

Abstract
Most GISTs contain KIT or PDGFRA kinase gain-of-function mutations, and therefore respond clinically to imatinib and other tyrosine kinase inhibitor (TKI) therapies. However, clinical progression subsequently results from selection of TKI-resistant clones, typically containing secondary mutations in the KIT kinase domain, which can be heterogeneous between and within GIST metastases in a given patient. TKI-resistant KIT oncoproteins require HSP90 chaperoning and are potently inactivated by HSP90-inhibitors, but clinical applications in GIST patients are constrained by the toxicity resulting from concomitant inactivation of various other HSP90 client proteins, beyond KIT and PDGFRA. To identify novel targets responsible for KIT oncoprotein function, we performed parallel genome-scale short hairpin RNA (shRNA)-mediated gene knockdowns in KIT-mutant GIST-T1 and GIST882. GIST cells were infected with a lentiviral shRNA pooled library targeting 11 194 human genes, and allowed to proliferate for 5~7 weeks, at which point assessment of relative hairpin abundance identified the HSP90-cofactor, CDC37, as one of the top six GIST-specific essential genes. Validations in treatment-naïve (GIST-T1, GIST882) vs. imatinib-resistant GISTs (GIST48, GIST430) demonstrated that: 1) CDC37 interacts with oncogenic KIT; 2) CDC37 regulates expression and activation of KIT and downstream signaling intermediates in GIST; and 3) unlike direct HSP90 inhibition, CDC37 knockdown accomplishes prolonged KIT inhibition (>20 days) in GIST. These studies highlight CDC37 as a key biologic vulnerability in both imatinib-sensitive and imatinib-resistant GIST. CDC37 targeting is expected to be selective for KIT/PDGFRA and a subset of other HSP90 clients, and thereby represents a promising strategy for inactivating the myriad KIT/PDGFRA oncoproteins in TKI-resistant GIST patients.
Keywords: CDC37, HSP90, GIST, targeted therapy, functional genomics, shRNA library
INTRODUCTION
Gastrointestinal stromal tumor (GIST) is the most common mesenchymal tumor of the gastrointestinal tract. Gain-of-function mutation of the receptor tyrosine kinases (RTKs) KIT or PDGFRA is a crucial oncogenic event in most GISTs, detectable in 85-90% of cases.1,2 Therapeutic targeting of the KIT/PDGFRA oncoproteins with imatinib enables clinical responses in 80% of inoperable GIST patients 3 and has also improved progression-free survival as an adjuvant treatment postoperatively in patients with high-risk localized GIST.4
Although dramatic therapeutic responses to imatinib are the norm in patients with inoperable GIST, clinical complete responses are rare (≤5%) and up to 90% of responding patients eventually develop secondary resistance, with median time to progression of two years with first-line TKI therapy.5,6 Clinical progression during imatinib therapy occurs typically at multiple metastatic sites due to a variety of molecular imatinib-resistance mechanisms of which KIT/PDGFRA kinase domain secondary mutations in the ATP-binding pocket or activation loop regions are the most frequent.7 Less common imatinib-resistance mechanisms include KIT gene amplification, and activation of kinases downstream of KIT/PDGFRA.8,9 Notably, at time of progression on imatinib there can be substantial heterogeneity in these molecular resistance mechanisms within and between metastases in an individual patient. The multikinase inhibitor, sunitinib, is the only currently approved therapy for advanced GIST following resistance to imatinib; although sunitinib is a potent inhibitor of imatinib-resistance caused by mutations in the KIT ATP-binding pocket, this therapy is less effective against imatinib-resistance mutations affecting the KIT kinase activation loop.10 Therefore, treatment of the entire spectrum of imatinib-resistance mutations, particularly those encoded by KIT exons 17 and 18, as well as PDGFRA exon 18 remains an urgent unmet medical need in GIST.
The extreme dependence of GIST cells upon KIT/PDGFRA activation is a striking example of oncogene addiction11, in which adaptations are required to optimize and stabilize the essential KIT/PDGFRA oncoproteins, creating secondary dependencies on proteins that are requisite for KIT/PDGFRA transforming activity. One such biologic dependency is HSP90 chaperoning, required for folding, localization and stabilization of the mutant KIT/PDGFRA oncoproteins in GIST.12 Preclinical validations have shown compelling responses to HSP90 inhibition in GIST, in vitro and in vivo: after HSP90 inhibition by a variety of compounds, KIT oncoproteins are rapidly degraded, with antiproliferative and pro-apopotic consequences.12,13 However, clinical translation of HSP90 targeting has been challenging, presumably because inhibition of HSP90 also targets HSP90-dependent, non-oncogenic, client proteins limiting the tolerance to chronic and potent HSP90 inhibition. These considerations, together with issues relating to trial design and patient selection criteria, might explain why initial clinical trials of ansamycin-analogue HSP90 inhibition in GIST – although showing evidence of biological activity – have had low response rates accompanied by toxicity.14 To identify candidate targets that might have greater specificity for KIT-mutant GIST, we undertook a genome-scale functional screen in which stable gene knockdowns were achieved by RNA interference.
RESULTS AND DISCUSSION
shRNA pooled library screen performance in GIST-T1 and GIST882 cells
Reproducibility of shRNA enrichment and depletion profiles was evaluated across the experimental replicates, as a quality control. The GIST882 and GIST-T1 screen replicates clustered closely within each cell line by both unsupervised and consensus clustering of shRNA depletion and enrichment profiles, attesting to the robustness of the screens. In addition, the GIST882 and GIST-T1 replicates clustered next to each other when compared to a reference dataset of 12 publicly available cancer cell line datasets,15 as well as after linkage analysis in the context of additional unpublished sarcoma cell lines (Suppl Fig. 1). A comprehensive list of the 11 194 ranked genes, along with annotation for the shRNA clones, is provided in Supplementary Table 1.
CDC37 is essential for GIST cell survival
In the pooled proliferation screens, cells carrying shRNAs that targeted proliferation-essential genes were depleted from the cell population over time. Scored according to the second best-scoring shRNA within each hairpin set, 25 out of 56 genes ranked in the top 0.5% of the distribution for both GIST882 and GIST-T1 (Table 1, left column). Of these 25 genes, 19 were also within the top 0.5% in at least 8 of 12 comparison non-GIST cancer lines15, and were thus identified as “commonly essential” genes not specific to GIST (Figure 1A). These genes were in functional categories known to be essential in cancer cell lines: regulation of mRNA splicing and processing, protein translation, and ribosome and proteasome structure and function. The other six genes were selectively essential for the two GIST cell lines versus the other lines (bold italic font, Table 1 left column): five of these encode mRNA processing proteins, whereas the remaining gene, CDC37, encodes an HSP90 co-factor. CDC37 is known to coordinate HSP90 chaperoning activity for a subset of HSP90 client proteins, including several kinases,17,18 by mechanisms involving CDC37 homodimerization, CDC37-HSP90 heterodimerization, and the formation of CDC37-kinase-HSP90 complexes.19 These observations suggest that CDC37 targeting might be a selective approach to derailing HSP90-mediated KIT oncoprotein chaperoning in GIST. The KIT oncogenic driver and the GIST-lineage-related transcription factor ETV1 also scored as essential genes in these primary screens and serve as positive controls (Figure 1B). In GIST-T1 cells, only one out of the five shRNAs targeting KIT was highly depleted, so KIT did not rank highly in the essential genes list; however, subsequent experiments showed that only the strongly depleted shRNA was highly effective at suppressing KIT in these cells (~70% knockdown) whereas the other four shRNAs produced <30% knockdown of KIT (Suppl. Fig. 2).
Table 1.
Top 0.5% essential genes according to the second best scoring hairpin in GIST882 and GIST-T1 (n=56 for each). Genes in bold font (upper fields) scored top 0.5% in the gene distribution in GIST lines but not in 12 non-GIST reference cancer cell lines of various lineages (described by Luo et al.15). Genes in regular font (lower fields) scored top 0.5% in both GIST and the 12 non-GIST reference set.
| GIST882 and GIST-T1 | GIST882 only | GIST-T1 only |
|---|---|---|
| CDC37 | UBC | PSMC4 |
| POLE | DYNC1H1 | EFTUD2 |
| RPS18 | VCP | PSMA2 |
| PSMA3 | HNRPK | PRPF3 |
| SFRS3 | PABPN1 | NCBP2 |
| EIF3S10 | SNRPD1 | ABCB7 |
| RPS13 | PSMC1 | EIF3S3 |
| RPS29 | FRAP1 | ATPBD1C |
| RPL23A | AFG3L2 | RPAP1 |
| PSMB2 | LOC375133 | RBM8A |
| RPL5 | PSMA6 | DDX48 |
| RPS9 | EIF3S5 | RNPS1 |
| RPS17 | TSG101 | COPS2 |
| RPL31 | PTPRCAP | RPS10 |
| ARCN1 | TUBB | MLXIP |
| PHB2 | RPS19 | ATP1A1 |
| RPS7 | KARS | POLR2F |
| RPS27A | SNRPD2 | RUVBL2 |
| RPS8 | PSMA1 | NUP205 |
| RPS15A | RPS14 | MKI67IP |
| PSMD1 | HNRPU | RAN |
| U2AF2 | RPS6 | RPS3A |
| CHD4 | RPS3 | RPS4X |
| AQR | ASCC3L1 | RPS11 |
| NHP2L1 | RPL34 | RPL7 |
| NDUFA4L2 | RPL6 | |
| RPSA | EIF5B | |
| USP39 | EIF2S2 | |
| PHB | SNRPE | |
| EIF1AX | HSPE1 | |
| TPR | U2AF1 | |
Figure 1. Primary shRNA pooled screen.

Development and applications of the 54K lentiviral shRNA pooled library from The RNAi Consortium (TRC) have been described previously.16 In brief, GIST cells were infected with a pool of 54 020 viruses targeting 11 194 genes and subjected to puromycin selection. Replicates of 20 million infected GIST-T1 and GIST882 cells were established after the infections and allowed to proliferate independently for 6-to-7 weeks. Genomic DNA was isolated from final harvests of cultured cells for shRNA amplification and massively-parallel sequencing as described previously.16 The 54 020 shRNAs were ranked by their relative depletion from the cell pool, and the corresponding 11 194 genes were then scored according to the rank of the second-most depleted shRNA (out of ~5 shRNAs targeting each gene), using the GENE-E program (http://www.broadinstitute.org/cancer/software/GENEE/download.html). (A) Most of the top 0.5% essential genes for GIST882 and GIST-T1 were commonly essential genes, based on their ranks in at least 8 of 12 non-GIST cancer cell lines of various lineages. However, six genes, including CDC37, were selectively essential in GIST882 and GIST-T1 compared to the non-GIST lines. (B) Gene ranks (red) and shRNA ranks (black) corresponding to CDC37, KIT, and ETV1 in GIST882 cells. Essential genes (oncogenes) rank on the top of the distribution.
CDC37 interacts with KIT in GIST, maintaining KIT expression and cell survival
CDC37 expression was demonstrated by immunoblotting in the GIST cell lines (Figure 2). Interaction between CDC37 and KIT in these GISTs was demonstrated by coimmunoprecipitations using KIT and CDC37 antibodies in KIT-positive GIST882 and GIST-T1 cells, with KIT-negative GIST48B cells serving as a negative control (Figure 2). CDC37 shRNA-mediated knockdowns resulted in >90% reduction of KIT expression and activation in the KIT-dependent GIST882, GIST430, and GIST-T1 lines (Figure 3A). KIT inhibition was associated with inactivation of downstream growth and survival signaling intermediates, including AKT. By contrast, AKT was not inhibited by CDC37 knockdown in the KIT-negative cell line GIST48B, suggesting that the observed inhibition of downstream signaling pathways in GIST882, GIST430, and GIST-T1 was KIT dependent (Figure 3A). CDC37 knockdown, unlike direct HSP90 inhibition21, resulted in persistent inhibition of KIT expression for more than 20 days, indicating that GIST cells have few compensatory pathways for CDC37 function. The abovementioned biochemical responses to CDC37 knockdown were accompanied by decreased GIST proliferation and survival as assessed by bright field microscopy and CellTiter-Glo assays (Figures 3B and 3C). Notably, these responses were also seen in GIST lines resistant to imatinib (GIST430) or to the ansamycin HSP90 inhibitor 17-AAG (GIST-T1/AAG and GIST882/AAG). CDC37 knockdown induced an increased sub-G0 cell cycle peak, consistent with a pro-apoptotic effect (Figure 3D).
Figure 2.
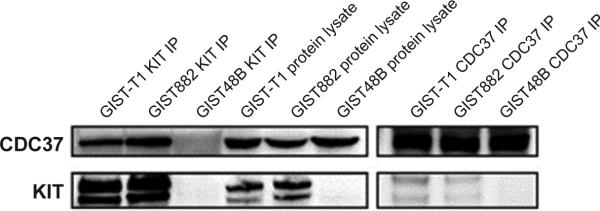
CDC37 expression and CDC37-KIT coimmunoprecipitations in GIST cell lines, demonstrating CDC37:KIT interaction. Whole cell lysates, electrophoresis and immunoblotting were carried out as described previously.20
Figure 3.

Persistent inhibition of KIT oncoprotein expression and phosphorylation, and decreased downstream signaling pathway activation in KIT-dependent GIST cells upon shRNA-mediated CDC37 knockdown. (A) Lentiviral constructs encoding shRNA specific sequences targeting CDC37 transcripts on the pLKO.1puro backbone were selected from the TRC library (TRCN0000116632: clone 1; and TRCN0000116633: clone 2; the TRC website is http://www.broadinstitute.org/rnai/trc/lib). Lentivirus preparations and lentiviral infections were performed as described previously.23 Cells were lysed for immunoblot analysis at 4, 10 and 20 days post-infection; whole cell lysates, electrophoresis and immunoblotting were carried out as described previously.20 (B) shRNA-mediated CDC37 knockdown inhibits viability of GISTs, including those resistant to imatinib (GIST430) and to the ansamycin-type HSP90-inhibitor 17-AAG (GIST882/AAG and GIST-T1/AAG). For cell viability evaluations by bright field microscopy, cell images were obtained using SPOT software (SPOT Imaging Solutions, Sterling Heights, MI) and a Nikon Eclipse TE2000-5 inverted microscope. For viability evaluations by ATP-based CellTiter-Glo luminescent assay (Promega, Madison, WI) (C), luciferase-catalyzed luciferin/ATP reactions were measured with a Veritas Microplate Luminometer (Turner Biosystems, Sunnyvale, CA) and normalized to the day 0 and pLKO control reads. (D) Increase in the number of cells in sub-G0 phase of the cell cycle (flow cytometry analysis of propidium iodide-stained DNA content as previously described), 12 10 days after lentiviral infection and puromycin selection.
shRNA-mediated CDC37 knockdown in GIST-T1 cells resulted in decreased tumor growth in vivo, with decreased tumor volume, overall decreased cellularity and decreased mitotic activity in mouse xenografts (Figure 4).
Figure 4.

shRNA-mediated CDC37 knockdown inhibits growth of GIST xenografts in mice. Athymic nude mice were injected subcutaneously with GIST-T1 cells expressing CDC37-targeting shRNA in one flank (shRNA1: TRCN0000116632; shRNA 2: TRCN0000116633; n=3 each) and empty pLKO.1 lentiviral vector in the other flank (n=3). 2×106 infected cells on puromycin selection were resuspended in BD Matrigel and implanted subcutaneously at each injection site. Tumor volume was evaluated weekly. Mice were euthanized by CO2 inhalation and necropsied six weeks after injection. (A, B) Tumors were resected and photographed, demonstrating significantly decreased tumor growth upon CDC37 knockdown. (C) Western blot confirming inhibition of CDC37 expression in GIST-T1 cells infected with CDC37 shRNA1 and shRNA2, compared to pLKO.1 lentiviral vector. (D) Histologic evaluation of formalin-fixed and paraffin-embedded (FFPE) samples after hematoxylin and eosin staining demonstrates sparsely cellular areas with no mitotic activity in CDC37-knockdown GIST xenografts, in comparison to highly cellular and mitotically active xenografts of pLKO.1 lentiviral infected cells.
Celastrol does not enable selective CDC37:HSP90 pharmacologic inhibition
Preclinical pharmacologic validations were attempted using the HSP90:CDC37 interface inhibitor celastrol.22 However, celastrol nonselectively inhibited viability of both KIT-dependent and KIT-independent (GIST48B) GIST cell lines (Suppl Fig. 3A), and celastrol did not result in dramatic dose-dependent inhibition of KIT expression or KIT activation, despite reduced AKT and MAPK phosphorylation (Suppl Fig. 3B). These findings underscore the protean effects of celastrol, which are reported to include proteasome inhibition,22 and which will likely preclude achieving CDC37:HSP90-inhibitory drug concentrations in the clinical setting. We therefore expect that effective clinical translation of the concepts in this report will require development of more selective CDC37 inhibitors. Opportunities for more selective CDC37 targeting might result from pharmacologic dysregulation of phosphorylation at CDC37 Ser13. This phosphorylation event – mediated by casein kinase 2 (CK2) – is requisite for CDC37 recruitment of kinase clients to the HSP90 complex, whereas CDC37 Ser13 dephosphorylation – mediated by protein phosphatase 5 (PP5) –is then necessary for priming CDC37 for tyrosine phosphorylation by YES1, ultimately enabling CDC37 and client release from the complex.19 Therefore, strategies targeting CK2, PP5, or YES1 might inhibit KIT-directed HSP90 functions in GIST.
In summary, we have performed unbiased genome-wide functional screens which identify CDC37 as a compelling therapeutic target in GIST. CDC37 interacts with KIT oncoproteins in GIST and is an essential enabler of KIT oncogenic function. Preclinical validations highlight the selectivity and efficacy of targeting KIT through CDC37 in GIST, with advantages compared to direct HSP90 targeting including persistent down-regulation of KIT expression and selectivity for only a subset of HSP90 client proteins. These observations suggest that selective targeting of CDC37 might be effective and less toxic than chronic HSP90 inhibition in GIST patients.
Supplementary Material
Acknowledgments
Financial support: Adrián Mariño-Enríquez, Wen-Bin Ou, Yuexiang Wang, Jonathan A. Fletcher and George D. Demetri, are supported by the GI SPORE 1P50CA12703-05, Virginia and Daniel K. Ludwig Trust for Cancer Research, Paul's Posse and Team Cesarini of the Pan Mass Challenge, LifeRaft Group and the GIST Cancer Research Fund. Adrián Mariño-Enríquez is also supported by a Sarcoma Alliance for Research Through Collaboration (SARC) Career Development Award. Wen-Bin Ou is also supported by Qianjiang Talents Project of Zhejiang (2012R10079), a grant from the Science and Technology Bureau of Jiaxing, Zhejiang (2012AY1039) and the Major Science and Technology Special Project of Zhejiang Province (2012C03007-4). The RNAi Consortium (TRC) supported the development of pooled screening methods used for these screens.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
Conflict of interest: Dr. George D. Demetri serves as consultant for Novartis, Pfizer, Sanofi-Aventis , Glaxo-Smith-Kline, Johnson & Johnson, Merrimack Pharma, Foundation Medicine, Merck, ZioPharm, N-of-One, Champions Biotechnology, Blueprint Medicines; he is a member of the Scientific Advisory Boards of ZioPharm, Kolltan Pharmaceuticals, N-of-One, and Blueprint Medicines; and he holds minor equity stakes at N-of-One, Champions Biotechnology, Kolltan Pharmaceuticals and Blueprint Medicines. Novartis, Pfizer, Sanofi-Aventis, Glaxo-Smith-Kline, Johnson & Johnson, Merck, and Amgen support clinical trials in the Center for Sarcoma and Bone Oncology, Dana-Farber Cancer Institute. Dr. Jonathan A. Fletcher has consulting arrangements with Novartis, Pfizer and Deciphera Pharmaceuticals. None of these relationships constitute a conflict of interest for this work. Adrián Mariño-Enríquez, Wen-Bin Ou, Glenn Cowley, Biao Luo, Anneliene H. Jonker, Mark Mayeda, Michael Okamoto, Grant Eilers, Jeffrey Czaplinski, Ewa Sicinska, Yuexiang Wang, Takahiro Taguchi and David E. Root have no potential conflicts of interest to declare.
REFERENCES
- 1.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–80. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 2.Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–10. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 3.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 4.DeMatteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, Demetri GD, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373(9669):1097–104. doi: 10.1016/S0140-6736(09)60500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: Randomised trial. Lancet. 2004;364(9440):1127–34. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 6.Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24(29):4764–74. doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- 7.Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11(11):4182–90. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 8.Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216(1):64–74. doi: 10.1002/path. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agaram NP, Wong GC, Guo T, Maki RG, Singer S, DeMatteo RP, et al. Novel V600E BRAF Mutations in Imatinib-Naive and Imatinib-Resistant Gastrointestinal Stromal Tumors. Genes Chromosomes Cancer. 2008;47(10):853–9. doi: 10.1002/gcc.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–9. doi: 10.1200/JCO.2007.15.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antonescu CR. The GIST paradigm: Lessons for other kinase-driven cancers. J Pathol. 2011;223(2):251–61. doi: 10.1002/path.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006;66(18):9153–61. doi: 10.1158/0008-5472.CAN-06-0165. [DOI] [PubMed] [Google Scholar]
- 13.Smyth T, Van Looy T, Curry JE, Rodriguez-Lopez AM, Wozniak A, Zhu M, et al. The HSP90 Inhibitor, AT13387, Is Effective against Imatinib-Sensitive and -Resistant Gastrointestinal Stromal Tumor Models. Mol Cancer Ther. 2012 doi: 10.1158/1535-7163.MCT-11-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demetri GD, Le Cesne A, Von Mehren M, Chmielowski B, Bauer S, Chow WA, et al. Final results from a phase III study of IPI-504 (retaspimycin hydrochloride) versus placebo in patients (pts) with gastrointestinal stromal tumors (GIST) following failure of kinase inhibitor therapies. [abstract]. 2010. Proceedings of the Gastrointestinal Cancers Symposium; Alexandria, VA. 2010 Jan 22; American Society of Clinical Oncology; [Google Scholar]
- 15.Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci U S A. 2008;105(51):20380–5. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A. 2011;108(30):12372–7. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaughan CK, Mollapour M, Smith JR, Truman A, Hu B, Good VM, et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of CDC37. Mol Cell. 2008;31(6):886–95. doi: 10.1016/j.molcel.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith JR, Workman P. Targeting CDC37: An alternative, kinase-directed strategy for disruption of oncogenic chaperoning. Cell Cycle. 2009;8(3):362–72. doi: 10.4161/cc.8.3.7531. [DOI] [PubMed] [Google Scholar]
- 19.Xu W, Mollapour M, Prodromou C, Wang S, Scroggins BT, Palchick Z, et al. Dynamic Tyrosine Phosphorylation Modulates Cycling of the HSP90-P50(CDC37)-AHA1 Chaperone Machine. Mol Cell. 2012:47. doi: 10.1016/j.molcel.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61(22):8118–21. [PubMed] [Google Scholar]
- 21.Chandarlapaty S, Sawai A, Ye Q, Scott A, Silinski M, Huang K, et al. SNX2112, a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER kinase-dependent cancers. Clin Cancer Res. 2008;14(1):240–8. doi: 10.1158/1078-0432.CCR-07-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D. Characterization of celastrol to inhibit HSP90 and CDC37 interaction. J Biol Chem. 2009;284(51):35381–9. doi: 10.1074/jbc.M109.051532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee CH, Ou WB, Marino-Enriquez A, Zhu M, Mayeda M, Wang Y, et al. 14-3-3 fusion oncogenes in high-grade endometrial stromal sarcoma. Proc Natl Acad Sci U S A. 2012;17(3):929–34. doi: 10.1073/pnas.1115528109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taguchi T, Sonobe H, Toyonaga S, Yamasaki I, Shuin T, Takano A, et al. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab Invest. 2002;82(5):663–5. doi: 10.1038/labinvest.3780461. [DOI] [PubMed] [Google Scholar]
- 25.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476(7360):346–50. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.