raf-1 cysteine-rich domain: a novel Ras and phospholipid binding site. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 8312–8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Horita D. A.; Waugh D. S.; Byrd R. A.; Morrison D. K. Solution structure and functional analysis of the cysteine-rich C1 domain of kinase suppressor of Ras (KSR). J. Mol. Biol. 2002, 315, 435–446. [DOI] [PubMed] [Google Scholar]
- Harjes E.; Harjes S.; Wohlgemuth S.; Muller K. H.; Krieger E.; Herrmann C.; Bayer P. GTP-Ras disrupts the intramolecular complex of C1 and RA domains of Nore1. Structure 2006, 14, 881–888. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay G.; Standaert M. L.; Sajan M. P.; Kanoh W.; Miura A.; Braun U.; Kruse F.; Leitges M.; Farese R. V. Protein kinase C-lambda knockout in embryonic stem cells and adipocytes impairs insulin-stimulated glucose transport. Mol. Endocrinol. 2004, 18, 373–383. [DOI] [PubMed] [Google Scholar]
- Fields A. P.; Regala R. P. Protein kinase Cι: human oncogene, prognostic marker, and therapeutic target. Pharmacol. Res. 2007, 55, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray N. R.; Kalari K. R.; Fields A. P. Protein kinase Cι expression and oncogenic signaling mechanisms in cancer. J. Cell. Physiol. 2011, 226, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Meco M. T.; Moscat J. The atypical PKCs in inflammation: NF-κB and beyond. Immunol. Rev. 2012, 246, 154–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Garcia L. A.; Schulze J. O.; Frohner W.; Zhang H.; Suss E.; Weber N.; Navratil J.; Amon S.; Hindie V.; Zeuzem S.; Jorgensen T. J. D.; Alzari P. M.; Neimanis S.; Engel M.; Biondi R. M. Allosteric regulation of protein kinase C PKCζ by the N-terminal C1 domain and small compounds to the PIF-Pocket. Chem. Biol. 2011, 18, 1463–1473. [DOI] [PubMed] [Google Scholar]
- Diaz-Meco M. T.; Municio M. M.; Sanchez P.; Lozano J.; Moscat J. λ-Interacting protein, a novel protein that specifically interacts with the zinc finger domain of the atypical protein kinase C isotype λ/ι and stimulates its kinase activity in vitro and in vivo. Mol. Cell. Biol. 1996, 16, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J.; Diaz-Meco M. T.; Albert A.; Campuzano S. Cell signaling and function organized by PB1 domain interactions. Mol. Cell 2006, 23, 631–640. [DOI] [PubMed] [Google Scholar]
- Diaz-Meco M. T.; Municio M. M.; Frutos S.; Sanchez P.; Lozano J.; Sanz L.; Moscat J. The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell 1996, 86, 777–786. [DOI] [PubMed] [Google Scholar]
- Marquez V. E.; Blumberg P. M. Diacylglycerol (DAG) and DAG-lactones as selective activators of protein kinase C (PK-C). Acc. Chem. Res. 2003, 36, 434–443. [DOI] [PubMed] [Google Scholar]
- Duan D.; Sigano D. M.; Kelley J. A.; Lai C. C.; Lewin N. E.; Kedei N.; Peach M. L.; Lee J.; Abeyweera T. P.; Rotenberg S. A.; Kim H.; Kim Y. H.; El Kazzouli S.; Chung J. U.; Young H. A.; Young M. R.; Baker A.; Colburn N. H.; Haimovitz-Friedman A.; Truman J. P.; Parrish D. A.; Deschamps J. R.; Perry N. A.; Surawski R. J.; Blumberg P. M.; Marquez V. E. Conformationally constrained analogues of diacylglycerol. 29. Cells sort diacylglycerol-lactone chemical zip codes to produce diverse and selective biological activities. J. Med. Chem. 2008, 51, 5198–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacro K.; Bienfait B.; Lee J.; Han K. C.; Kang J. H.; Benzaria S.; Lewin N. E.; Bhattacharyya D. K.; Blumberg P. M.; Marquez V. E. Conformationally constrained analogues of diacylglycerol (DAG). 16. How much structural complexity is necessary for recognition and high binding affinity to protein kinase C?. J. Med. Chem. 2000, 43, 921–944. [DOI] [PubMed] [Google Scholar]
- Lee J. Design and synthesis of bioisosteres of ultrapotent protein kinase C (PKC) ligand, 5-acetoxymethyl-5-hydroxymethyl-3-alkylidene tetrahydro-2-furanone. Arch. Pharmacal Res. 1998, 21, 452–457. [DOI] [PubMed] [Google Scholar]
- Choi Y.; Kang J. H.; Lewin N. E.; Blumberg P. M.; Lee J.; Marquez V. E. Conformationally constrained analogues of diacylglycerol. 19. Synthesis and protein kinase C binding affinity of diacylglycerol lactones bearing an n-hydroxylamide side chain. J. Med. Chem. 2003, 46, 2790–2793. [DOI] [PubMed] [Google Scholar]
- Kang J. H.; Siddiqui M. A.; Lewin N. E.; Sigano D. M.; Blumberg P. M.; Lee J.; Marquez V. E. Conformationally constrained analogues of diacylglycerol. 24. Asymmetric synthesis of a chiral R-DAG-lactone template as a versatile precursor for highly functionalized DAG-lactones. Org. Lett. 2004, 6, 2413–2416. [DOI] [PubMed] [Google Scholar]
- Nacro K.; Sigano D. M.; Yan S.; Nicklaus M. C.; Pearce L. L.; Lewin N. E.; Garfield S. H.; Blumberg P. M.; Marquez V. E. An optimized protein kinase C activating diacylglycerol combining high binding affinity (Ki) with reduced lipophilicity (log P). J. Med. Chem. 2001, 44, 1892–1904. [DOI] [PubMed] [Google Scholar]
- Sigano D. M.; Peach M. L.; Nacro K.; Choi Y.; Lewin N. E.; Nicklaus M. C.; Blumberg P. M.; Marquez V. M. Differential binding modes of diacylglycerol (DAG) and DAG lactones to protein kinase C (PK-C). J. Med. Chem. 2003, 46, 1571–1579. [DOI] [PubMed] [Google Scholar]
- Kang J. H.; Benzaria S.; Sigano D. M.; Lewin N. E.; Pu Y. M.; Peach M. L.; Blumberg P. M.; Marquez V. E. Conformationally constrained analogues of diacylglycerol. 26. Exploring the chemical space surrounding the C1 domain of protein kinase C with DAG-lactones containing aryl groups at the sn-1 and sn-2 positions. J. Med. Chem. 2006, 49, 3185–3203. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Goodman J. M.; Still W. C. An unbounded systematic search of conformational space. J. Comput. Chem. 1991, 12, 1110–1117. [Google Scholar]
- MacroModel, version 9.9; Schrödinger, LLC: New York, NY, 2011.
- Lewin N. E.; Blumberg P. M. [3H]Phorbol 12,13-dibutyrate binding assay for protein kinase C and related proteins. Methods Mol. Biol. 2003, 233, 129–156. [DOI] [PubMed] [Google Scholar]
- Wang Q. J.; Fang T. W.; Nacro K.; Marquez V. E.; Wang S.; Blumberg P. M. Role of the hydrophobic residues in the C1b domain of protein kinase C delta on ligand and phospholipid interactions. J. Biol. Chem. 2001, 276, 19580–19587. [DOI] [PubMed] [Google Scholar]