

Abstract
To assess the role of CCL2/MCP-1 in opiate drug abuse and HIV-1 comorbidity, the effects of systemic morphine and intrastriatal HIV-1 Tat on macrophage/microglial and astroglial activation were assessed in wild type and CCR2 null mice. Tat and/or morphine additively increased the proportion of CCL2 immunoreactive astroglia. The effects of morphine were prevented by naltrexone. Glial activation was significantly reduced in CCR2(−/−) versus wild-type mice following Tat or morphine plus Tat exposure. Thus, CCR2 contributes to local glial activation caused by Tat alone or in the presence of opiates, implicating CCR2 signaling in HIV-1 neuropathogenesis in drug abusers and non-abusers.
Keywords: AIDS, chemokines, μ-opioid receptors, drug abuse, astrocytes, microglia, CNS inflammation, monocyte chemoattractant protein-1/CCL2
1. Introduction
Opiates (substances derived from the opium poppy, such as heroin, morphine, or analogues such as oxycodone) are popular drugs of abuse, exacerbate the pathogenesis of human immunodeficiency type 1 (HIV-1) infection through preferential actions at endogenous μ-opioid receptors (MOR) (Gurwell et al., 2001; Hauser et al., 2005; Hauser et al., 2006). There is considerable evidence that opiate drugs can exacerbate HIV-1 encephalitis (HIVE) by modulating the expression and function of neuroimmune chemokines and their receptors (Rogers and Peterson, 2003). Importantly, the effects of opiates are not restricted to a particular cell type, and opiates per se directly affect the expression of chemokine ligands and receptors in neural (neurons, microglia, and astroglia) and non-neural cells (lymphocytes and monocytes/macrophages) (Hauser et al., 2005).
Chemokines mediate multiple inflammatory disorders in the CNS, including multiple sclerosis (Mahad and Ransohoff, 2003) and HIVE (Nath, 1999; Kaul et al., 2001). CC chemokine ligand 2 [CCL2, also known as monocyte chemoattractant protein-1 (MCP-1)], in particular, is a cofactor in HIV-1 pathogenesis and appears to initiate and sustain the neuroinflammatory process. CCL2 attracts and activates mononuclear phagocytes, as well as several other leukocyte types, to sites of CNS injury (McManus et al., 2000). CCL2 levels correlate with neurocognitive deficits accompanying HIV-1 or simian immunodeficiency virus infection (Sevigny et al., 2004; Mankowski et al., 2004; Avison et al., 2004; Chang et al., 2004). Moreover, mutations in CCL2 and CCR2 affect HIV pathogenesis (Gonzalez et al., 2002; Letendre et al., 2004; Singh et al., 2004). Thus, chemokines, including MCP-1 in particular, play an important role in neuroAIDS.
Although the cellular mechanisms by which opiates exacerbate the neuropathogenesis of HIV-1 are incompletely understood, there is emerging evidence that astroglia play a central role in the interaction. A subpopulation of astroglia express functional MOR (Eriksson et al., 1991; Stiene-Martin and Hauser, 1991; Hauser et al., 1996; Stiene-Martin et al., 1998; Stiene-Martin et al., 2001) and opiates markedly increase the production of chemokines by HIV-1 Tat exposed astrocytes (El-Hage et al., 2005). Astrocytes are an important source of chemokines in the CNS and express CCL2 (Ransohoff et al., 1993; Hayashi et al., 1995; Peterson et al., 1997; Oh et al., 1999) and variably express its cognate receptor CCR2 (Andjelkovic et al., 1999; Dorf et al., 2000), although CCR2 expression among astrocytes is heterogeneous and appears to be regulated by inflammation (Andjelkovic et al., 2002; Croitoru-Lamoury et al., 2003). HIV-1 Tat triggers CCL2 production and inflammatory cascades in astrocytes (Conant et al., 1998). Importantly, opiates enhance CCL2 release by Tat exposed astrocytes (El-Hage et al., 2005), and this results in a significantly increased motility of N9 microglial cells in vitro. Enhanced CCL2 release also likely contributes to macrophage/microglial chemotaxis in vivo (El-Hage et al., 2006).
Prompted by the importance of MCP-1 in neuroAIDS, and findings that opiates potentiate the production of MCP-1 in HIV-1 Tat-exposed astrocytes, we examined cellular changes in MCP-1 and glial activation in the striata of wild type and CCR2 null mice exposed to morphine and/or HIV-1 Tat. The results suggest that CCR2 contributes significantly to the activation of macrophages and glia seen within the brains of HIV-1 infected drug abusers and non-abusers.
2. Materials and Methods
ICR mice (Charles River Co., MA, USA) were used to assess changes in the proportion of CCL2 and/or glial fibrillary acidic protein- (GFAP) expressing cells following opiate and/or HIV-1 Tat1–72 (referred to as Tat) exposure (El-Hage et al., 2006). Wild type [CCR2(+/+)] and CCR2(−/−) mice on a C57Bl/J6 background were used to explore the role of CCR2 in mediating Tat or morphine and Tat-induced increases in macrophages/microglia or astroglia, which were maintained and characterized as previously described (Ambati et al., 2003). To assess the selectivity of Tat1–72, the effects of intrastriatal Tat1–72, a minimally toxic, deletion mutant variant of Tat1–72 (TatΔ31–61), and saline injections were compared in CCR2(+/+) and CCR2(−/−) C57Bl/J6 mice. In some cases, mice receiving Tat were also injected with sterile vehicle in the contralateral striatum. GFAP+ astroglia and F4/80+ macrophages/microglia were sampled as described below.
Time-release, vehicle (placebo implant), morphine (25 mg), and/or naltrexone (30 mg) pelleted implants (NIDA, Rockville, MD, USA) were used to continuously deliver drugs for 5 days. Pellets were surgically implanted subcutaneously under the subscapular skin using anesthesia and aseptic conditions as described before (El-Hage et al., 2006). Intrastriatal HIV-1 Tat1–72 (25 μg) or saline vehicle was injected at coordinates AP = +0.7 mm, ML = 2.0 mm and DV = −4.0 mm from bregma as previously described (El-Hage et al., 2006). Drug implants were administered 2 days after stereotaxic surgery and mice were euthanized by anesthesia overdose after 5 days of continuous drug exposure (El-Hage et al., 2006). Treatment groups consisted of mice receiving: (1) placebo implant, sham surgery (anesthesia only); (2) placebo implant, intrastriatal vehicle; (3) morphine implant (25 mg), intrastriatal vehicle; (4) morphine (25 mg) and naltrexone (30 mg) implants, intrastriatal vehicle; (5) placebo implant, intrastriatal Tat; (6) morphine implant (25 mg/kg), intrastriatal Tat; and (7) morphine (25 mg) and naltrexone (30 mg) implants, intrastriatal Tat.
Tissues were fixed in Zamboni’s modified phosphate-buffered paraformaldehyde. In frozen sections (10–18 μm thick), glial fibrillary acidic protein (GFAP), F4/80, MOR, and CCL2 were detected by indirect immunofluorescence as described before (El-Hage et al., 2006). Sections were counterstained with Hoechst 33342 (15 μg/ml in 0.1% BSA in PBS for 15 min at room temperature; Molecular Probes) to detect cell nuclei. Goat anti-CCR2 antibodies were obtained from Capralogics (Hardwick, MA, USA) and used at a 1:400 dilution. CCR2 primary antibodies were detected using biotinylated anti-Goat IgG (Vector Laboratories, Burlingame, CA) diluted 1:400 and visualized with StreptAvidin Alexa Fluor 488 (Molecular Probes/Invitrogen, Carlsbad, CA) diluted 1:500. A computer imaging system (Bioquant, Nashville, TN) and fluorescent microscope (Nikon Optiphot, Melville, NY) equipped with a motorized X-Y and Z-axes stage controller (Ludl Electronic Products, Ltd., Hawthorne, NY) were used to systematically, but arbitrarily, sample cells near (300 ± 100 μm) and distant (600 ± 100 μm) from the site of Tat injection within the striatum as previously described (El-Hage et al., 2006). The proportions of GFAP+ astroglia and F4/80+ macrophages/microglia near and distant from the intrastriatal injection site were assessed. Digital photomicrographs were prepared using a Leica, TCS SP confocal microscope. Statistical differences were assessed by using ANOVA and multiple comparisons assessed post-hoc using Duncan’s test.
3. Results
CCL2, μ-opioid receptor (MOR), and CCR2 immunoreactivity was colocalized with GFAP immunoreactivity in subpopulations astrocytes in mouse striata (Fig. 1). Relatively small subsets of astrocytes can express CCL2, MOR (Hauser et al., 1996; Stiene-Martin et al., 1998), or CCR normally, although the proportion of astrocytes possessing MOR (El-Hage et al., 2006) or CCL2 (see Fig. 2A) immunoreactivity can increase following combined morphine and Tat exposure (Fig. 1A–B). Similar to wild type mice, MOR immunoreactivity could be colocalized with GFAP in a subset of astrocytes in CCR2(−/−) mice (Fig. 1C). As expected, CCR2 immunoreactivity was present in wild type (Fig. 1D–F), but not CCR2 null, mice (Fig. 1G–I).
Fig. 1.

CCL2, μ-opioid receptor (MOR), and CCR2 immunocytochemical co-localization with glial fibrillary acidic protein (GFAP) in subpopulations of striatal astrocytes in wild type (A,B,D–F) and CCR2(−/−) mice (C,G–I). A small subpopulation of astrocytes normally express MOR (arrows in A), CCL2 (arrow in B), or CCR immunoreactivity (arrows in F), although the proportion possessing MOR (A) (El-Hage et al., 2006), or CCL2 (B), immunoreactivity increases following morphine and/or Tat exposure (see Fig. 2A) (scale bar A–B, = 25 μm; C = 15 μm). MOR could be localized in a subset of astrocytes in CCR2−/− mice (arrowheads in C delineate astrocytes with highly polarized MOR and GFAP cytoplasmic immunofluorescence; Hoechst counterstained nuclei). CCR2 immunoreactivity was present in wild type (D–F; arrows in F), but not in CCR2 (−/−) null (G–I) mice (D–I; scale bar = 20 μm).
Fig. 2.
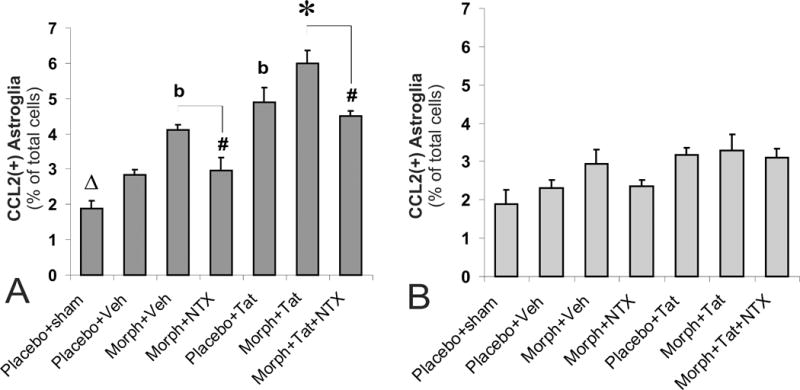
Effects of subcutaneous opiates and/or intrastriatal Tat on glial fibrillary acidic protein (GFAP) and CCL2 immunoreactivity at loci near (300 ± 100 μm) (A) and distant (600 ± 100 μm) (B) from the site of Tat injection in wild type mice. The proportion of CCL2 immunoreactive astrocytes increased significantly following morphine or Tat exposure (bP < 0.05 vs. vehicle-injected controls or morphine plus Tat treatment) (A), and showed additive increases with combined exposure (*P < 0.05 vs. other groups). The effects of morphine (alone or in combination with Tat) were antagonized by naltrexone (#P < 0.05 vs. morphine treatment) (A). Increases in CCL2+ astrocytes were seen following intrastriatal injection of vehicle compared to un-injected sham mice receiving placebo implants (ΔP < 0.05 vs. vehicle-injected, placebo controls) (A). The proportion of CCL2+ astrocytes was unchanged by morphine and/or Tat-treatment at locations distant from the site of injection (600 ± 100 μm) (B); n= 5–6 mice per group.
CCL2 immunoreactivity was detected minimally in astrocytes in sham-controls, but increased significantly following morphine or Tat exposure at 300 ± 100 μm (bP < 0.05; Fig. 2A). In combination, morphine and Tat caused marked increases in CCL2 expression by astrocytes compared to either substance alone (Fig. 2A; *P < 0.05 vs. other groups). The increases in CCL2+ astrocytes caused by morphine alone or in combination with Tat were antagonized by naltrexone (#P < 0.05 vs. morphine treatment; Fig. 2A). Interestingly, the introduction of a sterile needle alone was sufficient to induce increased CCL2 expression in astrocytes compared to sham-injected mice receiving placebo implants (ΔP < 0.05; Fig. 2A). For this reason, treatment groups were compared to vehicle-injected controls rather than to non-intrastriatally injected, control mice.
Using vehicle-injected mice as controls, no differences were observed in any experimental group at the farther distance of 600 ± 100 μm from the injection site (Fig. 2B). There was also no difference in the percentage of astrocytes expressing CCL2 between the sham-treated (anesthesia only) and vehicle injected-control mice. There was however, one additional and noteworthy finding at this distance. A comparison between both Tat containing regimens (that is, Tat alone or Tat plus morphine) versus sham-treated mice indicated a significant increase in the percentage of CCL2+ astrocytes. This is not indicated on the graph in Fig. 2B since, as discussed above, the sham-treated mice are not entirely appropriate controls. However, the observed differences suggest that there are subtle, reactive changes in glia that occur even at sites relatively distance to an application of Tat. These differences were not apparent in animals receiving morphine and were not reversible with naltrexone.
To characterize the role of CCR2 in mediating the response of macrophages/microglia and astroglia to HIV-1 Tat or the combined effects of opiates and Tat, wild type and CCR2 null mice were exposed to HIV-1 Tat intrastriatally in the presence of placebo or morphine implants (Fig. 3). Compared to wild type mice exposed to Tat or morphine and Tat, CCR2 deletion significantly reduced the proportion of GFAP+ astroglia (Fig. 3A) and F4/80+ macrophages/microglia (Fig. 3C) near (300 ± 100 μm) the Tat injection site. The proportion of astrocytes expressing MOR and/or GFAP in response to Tat or morphine plus Tat treatment appeared to be reduced, although this was not assessed quantitively. By contrast, Tat or morphine and Tat exposure did not significantly increase the proportion of GFAP+ astrocytes (Fig. 3B) or F4/80+ macrophages/microglia (Fig. 3D) compared to vehicle-injected controls distant (600 ±100 μm) from the injection epicenter.
Fig. 3.

CCR2 deletion (−/−) significantly reduced HIV-1 Tat or morphine plus Tat-induced increases in GFAP+ astroglia (A) and F4/80+ macrophages/microglia (C) compared to wild type (+/+) mice at sites near (300 ± 100 μm) the site of Tat injection. By contrast, at sites more distant from Tat injection (600 ± 100 μm), neither Tat nor morphine plus Tat significantly increased the proportion of astroglia (B) or macrophages/microglia (D); *P < 0.05, wild type versus CCR2(−/−) mice; n= 5 mice per group.
Intrastriatal Tat caused significant increases in the proportion of GFAP+ astroglia (Fig. 4A) and F4/80+ macrophages/microglia (Fig. 4B) compared to vehicle- or mutant TatΔ31–61- injected CCR2(+/+) mice, or Tat-injected CCR2(−/−) mice (*P < 0.05; ANOVA, followed post-hoc by Duncan’s test) (Fig 4). Significant glial changes were noted near (300 ± 100 μm), but not distant (600 ± 100 μm), from the site of injection (the 600 ± 100 μm findings are not shown). The absence of differences between vehicle-injected CCR2(+/+) and Tat-injected CCR2(−/−) mice indicates that the increases in macrophage/microglial and astroglial activation were mediated entirely by CCR2. Moreover, in wild type mice injected with ipsilaterally Tat and contralaterally with saline, the (contralateral) saline-injected side showed no additional increases in macrophage/microglial or astroglial numbers compared to mice injected with saline alone (Fig. 4). This suggests that intrastriatal Tat does not cause systemic inflammatory effects that influence reactive glial changes in the contralateral striatum.
Fig. 4.
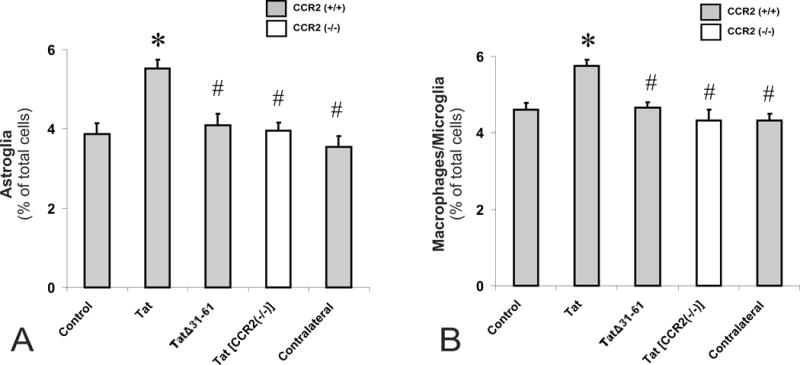
Intrastriatal HIV-1 Tat caused significant increases in the proportion of GFAP+ astroglia (A) and F4/80+ macrophages/microglia (B) (*P < 0.05 vs. vehicle controls) compared to vehicle- or mutant TatΔ31–61- injected CCR2(+/+) mice (TatΔ31–61), or Tat-injected CCR2(−/−)(Tat [CCR2(−/−)]) mice (#P < 0.05 vs. Tat-treatment) (A-B). Moreover, in wild type-mice injected with Tat ipsilaterally and saline in the contralateral striatum (Contralateral), there no additional increases in macrophage/microglial or astroglial numbers in the contralateral striatum compared to mice only receiving saline.
4. Discussion
Our findings indicate that the expression of CCL2 by astroglia is highly responsive to CNS insults. Astroglia appear to be significant contributors to morphine and/or Tat-induced increases in CCL2. First, a large majority of the CCL2+ cells were GFAP+ astroglia in the present study. Second, unlike macrophages/microglia (El-Hage et al., 2005), astroglia display synergistic increases in the release of CCL2 and other chemokines when exposed to Tat and morphine (El-Hage et al., 2005). To the contrary, microglia fail to show interactive increases in CCL2 (El-Hage et al., 2005) and opiates can limit the migration of monocyte/macrophages outside the CNS (Malik et al., 2002; El-Hage et al., 2006), although this is not a consistent feature (Singhal et al., 1996). A particular opiate effect is highly dependent on context (Hauser and Mangoura, 1998). Therefore, opiate-induced increases in CCL2 expression by astroglia may be a selective response not emulated by other cell types.
We additionally demonstrate that systemic morphine can increase the proportion of CCL2+ astrocytes near the site of Tat injection, suggesting that opiates can aggravate the proinflammatory effects of Tat. The increases in CCL2+ astrocytes confirm and extend in vitro observations indicating that morphine and Tat synergistically increase CCL2 mRNA and protein products in astrocytes (El-Hage et al., 2006). Based on immunocytochemical measures of the number of CCL2+ astroglia alone, we cannot infer the extent that CCL2 levels increase. Assuming that concurrent morphine and Tat exposure causes sustained approximately 120-fold increases in CCL2 release by astrocytes that are similar to those seen in vitro (El-Hage et al., 2005), then the local tissue concentrations in CCL2 are likely to be much greater than the levels predicted by an approximate doubling of CCL2+ cells seen in the present study. Somewhat unexpectedly, the introduction of a sterile needle (or perhaps the pressure of the injection) by itself is sufficient to increase the number of CCL2+ astrocytes. The ability of morphine to cause interactive increases in CCL2 expression by astroglia may not be an entirely Tat specific response. Notably, morphine alone increased the number of CCL2+ astrocytes at 300 ± 100 μm, but not at 600 ± 100 μm, from the needle tract in vehicle-injected controls, suggesting that systemic morphine was selectively modifying a focal inflammatory response. Moreover, this evidence suggests that, besides intensifying the response of glia to viral proteins, morphine can potentiate the innate response of the CNS to a wide variety of disease or injurious insults. Future studies will address whether the glial changes described at 7 days in the present study contribute to neurodegenerative changes at later times (El-Hage et al., 2006).
In an earlier study, we did not observe increases in glial numbers in ICR mice exposed to an identical 5 μg concentration of Tat as in C57Bl/J6 mice seen in the present study (El-Hage et al., 2006). Although it is assumed that increasing the dose of Tat would yield similar effects in ICR mice, qualitative strain differences in Tat sensitivity might underlie differences in the response of mice to HIV-1 Tat and further study is warranted. Tat causes dose-dependent increases in GFAP in the rat striatum (Bansal et al., 2000). Nevertheless, glial activation was seen in both ICR and C57Bl/6J mice after co-exposure to Tat and morphine, suggesting that morphine plays a significant regulatory role in Tat-induced responses that applies broadly across strains.
While astroglia appear to be an important source of CCL2, the cellular targets of CCL2 are more varied. Cells that express CCR2 in the CNS include macrophages/microglia, subpopulations of neurons and astrocytes (Simpson et al., 2000; Andjelkovic et al., 2002) and CCR2 expression is upregulated with inflammation (Banisadr et al., 2002; Mahad and Ransohoff, 2003).
Despite increases in CCL2+ astrocytes following Tat or opiate and Tat exposure (El-Hage et al., 2005; El-Hage et al., 2006), and reductions in glial activation in CCR2 null mice, it cannot be assumed that CCL2 is the only ligand that activates CCR2 in the present study. Though CCL2 is the principal endogenous CCR2 agonist, alternative chemokines can also activate CCR2 and may contribute to our findings. For example, CCL7 (MCP-3), CCL8 (MCP-2), CCL13 (MCP-4), and CCL12 (MCP-5) can activate CCR2 in mice (Ma et al., 2002; Mantovani et al., 2004). We previously found that, in addition to CCL2, the release of CCL5 and CCL12 are markedly increased in opiate and/or Tat-exposed astrocytes (El-Hage et al., 2005), and concluded that CCL5, CCL12 or perhaps another chemokine contributed directly to opiate and HIV-1-induced reactive glial changes (El-Hage et al., 2006). CCR5 is upregulated in glia with HIV or SIV infection (Petito et al., 2001; Croitoru-Lamoury et al., 2003), suggesting Tat might increase CCL5-CCR5 signaling. By contrast, in monocytes or Chinese hamster ovary (CHO) cells, MOR or δ-opioid receptor activation causes heterologous desensitization of CCR5 and decreases infectivity by R5 HIV-1 strains (Szabo et al., 2003), suggesting that opiates attenuate CCL5-CCR5 signaling.
The present study demonstrates that CCR2 signaling contributes significantly to Tat or opiate and Tat-induced macrophage and glial activation, and further suggests that the reactive glial changes caused by HIV-1 Tat are mediated entirely by CCR2. Tat-induced glial changes were completely abolished in CCR2(−/−) mice and equivalent to those seen in saline-injected control mice. This result was unexpected because HIV-1 Tat increases the expression of chemokines besides CCL2 in astroglia, including CCL5/RANTES (El-Hage et al., 2005) and perhaps other chemokines and/or chemokine receptors (Mahajan et al., 2005), that presumably would maintain chemotaxis in the absence of CCR2. Moreover, in previous studies, the use of anti-MCP-1 immunoneutralizing antibodies, or CCL2(−/−) astrocytes, failed to completely block microglial chemotaxis in response to conditioned medium from Tat- or morphine and Tat-exposed astrocytes (El-Hage et al., 2006). In addition, the absence of glial changes in CCR2(−/−) mice suggest that the alternative CCL2 receptor, L-CCR (Zuurman et al., 2003; Brouwer et al., 2004), is not involved. Collectively, the above findings suggest that an alternative CCR2 chemokine agonist, such as CCL12, is likely to have been acting together with CCL2 to enhance morphine and Tat-induced microglial chemotaxis in the absence of CCL2 in our earlier studies (El-Hage et al., 2006). Further studies are needed to assess whether CCL12 is involved and whether the synergistic effect of morphine is additionally mediated by chemokines besides CCL2.
We conclude that CCR2 contributes significantly to the activation of macrophages and glia seen in the CNS of HIV-1 infected drug abusers and non-abusers. As noted earlier, CCL2 levels are highly correlated with neurocognitive deficits accompanying HIV-1 or simian immunodeficiency virus infection (Sevigny et al., 2004; Mankowski et al., 2004; Avison et al., 2004; Chang et al., 2004), and CCL2 levels are markedly increased by substance abuse in HIV-1 infected individuals (Hauser et al., 2005; Hauser et al., 2006). Therapeutic strategies that interfere with endogenous CCR2 ligand-receptor interactions would likely be beneficial to HIV-1 infected individuals regardless of whether or not they abuse opiate drugs.
Acknowledgments
This work was supported by NIH grants DA13278, DA19398, P20RR015592, and EY15422. We thank Dr. Avindra Nath for providing HIV-1 Tat protein and expert guidance, and Mr. Kenneth Martin for expert technical assistance.
Abbreviations
- CCL
CC or beta chemokine ligand
- CCR
CC or beta chemokine receptor
- GFAP
glial fibrillary acidic protein
- HIV-1
human immunodeficiency virus type 1
- HIVE
human immunodeficiency virus encephalitis
- MCP-1 or CCL2
monocyte chemoattractant protein-1
- MOR
μ-opioid receptor
- RANTES or CCL5
regulated on activation normal T cell expressed and secreted
- Tat
transactivator of transcription
References
- Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003;9:1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- Andjelkovic AV, Kerkovich D, Shanley J, Pulliam L, Pachter JS. Expression of binding sites for beta chemokines on human astrocytes. Glia. 1999;28:225–235. [PubMed] [Google Scholar]
- Andjelkovic AV, Song L, Dzenko KA, Cong H, Pachter JS. Functional expression of CCR2 by human fetal astrocytes. J Neurosci Res. 2002;70:219–231. doi: 10.1002/jnr.10372. [DOI] [PubMed] [Google Scholar]
- Avison MJ, Nath A, Greene-Avison R, Schmitt FA, Bales RA, Ethisham A, Greenberg RN, Berger JR. Inflammatory changes and breakdown of microvascular integrity in early human immunodeficiency virus dementia. J Neurovirol. 2004;10:223–232. doi: 10.1080/13550280490463532. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Queraud-Lesaux F, Boutterin MC, Pelaprat D, Zalc B, Rostene W, Haour F, Parsadaniantz SM. Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. J Neurochem. 2002;81:257–269. doi: 10.1046/j.1471-4159.2002.00809.x. [DOI] [PubMed] [Google Scholar]
- Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM. Neurotoxicity of HIV-1 proteins gp120 and tat in the rat striatum. Brain Res. 2000;879:42–49. doi: 10.1016/s0006-8993(00)02725-6. [DOI] [PubMed] [Google Scholar]
- Brouwer N, Zuurman MW, Wei T, Ransohoff RM, Boddeke HW, Biber K. Induction of glial L-CCR mRNA expression in spinal cord and brain in experimental autoimmune encephalomyelitis. Glia. 2004;46:84–94. doi: 10.1002/glia.10352. [DOI] [PubMed] [Google Scholar]
- Chang L, Ernst T, St HC, Conant K. Antiretroviral treatment alters relationship between MCP-1 and neurometabolites in HIV patients. Antivir Ther. 2004;9:431–440. doi: 10.1177/135965350400900302. [DOI] [PubMed] [Google Scholar]
- Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci U S A. 1998;95:3117–3121. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croitoru-Lamoury J, Guillemin GJ, Boussin FD, Mognetti B, Gigout LI, Cheret A, Vaslin B, Le GR, Brew BJ, Dormont D. Expression of chemokines and their receptors in human and simian astrocytes: evidence for a central role of TNF alpha and IFN gamma in CXCR4 and CCR5 modulation. Glia. 2003;41:354–370. doi: 10.1002/glia.10181. [DOI] [PubMed] [Google Scholar]
- Dorf ME, Berman MA, Tanabe S, Heesen M, Luo Y. Astrocytes express functional chemokine receptors. J Neuroimmunol. 2000;111:109–121. doi: 10.1016/s0165-5728(00)00371-4. [DOI] [PubMed] [Google Scholar]
- El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia. 2005;50:91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, Bruce-Keller AJ, Hauser KF. HIV Tat1–72 and Opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia. 2006;53:132–146. doi: 10.1002/glia.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PS, Hansson E, Rönnbäck L. Mu and delta opiate receptors in neuronal and astroglial primary cultures from various regions of the brain-coupling with adenylate cyclase, localisation on the same neurones and association with dopamine (D1) receptor adenylate cyclase. Neuropharmacology. 1991;30:1233–1239. doi: 10.1016/0028-3908(91)90170-g. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Rovin BH, Sen L, Cooke G, Dhanda R, Mummidi S, Kulkarni H, Bamshad MJ, Telles V, Anderson SA, Walter EA, Stephan KT, Deucher M, Mangano A, Bologna R, Ahuja SS, Dolan MJ, Ahuja SK. HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc Natl Acad Sci U S A. 2002;99:13795–13800. doi: 10.1073/pnas.202357499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurwell JA, Nath A, Sun Q, Zhang J, Martin KM, Chen Y, Hauser KF. Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience. 2001;102:555–563. doi: 10.1016/s0306-4522(00)00461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser KF, El-Hage N, Buch S, Berger JR, Tyor WR, Nath A, Bruce-Keller AJ, Knapp PE. Molecular targets of opiate drug abuse in neuroAIDS. Neurotox Res. 2005;8:63–80. doi: 10.1007/BF03033820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser KF, El-Hage N, Buch S, Tyor WR, Nath A, Bruce-Keller AJ, Knapp PE. Impact of opiate-HIV-1 interactions on neurotoxic signaling. J Neuroimmune Pharmacol. 2006;1:98–105. doi: 10.1007/s11481-005-9000-4. [DOI] [PubMed] [Google Scholar]
- Hauser KF, Mangoura D. Diversity of the endogenous opioid system in development: novel signal transduction translates multiple extracellular signals into neural cell growth and differentiation. Perspect Dev Neurobiol. 1998;5:437–449. [PubMed] [Google Scholar]
- Hauser KF, Stiene-Martin A, Mattson MP, Elde RP, Ryan SE, Godleske CC. μ-Opioid receptor-induced Ca2+ mobilization and astroglial development: Morphine inhibits DNA synthesis and stimulates cellular hypertrophy through a Ca2+-dependent mechanism. Brain Res. 1996;720:191–203. doi: 10.1016/0006-8993(96)00103-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Luo Y, Laning J, Strieter RM, Dorf ME. Production and function of monocyte chemoattractant protein-1 and other beta-chemokines in murine glial cells. J Neuroimmunol. 1995;60:143–150. doi: 10.1016/0165-5728(95)00064-9. [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Letendre S, Marquie-Beck J, Singh KK, de Almeida S, Zimmerman J, Spector SA, Grant I, Ellis R. The monocyte chemotactic protein-1 -2578G allele is associated with elevated MCP-1 concentrations in cerebrospinal fluid. J Neuroimmunol. 2004;157:193–196. doi: 10.1016/j.jneuroim.2004.08.028. [DOI] [PubMed] [Google Scholar]
- Ma M, Wei T, Boring L, Charo IF, Ransohoff RM, Jakeman LB. Monocyte recruitment and myelin removal are delayed following spinal cord injury in mice with CCR2 chemokine receptor deletion. J Neurosci Res. 2002;68:691–702. doi: 10.1002/jnr.10269. [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE) Semin Immunol. 2003;15:23–32. doi: 10.1016/s1044-5323(02)00125-2. [DOI] [PubMed] [Google Scholar]
- Mahajan SD, Aalinkeel R, Reynolds JL, Nair BB, Fernandez SF, Schwartz SA, Nair MP. Morphine exacerbates HIV-1 viral protein gp120 induced modulation of chemokine gene expression in U373 astrocytoma cells. Curr HIV Res. 2005;3:277–288. doi: 10.2174/1570162054368048. [DOI] [PubMed] [Google Scholar]
- Malik AA, Radhakrishnan N, Reddy K, Smith AD, Singhal PC. Morphine-induced macrophage apoptosis modulates migration of macrophages: use of in vitro model of urinary tract infection. J Endourol. 2002;16:605–610. doi: 10.1089/089277902320913314. [DOI] [PubMed] [Google Scholar]
- Mankowski JL, Queen SE, Clements JE, Zink MC. Cerebrospinal fluid markers that predict SIV CNS disease. J Neuroimmunol. 2004;157:66–70. doi: 10.1016/j.jneuroim.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- McManus CM, Weidenheim K, Woodman SE, Nunez J, Hesselgesser J, Nath A, Berman JW. Chemokine and chemokine-receptor expression in human glial elements: induction by the HIV protein, Tat, and chemokine autoregulation. Am J Pathol. 2000;156:1441–1453. doi: 10.1016/S0002-9440(10)65013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A. Pathobiology of human immunodeficiency virus dementia. Semin Neurol. 1999;19:113–127. doi: 10.1055/s-2008-1040830. [DOI] [PubMed] [Google Scholar]
- Oh JW, Schwiebert LM, Benveniste EN. Cytokine regulation of CC and CXC chemokine expression by human astrocytes. J Neurovirol. 1999;5:82–94. doi: 10.3109/13550289909029749. [DOI] [PubMed] [Google Scholar]
- Peterson PK, Hu S, Salak-Johnson J, Molitor TW, Chao CC. Differential production of and migratory response to beta chemokines by human microglia and astrocytes. J Infect Dis. 1997;175:478–481. doi: 10.1093/infdis/175.2.478. [DOI] [PubMed] [Google Scholar]
- Petito CK, Roberts B, Cantando JD, Rabinstein A, Duncan R. Hippocampal injury and alterations in neuronal chemokine co-receptor expression in patients with AIDS. J Neuropathol Exp Neurol. 2001;60:377–385. doi: 10.1093/jnen/60.4.377. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, Estes ML, Thomas DM, Tuohy VK. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J. 1993;7:592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]
- Rogers TJ, Peterson PK. Opioid G protein-coupled receptors: signals at the crossroads of inflammation. Trends Immunol. 2003;24:116–121. doi: 10.1016/s1471-4906(03)00003-6. [DOI] [PubMed] [Google Scholar]
- Sevigny JJ, Albert SM, McDermott MP, McArthur JC, Sacktor N, Conant K, Schifitto G, Selnes OA, Stern Y, McClernon DR, Palumbo D, Kieburtz K, Riggs G, Cohen B, Epstein LG, Marder K. Evaluation of HIV RNA and markers of immune activation as predictors of HIV-associated dementia. Neurology. 2004;63:2084–2090. doi: 10.1212/01.wnl.0000145763.68284.15. [DOI] [PubMed] [Google Scholar]
- Simpson J, Rezaie P, Newcombe J, Cuzner ML, Male D, Woodroofe MN. Expression of the beta-chemokine receptors CCR2, CCR3 and CCR5 in multiple sclerosis central nervous system tissue. J Neuroimmunol. 2000;108:192–200. doi: 10.1016/s0165-5728(00)00274-5. [DOI] [PubMed] [Google Scholar]
- Singh KK, Ellis RJ, Marquie-Beck J, Letendre S, Heaton RK, Grant I, Spector SA. CCR2 polymorphisms affect neuropsychological impairment in HIV-1-infected adults. J Neuroimmunol. 2004;157:185–192. doi: 10.1016/j.jneuroim.2004.08.027. [DOI] [PubMed] [Google Scholar]
- Singhal PC, Shan ZH, Garg P, Sharma K, Sharma P, Gibbons N. Morphine modulates migration of monocytes. Nephron. 1996;73:526–531. doi: 10.1159/000189135. [DOI] [PubMed] [Google Scholar]
- Stiene-Martin A, Hauser KF. Glial growth is regulated by agonists selective for multiple opioid receptor types in vitro. J Neurosci Res. 1991;29:538–548. doi: 10.1002/jnr.490290415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiene-Martin A, Knapp PE, Martin KM, Gurwell JA, Ryan S, Thornton SR, Smith FL, Hauser KF. Opioid system diversity in developing neurons, astroglia, and oligodendroglia in the subventricular zone and striatum: impact on gliogenesis in vivo. Glia. 2001;36:78–88. [PMC free article] [PubMed] [Google Scholar]
- Stiene-Martin A, Zhou R, Hauser KF. Regional, developmental, and cell cycle-dependent differences in μ, δ, and κ–opioid receptor expression among cultured mouse astrocytes. Glia. 1998;22:249–259. [PMC free article] [PubMed] [Google Scholar]
- Szabo I, Wetzel MA, Zhang N, Steele AD, Kaminsky DE, Chen C, Liu-Chen LY, Bednar F, Henderson EE, Howard OM, Oppenheim JJ, Rogers TJ. Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J Leukoc Biol. 2003;74:1074–1082. doi: 10.1189/jlb.0203067. [DOI] [PubMed] [Google Scholar]
- Zuurman MW, Heeroma J, Brouwer N, Boddeke HW, Biber K. LPS-induced expression of a novel chemokine receptor (L-CCR) in mouse glial cells in vitro and in vivo. Glia. 2003;41:327–336. doi: 10.1002/glia.10156. [DOI] [PubMed] [Google Scholar]