

Sil1 is a nucleotide exchange factor for the ER chaperone BiP, and mutations in this gene lead to Marinesco–Sjögren syndrome, a multisystem disorder. Effects of loss of Sil1 on biosynthesis and secretion of antibodies, a well-characterized BiP client, are determined in a mouse model for this disease and patient-derived lymphoblastoid cell lines.
Abstract
Sil1 is a nucleotide exchange factor for the endoplasmic reticulum chaperone BiP, and mutations in this gene lead to Marinesco–Sjögren syndrome (MSS), a debilitating autosomal recessive disease characterized by multisystem defects. A mouse model for MSS was previously produced by disrupting Sil1 using gene-trap methodology. The resulting Sil1Gt mouse phenocopies several pathologies associated with MSS, although its ability to assemble and secrete antibodies, the best-characterized substrate of BiP, has not been investigated. In vivo antigen-specific immunizations and ex vivo LPS stimulation of splenic B cells revealed that the Sil1Gt mouse was indistinguishable from wild-type age-matched controls in terms of both the kinetics and magnitude of antigen-specific antibody responses. There was no significant accumulation of BiP-associated Ig assembly intermediates or evidence that another molecular chaperone system was used for antibody production in the LPS-stimulated splenic B cells from Sil1Gt mice. ER chaperones were expressed at the same level in Sil1WT and Sil1Gt mice, indicating that there was no evident compensation for the disruption of Sil1. Finally, these results were confirmed and extended in three human EBV-transformed lymphoblastoid cell lines from individuals with MSS, leading us to conclude that the BiP cofactor Sil1 is dispensable for antibody production.
INTRODUCTION
It has been estimated that one-third of the human genome encodes proteins that will populate the single-membrane-bound organelles of the cell or that will be secreted or expressed at the cell surface. These proteins are translocated into the endoplasmic reticulum (ER) lumen as they are synthesized and often undergo modifications and begin to fold cotranslationally. The proper maturation of these proteins is both assisted and monitored by the resident molecular chaperones of this organelle to prevent off-pathway folding, which might lead to aggregation, and to ensure that only those molecular forms of the newly synthesized proteins that can pass ER quality control measures are permitted to leave the ER for their proper destination (Ellgaard and Helenius, 2003; Braakman and Bulleid, 2011). Until that time, nascent proteins are retained in the ER via their interaction with molecular chaperones, and those proteins that ultimately fail to mature properly are retrotranslocated to the cytosol where they are marked for degradation by the ubiquitin proteasome system. Two major chaperone families exist in the ER—the Hsp70 family member BiP and its cofactors, and the lectin chaperones, calnexin and calreticulin and their attendant cofactors.
Like other Hsp70 family members, BiP is composed of an N-terminal nucleotide-binding domain (NBD) and a C-terminal substrate-binding domain (SBD) that communicate with each other via a linker region. The binding of BiP to substrates is regulated by its nucleotide-bound state (Wei et al., 1995; Hendershot et al., 1996; Marcinowski et al., 2011, 2013). When ATP occupies the NBD, the lid of the SBD is open, allowing ready access to extended regions of nascent proteins or substrates, but this open conformation does not promote stable binding. However, the binding of substrate to the SBD triggers ATP hydrolysis, which in turn induces at least partial closing of the lid over the bound substrate and stabilizes this association (Hendershot et al., 1996; Marcinowski et al., 2013). The substrate does not fold while bound to BiP but is protected from off-pathway intermediates or aggregation. The lid of the SBD must be reopened to release the substrate so it can fold, which requires the bound ADP to be exchanged for ATP. Although three different structurally unrelated groups of nucleotide exchange factors (NEFs) exist in the cytosol of eukaryotic organisms, only two types, each with a single representative, have been identified in the ER: the HSPBP1 orthologue BAP/Sil1 (Kabani et al., 2000; Chung et al., 2002) and the large Hsp70 member Grp170 (Steel et al., 2004; Weitzmann et al., 2006). Whereas Sil1 seems to function only as a NEF, the large Hsp70 proteins, including Grp170, also bind directly to substrates (Lin et al., 1993; Spee et al., 1999; Buck et al., 2013; Behnke and Hendershot, 2014), making their functional activity more complex and less well understood. Structural analysis of yeast Sil1p reveals that it binds to the NBD of BiP and disrupts a hydrogen bond between the two lobes, which results in a rearrangement of the NBD that induces the release of ADP from BiP (Yan et al., 2011). Based on homology mapping with the cytosolic orthologue HspBP1, exons 6 and 9 of Sil1 are predicted to constitute the major interaction site with the NBD of BiP, with exon 10 providing a minor interaction (Senderek et al., 2005).
Mutations in the Sil1 gene have been found in more than half of the cases of Marinesco–Sjögren syndrome (MSS; Anttonen et al., 2005; Senderek et al., 2005), an autosomal recessive disease characterized by multisystem defects, including ataxia due to Purkinje cell loss, cataracts, and progressive myopathy (Sjogren, 1950; Krieger et al., 2013). MSS-associated mutations occur throughout the Sil1 gene and most lead to the disruption of significant portions of the protein (Anttonen et al., 2008; Horvers et al., 2013), although the fate of most of these fragments has not been characterized. In fact, it remains unclear whether the associated pathologies arise from reduced maturation of essential secretory pathway proteins, depletion of BiP stores resulting in activation of the ER stress response, toxicity due to aggregation of secretory pathway proteins, or even perhaps of the mutant Sil1 protein itself. Three MSS-associated mutations have been identified that disrupt only the last four or five amino acids, which were shown to reduce the solubility of the mutant proteins and decrease their stability, leading to significantly diminished expression of mutant Sil1 (Anttonen et al., 2008; Howes et al., 2012). Although a Sil1-null mouse is not available, the gene has been disrupted spontaneously by transposon insertion, Sil1wz, and by gene-trap methodology, Sil1Gt (Zhao et al., 2005). In both cases, the Sil1 gene is disrupted between exons 7 and 8, resulting in loss of amino acids 261–465 of the Sil1 protein. The resulting mice are referred to as woozy mice and have been reported to phenocopy some of the pathologies associated with MSS, including cerebellar degeneration resulting in ataxia (Zhao et al., 2005) and a progressive myopathy (Roos et al., 2014). Examination of cerebella from these mice revealed the presence of protein aggregates and activation of the ER stress response, which is induced by the accumulation of unfolded proteins in this organelle.
To begin to understand the effects of Sil1 gene disruption on secretory pathway protein maturation, we chose to examine the assembly and secretion of immunoglobulins, which are the best studied BiP substrates (Haas and Wabl, 1983; Bole et al., 1986; Hendershot et al., 1987; Lee et al., 1999; Feige et al., 2009). Immunoglobulin (Ig) is a heteromeric protein composed of two heavy chains (HCs) and two light chains (LCs) that are combined in the ER. BiP binds to all the assembly intermediates and prevents their secretion but does not interact with completely assembled complexes (Bole et al., 1986; Hendershot et al., 1987). Coexpression of BiP mutants that cannot be released from Ig heavy chains with ATP prevents the assembly and secretion of antibodies (Hendershot et al., 1996; Vanhove et al., 2001), suggesting that a NEF might be required for this process normally. Finally, we considered that the incredibly large quantities of antibodies produced by plasma cells, which put very high demands on ER homeostasis, would make these cells particularly sensitive to disruptions in the chaperone network. Together these points argue that antibodies would represent a good model to examine the requirements for a nucleotide exchange factor in BiP-mediated folding. The availability of a mouse model with a disrupted Sil1 gene and Epstein–Barr virus (EBV)–transformed B lymphoblastoid cell lines (LBLs) from individuals with MSS provide important biological tools for examining the effect of Sil1 protein loss on antibody assembly and secretion both in vivo and ex vivo, which in addition to establishing the requirement for Sil1 in Ig assembly and secretion, could also shed light on humoral immune function in patients.
RESULTS
Detection of disrupted Sil1 transcripts in woozy mice
The Sil1 gene has been disrupted beyond exon 7 in woozy mice by either a spontaneous insertion of an ETn retrotransposon, Sil1wz, or the insertion of the β-geo gene-trap cassette, Sil1Gt (Zhao et al., 2005). In both cases, exon 7 is spliced to the inserted gene, generating a fusion protein that should possess the N-terminal 260 amino acids (out of a total 465 amino acids) encoded by the Sil1 gene followed by either 32 amino acids of the transposon or a CD4 transmembrane region and a β-geo cassette, respectively (Supplemental Figure S1). In spite of the very different fusion proteins generated in these two woozy mice, the phenotypes appear to be very similar, suggesting that both may lead to a loss of functional Sil1 protein. Of importance, the chimeric product of neither Sil1 disruption has been examined, but a truncated version of Sil1 possessing only the N-terminal 260 amino acids was expressed in COS-1 cells. This mutant is less stable and binds BiP with reduced affinity compared with the wild-type Sil1 protein (Zhao et al., 2005). Because Sil1 expression had not been previously measured in plasmablasts and published reports suggest that levels can vary significantly between various tissues in humans (Chung et al., 2002) and mice (Ittner et al., 2014), we first examined Sil1 transcript levels in day 4 lipopolysaccharide (LPS)–stimulated splenic B cells from wild-type and woozy mice by real-time PCR. Primers were selectively designed to amplify regions that are either shared by the Sil1WT and Sil1Gt transcripts (primer pair 1) or are present exclusively in Sil1WT transcripts (primer pair 2; (Supplemental Figure S1). For reasons that are unclear, we found that Sil1Gt transcripts were present at only one-third of the level of the wild-type Sil1 transcripts. As expected, the Sil1 transcripts from the Sil1Gt mouse could not be amplified with the primer pair extending across exons 9 and 10, demonstrating that the plasmablasts are indeed derived from Sil1Gt homozygous mice as opposed to misgenotyped heterozygous mice. All mice used for immunizations and LPS stimulation assays were genotyped at least three times to ensure the accuracy of the genotypes being used.
Characterization of immune populations in wild-type and woozy mice
We first examined the effects of Sil1 disruption on the distribution of cellular subpopulations present in the spleen and thymus. The relative percentage of CD3+/CD4+ T helper and CD3+/CD8+ cytotoxic T-lymphocytes, B-lymphocytes (B220+), macrophages (Mac1+), granulocytes (Gr1+), and natural killer cells (NK1.1+) were very similar between wild-type and Sil1Gt mice (Figure 1A). Thymocytes were stained for a variety of markers to identify the indicated developmental stages of T-cell populations in the Sil1Gt mouse compared with their wild-type littermates (Figure 1, B and C). We observed no significant differences in the numbers of any of the developmental stages of T-cells. Thus disruption of the Sil1 gene did not appear to affect the maturation or survival of any of the indicated immune populations.
FIGURE 1:
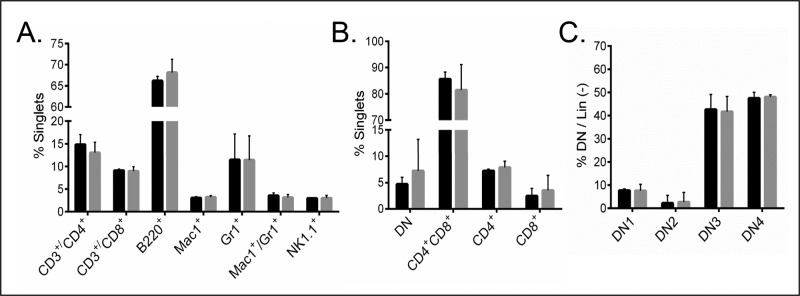
Disruption of Sil1 gene does not affect splenocyte or thymocyte populations. (A) Single-cell splenocytes from wild-type (black bars) and woozy (gray bars) mice were stained with the indicated antibodies, subjected to fluorescence-activated cell sorting analyses, and indicated populations were expressed as percentage of nucleated, singlet splenic cells. (B, C) Thymocytes from wild-type and woozy mice were stained to identify developmental subpopulations and similarly portrayed. Error bars indicate mean ± SD.
Sil1Gt mice produce normal levels of antigen-specific antibodies in response to immunization with a T-dependent antigen
Although the number of mature B- and T-cells in the woozy mouse appeared normal, we examined their ability to respond to immunization with the T-dependent antigen 4-hydroxy-3-nitrophenylacetyl conjugated to keyhole limpet hemocyanin (NP15-KLH). Ten wild-type and 10 woozy mice were immunized and boosted 2 wk later. Sera were obtained at the indicated days postimmunization, and NP15-KLH–specific antibodies were quantified to detect differences in either the magnitude or kinetics of antibody production. We chose to measure three antibody isotypes, including two that assemble via the H → HL → H2L2 pathway (IgM and IgG2b) and one that assembles through H → H2 → H2L → H2L2 intermediates (IgG1). Because heavy chains remain incompletely folded until they associate with light chains (Feige et al., 2009), we reasoned that antibodies assembling through the latter pathway might be more dependent on BiP and as a result show a greater need for functional Sil1. Somewhat surprisingly, we found that there was no significant difference in either the magnitude or kinetics of producing IgM, IgG2b, or IgG1 antibodies to NP15-KLH (Figure 2). This argued that the loss of Sil1 function did not prevent the secretion of antibodies regardless of their route of assembly.
FIGURE 2:
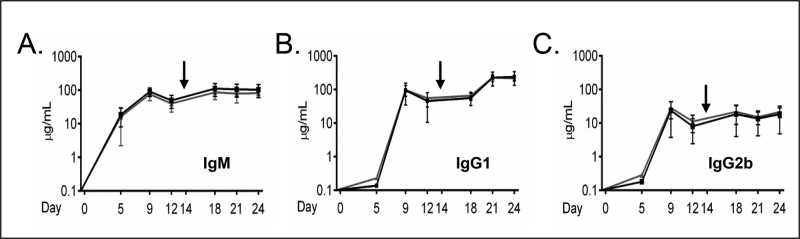
Sil1Gt mice respond to T-dependent antigens similarly to age-matched Sil1WT mice. Sil1WT (black line) and Sil1Gt (gray line) mice were challenged with NP15-KLH as described, and serum was collected at the indicated intervals. Black arrow indicates the booster immunization. Antigen-specific responses comprised of IgM (A), IgG1 (B), and IgG2b (C) were determined by ELISA. Each point displays the median value. Ten Sil1WT and Sil1Gt mice each were immunized and assayed. Error bars indicate interquartile range of values.
LPS-stimulated B cells from Sil1Gt mice synthesize and secrete IgM antibodies in similar amounts compared to wild-type littermates
We next examined Ig synthesis in purified murine splenic B cells stimulated with LPS ex vivo. This allowed us to examine directly Ig secretion over a shorter time span and determine whether unassembled Ig chains accumulated in the woozy splenocytes. Isolated splenic B cells were cultured with LPS, and cells and culture supernatant were isolated every day for 4 d. There was no significant difference in the growth rate of stimulated B cells from woozy mice compared with their wild-type littermates (unpublished data). Western blot analysis of cell lysates revealed robust and equivalent up-regulation of the secretory type of IgM heavy chains (μs) and κ light chain in both wild-type and woozy mice (Figure 3A). When culture supernatants were analyzed for secreted IgM, similar levels of μ heavy and κ light chains were readily detected after 2 d of stimulation and continued to increase throughout the course of the experiment in both cases (Figure 3B).
FIGURE 3:

Disruption of Sil1 gene does not adversely affect IgM biosynthesis or secretion in response to ex vivo LPS stimulation. Isolated splenocytes were cultured with LPS for the indicated times, and lysates corresponding to equivalent numbers of cells were analyzed under reducing (A) or nonreducing (C) conditions, followed by blotting with the indicated immune reagents. Actin served as a control for loading. (B) Culture supernatants corresponding to an equivalent number of cells for each day were immunoprecipitated with anti–μ HC antibody, analyzed by reducing SDS–PAGE, and probed with the indicated antibodies. The center lane contains only the immunoprecipitating antibody (Ppt Ab), which is recognized by the secondary antibody used to identify the mouse κ LC. Its mobility is designated with asterisks on the left.
We determined the assembly status of IgM present in the splenocytes after 3 and 4 d of LPS stimulation on nonreducing gels. Under steady-state conditions, the μ heavy chains in the cells from both wild-type and woozy mice were present predominantly as H2L2 monomers and H2L2 polymers, which are likely to be pentamers, although a polymeric intermediate can be detected with both anti-μ and anti-κ antisera (Figure 3C). Trace amounts of HL hemimers and LC dimers were detected. However, there was no evidence that unassembled or incompletely assembled Ig molecules accumulated in plasmablasts from the woozy mice to a greater extent than observed in the wild-type cells. To assess whether unassembled IgM was aggregating in these cells, we examined the NP-40–insoluble fraction for the presence of IgM. Very small amounts of insoluble κ light chains and μ heavy chains could be detected in the differentiating splenic B cells, but the levels of both were very similar in the two genotypes (Supplemental Figure S2A). Thus we found no evidence that the woozy mice were compromised in their ability to assemble or secrete antibodies, suggesting that Sil1 was not required to release BiP from Ig heavy chains.
Sil1 disruption does not affect chaperone levels in LPS-stimulated B cells
Our examination of day 4 LPS-simulated splenic B cells revealed that Sil1 transcripts were present in wild-type cells and in lesser amounts in the woozy cells (Supplemental Figure S1). Most ER molecular chaperones and resident proteins are normally up-regulated during plasma cell differentiation (Wiest et al., 1990; van Anken et al., 2003). This occurs via activation of a limited unfolded protein response (UPR; Gass et al., 2002; Iwakoshi et al., 2003), which serves to maintain homeostasis in the dramatically expanded ER in preparation for the massive production of antibodies. However, because Sil1 is not induced by the UPR (Chung et al., 2002), we considered the possibility that the Sil1 protein expression remained unchanged during plasma cell differentiation and therefore did not contribute significantly to BiP-mediated folding in these cells. To address this possibility, we examined Sil1 protein levels in the wild-type and woozy mice over the course of LPS-induced differentiation. Although basal levels were quite low in the unstimulated splenic B cells, Sil1 protein became readily detectable over the course of differentiation in the wild-type plasmablasts (Figure 4A). However, when the differentiating woozy splenocytes were examined with this antibody, we were unable to observe a unique band in the region corresponding to the predicted molecular weight for the chimeric Sil1Gt protein (Supplemental Figure S2B), suggesting that it was unstable.
FIGURE 4:

Chaperone levels are unaltered during plasmablast differentiation in Sil1Gt mice. Direct cell lysates were prepared, electrophoresed under reducing conditions as described in Figure 3, and probed with the indicated antibodies to molecular chaperones (A, B) and BiP cofactors (C–E). Because samples were run on different gels, a loading control is included for each. The anti-Sil1 antibody used in A was purchased from Proteintech. The anti-calreticulin antibody (CRT) used in B detected an additional, unknown band only in the unstimulated splenocytes, which is indicated with an asterisk.
The loss of Sil1 function in mouse Purkinje cells leads to activation of the UPR, which results in higher-than-normal levels of some ER chaperones, including Grp170, and eventually to cell death (Zhao et al., 2005). Both of these consequences could be prevented by transgenic overexpression of Grp170 (Zhao et al., 2010), which has both nucleotide exchange (Steel et al., 2004; Weitzmann et al., 2006) and chaperone activity (Park et al., 2003; Behnke and Hendershot, 2014). Thus we sought to determine whether the loss of Sil1 might be compensated by an even greater increase in expression of Grp170 or other chaperones during plasma cell differentiation. When cell lysates from wild-type and woozy LPS-stimulated splenic B cells were examined, the up-regulation of Grp170 and other chaperones like Grp94, calnexin, and calreticulin, all of which have been reported to bind μ heavy chains (Melnick et al., 1992; Lin et al., 1993; David et al., 1993), appeared to be very similar (Figure 4, A and B). Thus, unlike the Purkinje cells of the woozy mouse, there was no compensating overexpression of Grp170 or any other ER chaperone examined.
The ER also possesses a number of DnaJ-like cofactors that serve to bind unfolded protein substrates and deliver them to BiP (Otero et al., 2010). A previous study reported that deletion of one of the ER-localized ERdj proteins in the Sil1Gt mice partially suppressed the Purkinje defects associated with Sil1 disruption (Zhao et al., 2010), perhaps due to decreased transfer of substrates to BiP. Therefore we compared levels of several of the resident ER DnaJ family proteins during differentiation. All three ERdj proteins examined were similarly up-regulated in wild-type mice and their woozy littermates in response to plasma cell differentiation (Figure 4, C–E). This includes ERdj3, which is specific for Ig heavy chains (Shen and Hendershot, 2005), and ERdj5, which also interacts with unassembled Ig heavy chains (our unpublished data). Thus there is no evidence that the loss of Sil1 function is compensated by increasing Grp170 levels or decreasing these ERdj BiP cofactors.
Immunoglobulin synthesis is not relegated to a different chaperone system in Sil1-disrupted splenic B cells during differentiation
Although we saw no evidence that woozy mice compensated for the loss of Sil1 function by abnormally up-regulating Grp170 or altering the expression of other ER resident proteins, it was conceivable that in the absence of Sil1, immunoglobulin synthesis was controlled by another ER chaperone. Of note, Grp94 (Melnick et al., 1992) and Grp170 (Lin et al., 1993; Behnke and Hendershot, 2014) can bind directly to immunoglobulin chains as a chaperone, and the lectin chaperones have also been demonstrated to interact with μ heavy chains (David et al., 1993). To determine whether IgM antibodies were relying on another chaperone for its assembly, we performed pulse-chase experiments on splenic B cells that were stimulated with LPS for 2 d and prepared cell lysates under conditions most conducive to maintaining association of client proteins for each of the chaperones. For BiP and Grp170, lysates were prepared with apyrase or Mg2+-ATP respectively, whereas for Grp94, calnexin, and calreticulin, no supplements were added. When the pulse-labeled samples were examined (Figure 5, A and B), there was no evidence of μ heavy chains coprecipitating with Grp170, Grp94, calnexin, or calreticulin in either genotype above the background observed with protein A agarose, and consequently no μ heavy chains were coisolated with the chaperones immunoprecipitated from chase samples (Figure 5, C and D). This strongly argues that IgM antibodies were not utilizing one of these chaperones in the Sil1Gt cells. The fact that μ heavy chains and BiP migrate identically on these gels made it impossible to determine whether the heavy chains were interacting with BiP in both genetic backgrounds, although the signal in the BiP-immunoprecipitated material appeared slightly sharper and less strong in the chase sample compared with the pulse. To determine whether μ heavy chains were interacting with BiP in the Sil1Gt cells, we performed immunoprecipitation coupled to Western blotting assays. We found the secretory form of μ (μs) coimmunoprecipitated with BiP, and correspondingly BiP was coisolated with anti-μ precipitated material in plasmablasts from both wild-type and woozy mice (Supplemental Figure S3). Thus, although Sil1 was dispensable for IgM production, it was not because BiP was not used during Ig assembly in the Sil1Gt cells. Together these data demonstrate there is no evidence that antibodies are dispatched to a different chaperone system when Sil1 is not available to release BiP from Ig heavy chains, nor do unassembled heavy chains remain bound to BiP in these cells.
FIGURE 5:

Loss of Sil1 does not redirect IgM assembly to another chaperone system. LPS- stimulated splenic B cells from day 2 were pulse labeled with [35S]methionine and cysteine and chased for 0 or 1 h. (A, C) Aliquots of the cells were lysed with apyrase (lanes 1–3 and 8–10), without apyrase (lanes 4–7 and 11–14), or (B, D) with the addition of Mg2+-ATP (lanes 1–3 and 5–7). The lysates and media (m) were immunoprecipitated with the indicated antibodies and analyzed by reducing SDS–PAGE. The migration of these chaperones is denoted with the following symbols: BiP (*), Grp94 (§), calreticulin (‡), and calnexin (#); mobility of heavy and light chains is indicated.
Antibody assembly and secretion are normal in lymphoblastoid lines from individuals with MSS
We were surprised to find that there was no aspect of antibody production in vivo or ex vivo that was affected by Sil1 disruption, even though these woozy mice have profound defects in a variety of other tissues (Zhao et al., 2010; Roos et al., 2014; Ittner et al., 2014). Because the immune systems of mice and humans do not always function identically (Conley et al., 2000), we obtained lymphoblastoid lines produced from three related individuals with MSS and two nonaffected relatives (Ezgu et al., 2014). In sequencing genomic DNA to verify the alteration of the Sil1 gene, we discovered a novel mutation in the 5′ GT-rich region of the splice donor site of intron 9 (c.1029+6T>C) in addition to the previously reported c.1030-18G>A mutation, 18 bases upstream of exon 10 (Ezgu et al., 2014; Supplemental Figure S4A). Sequencing of cDNA confirmed that this resulted in aberrant splicing of exon 9 causing a frameshift after Lys-343 and a premature stop 13 amino acids later (Supplemental Figure S4B). Western blot analyses of these lines revealed that all three of the MSS cell lines produced both heavy and light chains, which would allow us to examine Ig assembly and secretion (Supplemental Figure S4C). Using both our polyclonal antiserum raised to the full-length Sil1 protein and an antibody that was raised to a region of Sil1 that is retained in the predicted MSS protein, we were unable to detect the mutant Sil1 protein under steady-state conditions in any of the MSS lines (Supplemental Figure S5A), suggesting that the mutant protein is either not readily translated or is unstable. To test this possibility, we incubated a control and MSS patient–derived lymphoblastoid cell line with and without MG132 to prevent proteasomal degradation. A faint band corresponding to the predicted size of the Sil1 mutant protein could be detected only after inhibiting degradation, indicating that the truncated MSS-associated protein was unstable (Supplemental Figure S5B). There was no visible change in the expression of Grp170, BiP, or calreticulin (Supplemental Figure S4D), which is in keeping with our data from the LPS-stimulated murine plasmablasts (Figure 4). To examine Ig assembly and secretion, we chose MSS lines #1 and #2, which both produced IgG antibodies that assemble through the H → H2 → H2L → H2L2 pathway, as well as one of the control lines that used the same Ig assembly pathway. Pulse-chase experiments revealed that both MSS cell lines were able to readily assemble and secrete IgG antibodies and that there was no evidence of incompletely assembled precursor molecules accumulating in the cells (Figure 6). Together these data indicate that Sil1 protein/function is not required for BiP release from antibodies during their production.
FIGURE 6:

B lymphoblastoid cell lines produced from individuals with MSS synthesize and secrete IgG antibodies normally. One control and two LBL lines from MSS individuals that produce IgG were pulse labeled with [35S]methionine and cysteine and chased for the indicated times. Cell lysates were immunoprecipitated with anti–γ heavy chain and electrophoresed under reducing (A) or nonreducing (C) conditions. Culture supernatants were similarly immunoprecipitated and analyzed under reducing conditions (B).
DISCUSSION
Sil1 is not required for Ig assembly or secretion
Mutations in Sil1 in individuals with MSS results in multiple system defects (Superneau et al., 1987; Torbergsen et al., 1991; Horvers et al., 2013; Barends et al., 2013), many of which are phenocopied by disrupting this gene in mice (Zhao et al., 2005; Roos et al., 2014). To begin to understand the consequences of losing Sil1 function on the release of substrates from BiP, we turned to immunoglobulins, which can be studied in vivo and ex vivo, allowing greater scrutiny of the process. Surprisingly, we found no evidence that B-cell development was affected, nor was there a decrease in either the kinetics or magnitude of antibodies produced in response to antigen stimulation in vivo or LPS stimulation of purified splenic B cells for any isotype analyzed. This was not due to the fact that plasma cells do not make Sil1 protein, as we observed increases in this NEF during the course of differentiation in wild-type plasmablasts, and lymphoblastoid lines from control individuals had readily detectable amounts of Sil1. We did not detect any unique changes in the complement of ER chaperones induced during plasma cell differentiation or any evidence that another chaperone system was used to support antibody synthesis in the absence of Sil1. Contrary to our findings, one recent report on an individual with MSS noted decreased levels of serum IgG2 and IgA but not IgM (Hasegawa et al., 2014). However, based on the fact that recurrent infections are not included in common symptoms experienced by MSS patients (Krieger et al., 2013) and on our data on normal Ig production in LBL cell lines from affected individuals, it seems more likely that the reduced levels of Ig in this patient were unrelated to the loss of Sil1 function.
How is BiP released from Ig subunits to allow their assembly when Sil1 is absent?
An obvious candidate for releasing BiP is Grp170, the resident ER large Hsp70 family member, which also possesses nucleotide exchange activity (Weitzmann et al., 2006). A recent study in yeast on the cytosolic orthologues of Grp170 and Sil1—Sse1 and Fes1, respectively—found that Sse1 was more critical for both protein folding and degradation, whereas neither protein was required for renaturation of heat-aggregated proteins (Abrams et al., 2014). However, we did not detect an increase in Grp170 expression in the woozy splenic B cells above that which normally occurs during plasma cell differentiation, which is in contrast to the findings with Purkinje cells (Zhao et al., 2010) and muscle tissue (Roos et al., 2014) from woozy mice. It is possible that sufficient levels of Grp170 are already present in the plasmablasts to induce the release of BiP from assembling antibodies. There are no data to establish relative levels of Grp170 versus Sil1 in a variety of tissues to assess whether the loss of Sil1 might be felt more acutely in some tissues than in others. A previous analysis of tissue blots from our lab found that Sil1 transcript levels were particularly high in placenta, liver, kidney, skeletal muscle, and heart (Chung et al., 2002). With the exception of a progressive myopathy, these represent tissues that are not usually affected in MSS patients (Horvers et al., 2013; Krieger et al., 2013). A closer examination of relative levels of Sil1 and Grp170 in tissues that are affected by Sil1 loss compared with those that are not might shed light on this possibility. Alternatively, it is conceivable that additional BiP nucleotide exchange factors exist in the ER that have not yet been identified, which could complement the loss of Sil1 in plasma cells and other nonaffected tissue. The eukaryotic cytosol expresses a third family of NEFs, the Bag1 proteins (Hohfeld and Jentsch, 1997), but there are no obvious ER orothologues in the mammalian databases. Finally, we considered the possibility that the association of LC is sufficient to bypass the need for BiP or to displace wild-type BiP without requiring nucleotide exchange. In the case of IgM and IgG2b, for which LC can bind the HC cotranslationally, driving assembly through the H → HL → H2L2 pathway, it is conceivable that the requirement for BiP, and thus for a nucleotide exchange factor, is relatively small. However, we also found that both mouse and human IgG subclasses that combine through the H → H2 → H2L → H2L2, which interact with BiP throughout the assembly process, were equally unaffected, making this possibility less viable but as yet untested in vitro.
Why do Sil1-disrupted mice and MSS patients have defects that appear to affect only some systems?
The dependence of only some tissues on Sil1 remains one of the puzzling manifestations of the disease and of the phenotypes observed in woozy mice. In fact, it appears that highly secretory organs such as pancreas, liver, and plasma cells, where one might expect adverse consequences when a component of the chaperone machinery is lost, are relatively less affected by Sil1 loss. Instead the effects are greatest in the Purkinje cells of the cerebellum, the eye lens, and skeletal muscle (Krieger et al., 2013), which are not associated with the production of large amounts of secretory proteins that are BiP substrates. Perhaps the more classic secretory tissues are better able to respond to or accommodate changes in the client load of the ER. It is conceivable that there is some BiP:client specificity for the two NEFs of the ER, but this has yet to be demonstrated in any organism, organelle, or system.
Another interesting possibility is that the various clinical symptoms associated with MSS represent a progressive inability to maintain ER homeostasis with aging, which might occur at distinct times in various tissues, a phenomenon referred to as proteostasis collapse (Ben-Zvi et al., 2009). In support of this possibility, the Purkinje cell loss leading to ataxia and the progressive myopathy associated with MSS and observed in woozy mice can occur with quite different kinetics. Although we have not rigorously examined immune responses in aged Sil1Gt mice, we quantified splenic populations in a 22-mo-old mouse and found no significant differences in any subpopulations (unpublished data). However, one of our aged Sil1Gt mice experienced a severe infection with Pasteurella pneumotropica, which usually occurs only in immunodeficient mice (Moore and Aldred, 1978). As sufficient numbers of our Sil1Gt mice age, we intend to revisit the ability of older mice to produce antibodies.
In summary, we conducted a rigorous study of the effects of Sil1 loss on the synthesis of antibodies, the best-characterized BiP-dependent substrate. We found no change in either the magnitude or kinetics of antigen- or LPS-stimulated production of antibodies in the Sil1Gt mice compared with age-matched littermates. Furthermore, there was no evidence that the other ER NEF, Grp170, was abnormally up-regulated or that a different chaperone system was used to synthesize antibodies. Similar data were obtained with lymphoblastoid cell lines produced from individuals with MSS. We conclude that although loss of Sil1 in humans and mice can cause serious multisystem defects, these are not extended to the production of antibodies.
MATERIALS AND METHODS
Generation of wild-type and Sil1-disrupted mice
The Sil1 gene-trap mouse strain B6;129Sil1Gt (pGT2TMpfa)1Slac was kindly provided by Susan Ackerman (HHMI and The Jackson Labs, Bar Harbor, ME). Sil1Gt heterozygous mice were crossed to generate homozygous woozy mice and wild-type littermates. At 3 wk of age, DNA was isolated from mouse tail snips and genotyped as reported (Zhao et al., 2005). The primers for the wild-type allele were 5′-CTATAGCCCGCCTCAGGAGTGTTGTA-3′ and 5′-ATTGCCCCATAGAGCCATTTGTTGT-3′, and those for the disrupted allele were 5′-CACCGGATGCAGAAAAGCCACAAT-3′ and 5′-GCAACTCGCCGCACATCTGAACTT-3′. Mutant mice were born at expected Mendelian ratios. The breeding and housing of mice were approved by the Animal Use and Care Committee and Animal Research Center at St. Jude Children's Research Hospital, which adhere to National Institutes of Health guidelines.
Detection of mutant transcripts and protein in woozy mice
To determine the relative transcript levels of Sil1 in Sil1Gt mice compared with wild-type littermates, we isolated total RNA from wild-type and mutant plasmablasts after 4 d of LPS stimulation using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA) according to manufacturer's instructions. Real-time PCR was performed with 2 μg of total RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and a PTC-200 Thermal Cycler (MJ Research, Watertown, MA). Quantitative Real Time PCR (qPCR) primers were obtained from Invitrogen. The primer pairs used were as follows: Sil1 FP1, 5′-AGGAATTTGATGAGCTGAACGT-3′, and Sil1 RP1, 5′-TCCTGTGCGTTATCCATCTGA-3′, which produces a fragment that extends across exon 6 and the exon 6–exon 7 boundary and detects both Sil1WT and Sil1Gt transcripts; Sil1 FP2, 5′-CTGCGCCACTTCCCCTAT-3′, and Sil1 RP2, 5′-GCAAACATCTTCTCAGTGACCA-3′, which produces a fragment that extends across exon 9 and the exon 9–exon 10 boundary and detects only Sil1WT transcripts; and HPRT1 FP, 5′-AGGGAGAGCGTTGGGCTTACCT-3′, and HPRT1 RP, 5′-CCTGGTTCATCATCGCTAATCACGA-3′. The qPCR was performed with 7900HT Fast Real-Time PCR System (Applied Biosystems) using the Power SYBR Green PCR Master Mix (Applied Biosystems). All qPCRs were carried out in triplicate. The signal obtained for Sil1 mRNA with both primer pairs was normalized to that obtained for HPRT1 mRNA, which served as an internal control. The value for wild-type Sil1 mRNA was set to 1, and the value for Sil1Gt mRNA is presented as a fraction of this number.
Characterization of immune cell populations in spleen and thymus
Murine spleen and thymus were excised from 3- to 6-mo-old male C57BL/6 Sil1WT and Sil1Gt mice. Splenic cell suspensions were stained with CD3-APC, CD4-FITC, CD8-APC-Cy7, NK1.1-PE, B220-e605, Gr1-PE-Cy7, Mac1-Alexa 700, and 4′,6-diamidino-2-phenylindole (DAPI). Splenic subpopulations were analyzed as follows: CD3+/CD4+, CD3+/CD8+, B220+, Mac1+, Gr1+, Mac1+/Gr1+, and NK1.1+ cells. Thymic cell suspensions were stained with Lin (B220/Mac1/Gr1)–fluorescein isothiocyanate, CD3-PE, CD4-PerCP-Cy5.5, CD44-PE-Cy7, CD25-APC, CD8-Alexa 700, and DAPI. T-cell subpopulations were identified as follows: double negative, DN (CD4−/CD8−), DN1 (CD4−/CD8−/CD25−/CD44+), DN2 (CD4−/CD8−/CD25+/CD44+), DN3 (CD4−/CD8−/CD25+/CD44−), DN4 (CD4−/CD8−/CD25−/CD44−), DP (CD4+/CD8+), CD4SP (CD4+/CD8−), and CD8SP (CD4−/CD8+). Staining and flow cytometry were performed by the SJCRH Flow Cytometry and Cell Sorting shared resource on a LSR II Flow Cytometer (BD Biosciences, San Jose, CA) and analyzed using BD FACSDiva software (BD Biosciences). All antibodies were procured from BD Biosciences.
Immunization protocol and enzyme-linked immunosorbent assay for antigen-specific response
Ten each of Sil1WT and Sil1Gt mice were immunized with a 1:1 homogenate of the T-dependent antigen NP15-KLH (Biosearch Technologies, Novato, CA) in Freund's complete adjuvant, injected intraperitoneally, and boosted after 2 wk with NP15-KLH in Freund's incomplete adjuvant. Blood was collected on the indicated days by alternating retro-orbital and tail-vein bleedings. Antigen-specific serum IgM, IgG1, and IgG2b antibody titers were determined by direct enzyme-linked immunosorbent assay (ELISA). Bound antibody was detected with goat anti-mouse isotype–specific, biotin-conjugated antibodies (Southern Biotech, Birmingham, AL) and streptavidin-conjugated alkaline phosphatase (Jackson ImmunoResearch, West Grove, PA). Absorbances at 410 nm were read on a Benchmark Plus plate reader after incubation with phosphatase substrate (Thermo Scientific, Waltham, MA). Serum concentrations were determined from standard curves with purified mouse IgM, IgG1, or IgG2b proteins (Pharmingen, BD Biosciences). Only triplicates with a coefficient of variation of <10% were used to calculate serum concentrations.
Ex vivo stimulation of purified splenocytes with LPS
Spleens were harvested from three 10- to 12-wk-old male Sil1WT and three age-matched male Sil1Gt mice, and single-cell suspensions were prepared. After lysing of red blood cells, the non–B cells were immune depleted with antibodies to cell surface markers TER-119, CD4, CD8, Gr1, Mac1, and NK1.1 using an AutoMACS cell separator (Miltenyi Biotec, San Diego, CA). Aliquots of the resulting samples were stained with anti-B220 conjugated to allophycocyanin (APC) and analyzed on a LSR II Flow Cytometer using BD FACSDiva software to determine the purity of remaining B cells, which was >80% for all experiments. All reagents used were procured from BD Biosciences. The purified primary B cells were seeded at an initial cell density of 1 × 106 cells/ml in RPMI 1640 supplemented with 20% (vol/vol) FBS Premium (Atlanta Biologicals, Atlanta, GA), 2 mM l-glutamine, 100 U/ml penicillin/streptomycin antibiotics, 1× (vol/vol) MEM Non-Essential Amino Acids (Life Technologies, Grand Island, NY), and 55 μM 2-mercaptoethanol (Life Technologies) at 8% CO2 and induced to differentiate using 50 μg/ml LPS (Escherichia coli 055:B5; Sigma-Aldrich, St. Louis, MO). We harvested 4 × 106 cells each day along with an equivalent volume of culture supernatant. Medium was changed daily, and cells were maintained at a density of <2 × 106 cells/ml.
EBV-transformed lymphoblastoid cell lines
EBV-transformed peripheral blood lymphocytes from three related individuals with MSS and two unaffected family members were kindly provided by William R. Wilcox (Emory University, Atlanta, GA). The resulting cell lines were cultured in RPMI 1640 (Cellgro, Manassas, VA) supplemented with 10% (vol/vol) FBS Premium (Atlanta Biologicals), 2 mM l-glutamine, 100 U/ml penicillin/streptomycin antibiotics, and Gentamicin Reagent Solution (10 μg/ml) at 37°C in 5% CO2.
Western blotting
Nonidet P-40 cell lysates from B lymphoblastoid lines and LPS-stimulated plasmablasts were produced as previously described (Behnke and Hendershot, 2014). The resulting clarified lysates were designated as the NP-40–soluble fraction. The NP-40–insoluble pellets were dissolved in 50 μl Tris-SDS buffer and prepared as described (Okuda-Shimizu et al., 2007). For direct Western blotting, the NP-40–soluble and –insoluble fractions were normalized for equivalent cell number (plasmablasts) or protein amount (lymphoblastoid cells) and electrophoresed on 10% SDS–polyacrylamide gels under reducing or nonreducing conditions as indicated. In cases in which IgM secretion was analyzed, a corresponding amount of culture supernatant was first immunoprecipitated with goat anti–mouse μ and CaptivA PriMAB protein A agarose beads (RepliGen, Waltham, MA). For experiments to determine whether the mutant Sil1 protein made in LBLs produced from individuals with MSS was unstable, cells were treated with 10 μM MG132 (Calbiochem, San Diego, CA) for 5 h or left untreated. In all cases, after electrophoresis on SDS–polyacrylamide gels, the separated proteins were transferred to Immun-Blot PVDF membrane (Bio-Rad, Hercules, CA) and incubated with antibodies specific for the indicated proteins. Antisera to rodent BiP (Hendershot et al., 1995), Sil1 (Chung et al., 2002), ERdj3 (Shen and Hendershot, 2005), Grp170 (Behnke and Hendershot, 2014), and Grp94 and calnexin (Lawson et al., 1998) were produced in our lab. The anti–human BiP antiserum was kindly provided by Ineke Braakman (Utrecht University, Utrecht, Netherlands), and the anti-ERdj1 antiserum was a generous gift from Richard Zimmermann (Universität des Saarlandes, Homburg, Germany). Antibodies to human Ig κ LC, calreticulin, Hsc70, and actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and the anti-ERdj5 was obtained from Abnova (Walnut, CA). Two anti-Sil1 reagents were purchased—one from LifeSpan Biosciences (Seattle, WA), which was raised to a fragment that is preserved in the MSS mutant Sil1 proteins studied here, and the other from Proteintech (Chicago, IL), which was produced against full-length human Sil1 protein. Goat anti–mouse μ HC and κ LC and anti–human μ HC, γ HC, and λ LC antibodies were obtained from Southern Biotech, and horseradish peroxidase–conjugated, species-specific secondary immune reagents were purchased from Santa Cruz Biotechnology.
Metabolic labeling
We pulse-labeled 4 × 106 plasmablasts from day 2 and 3 LPS cultures with 300 μCi of EasyTag EXPRESS35S Protein Labeling Mix (PerkinElmer, Waltham, MA) for 20 min and either lysed them immediately (0-h chase) or recultured them for 1 h in complete medium supplemented with 2 mM unlabeled l-Cys and l-Met (1-h chase). In the case of anti-BiP immunoprecipitations, 10 U/ml apyrase (Sigma-Aldrich) was added to the lysing buffer, whereas for anti-Grp170, the lysis buffer was supplemented with 2 mM Mg2+-ATP (Sigma-Aldrich) and 25 mM KCl (Thermo Fisher Scientific, Waltham, MA) as described (Behnke and Hendershot, 2014). Antisera to calreticulin (Abcam) and calnexin were added directly to unsupplemented cell lysates. In all cases, immune complexes were isolated with protein A agarose beads, and isolated proteins were subjected to reducing SDS–PAGE. The resulting gels were incubated in Amplify (GE Healthcare, Pittsburgh, PA), supplemented with 3% glycerol, for 30 min at room temperature before they were dried and exposed to film (Fujifilm, Elmsford, NY). To determine whether Sil1-deficient lymphoblastoid cells exhibit a difference in their ability to synthesize and secrete antibodies, we performed pulse-chase experiments. We labeled 6 × 106 lymphoblastoid cells with 400 μCi of [35S]methionine and cysteine for 30 min and chased them in complete medium for 0, 1.5, or 3 h, respectively. Postlysis, the NP-40–soluble fraction and an equivalent amount of culture supernatant were first immunoprecipitated for γ HC and then analyzed on reducing or nonreducing SDS–polyacrylamide gels as indicated.
Sil1 sequencing
Sequence analysis was performed for the human Sil1 gene (GenBank NM_001037633) and transcript from control and MSS-derived lymphoblastoid cells. Genomic DNA (gDNA) and RNA were extracted using the DNeasy Blood & Tissue Kit (Qiagen) and RNeasy Plus Mini Kit (Qiagen), respectively, according to the manufacturer's instructions. Using gDNA as template, we PCR-amplified regions spanning exon 9 and the 5′ region of intron 9 using primer pairs 5′-GGGACTAATTTGGGGCACCT-3′ and 5′-CCTCAGCCTTTTGTCCCCTC-3′, and the 3′ region of intron 9 and the translated region of exon 10 using primer pairs 5′-TGGGGATAGAGGAAGAGGTGG-3′ and 5′-GTCCTCCTGCCTGAGAAGC-3′. We used 2 μg of total RNA to synthesize cDNA as described. We PCR-amplified the region spanning exons 9 and 10 using primer pairs 5′-AAGAAGAAGGTCCTGTTTGCAC-3′ and 5′-GAAGTAGCCCTCGTCCTCAC-3′ to verify the frameshift mutation caused by c.1030-18G>A (Senderek et al., 2005; Ezgu et al., 2014). Primer pairs were removed using the Wizard SV Gel and PCR Clean-up System (Promega, Madison, WI), and products were sequenced using Big Dye Terminator (v3.1) Chemistry on Applied Biosystem 3730XL DNA Analyzers by the Hartwell Center, St. Jude Children's Research Hospital.
Statistical analysis
Statistical analyses were computed by Student's t test using Prism 6 (GraphPad Software, La Jolla, CA). Error bars represent mean ± SD, except in Figure 2, where error bars indicate interquartile range of values. Statistical significance was concluded at p < 0.05.
Supplementary Material
Acknowledgments
We thank members of the Hendershot lab for helpful scientific discussions, the Veterinary Pathology Core for help in harvesting mouse tissues, and the Flow Cytometry and Cell Sorting Resource for assistance in the analysis of mouse lymphoid populations. This work was funded by National Institutes of Health Grant R01 GM54068 awarded to L.M.H. and by the American Lebanese Syrian Associated Charities of St. Jude Children's Research Hospital.
Abbreviations used:
- EBV
Epstein–Barr virus
- ER
endoplasmic reticulum
- HC
heavy chain
- LBL
lymphoblastoid cell line
- LC
light chain
- LPS
lipopolysaccharide
- MSS
Marinesco–Sjögren syndrome
- NBD
nucleotide-binding domain
- NEF
nucleotide exchange factor
- NP15-KLH
4-hydroxy-3-nitrophenylacetyl conjugated to keyhole limpet hemocyanin.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-09-1392) on December 3, 2014.
*Present address: Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, TN 37232.
REFERENCES
- Abrams JL, Verghese J, Gibney PA, Morano KA. Hierarchical functional specificity of cytosolic heat shock protein 70 (Hsp70) nucleotide exchange factors in yeast. J Biol Chem. 2014;289:13155–13167. doi: 10.1074/jbc.M113.530014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttonen AK, Mahjneh I, Hamalainen RH, Lagier-Tourenne C, Kopra O, Waris L, Anttonen M, Joensuu T, Kalimo H, Paetau A, et al. The gene disrupted in Marinesco-Sjogren syndrome encodes SIL1, an HSPA5 cochaperone. Nat Genet. 2005;37:1309–1311. doi: 10.1038/ng1677. [DOI] [PubMed] [Google Scholar]
- Anttonen AK, Siintola E, Tranebjaerg L, Iwata NK, Bijlsma EK, Meguro H, Ichikawa Y, Goto J, Kopra O, Lehesjoki AE. Novel SIL1 mutations and exclusion of functional candidate genes in Marinesco-Sjogren syndrome. Eur J Hum Genet. 2008;16:961–969. doi: 10.1038/ejhg.2008.22. [DOI] [PubMed] [Google Scholar]
- Barends TR, Brosi RW, Steinmetz A, Scherer A, Hartmann E, Eschenbach J, Lorenz T, Seidel R, Shoeman RL, Zimmermann S, et al. Combining crystallography and EPR: crystal and solution structures of the multidomain cochaperone DnaJ. Acta Crystallogr D Biol Crystallogr. 2013;69:1540–1552. doi: 10.1107/S0907444913010640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke J, Hendershot LM. The large hsp70 grp170 binds to unfolded protein substrates in vivo with a regulation distinct from conventional hsp70s. J Biol Chem. 2014;289:2899–2907. doi: 10.1074/jbc.M113.507491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci USA. 2009;106:14914–14919. doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bole DG, Hendershot LM, Kearney JF. Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J Cell Biol. 1986;102:1558–1566. doi: 10.1083/jcb.102.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- Buck TM, Plavchak L, Roy A, Donnelly BF, Kashlan OB, Kleyman TR, Subramanya AR, Brodsky JL. The Lhs1/GRP170 chaperones facilitate the endoplasmic reticulum-associated degradation of the epithelial sodium channel. J Biol Chem. 2013;288:18366–18380. doi: 10.1074/jbc.M113.469882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KT, Shen Y, Hendershot LM. BAP, a mammalian BiP associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J Biol Chem. 2002;277:47557–47563. doi: 10.1074/jbc.M208377200. [DOI] [PubMed] [Google Scholar]
- Conley ME, Rohrer J, Rapalus L, Boylin EC, Minegishi Y. Defects in early B-cell development: comparing the consequences of abnormalities in pre-BCR signaling in the human and the mouse. Immunol Rev. 2000;178:75–90. doi: 10.1034/j.1600-065x.2000.17809.x. [DOI] [PubMed] [Google Scholar]
- David V, Hochstenbach F, Rajagopalan S, Brenner MB. Interaction with newly synthesized and retained proteins in the endoplasmic reticulum suggests a chaperone function for human integral membrane protein IP90 (calnexin) J Biol Chem. 1993;268:9585–9592. [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Ezgu F, Krejci P, Li S, de Sousa C, Graham JM Jr, Hansmann I, He W, Porpora K, Wand D, Wertelecki W, et al. Phenotype-genotype correlations in patients with Marinesco-Sjogren syndrome. Clin Genet. 2014;86:74–84. doi: 10.1111/cge.12230. [DOI] [PubMed] [Google Scholar]
- Feige MJ, Groscurth S, Marcinowski M, Shimizu Y, Kessler H, Hendershot LM, Buchner J. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol Cell. 2009;34:569–579. doi: 10.1016/j.molcel.2009.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass JN, Gifford NM, Brewer JW. Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J Biol Chem. 2002;277:49047–49054. doi: 10.1074/jbc.M205011200. [DOI] [PubMed] [Google Scholar]
- Haas IG, Wabl M. Immunoglobulin heavy chain binding protein. Nature. 1983;306:387–389. doi: 10.1038/306387a0. [DOI] [PubMed] [Google Scholar]
- Hasegawa S, Imai K, Yoshida K, Okuno Y, Muramatsu H, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Kojima S, et al. Whole-exome sequence analysis of ataxia telangiectasia-like phenotype. J Neurol Sci. 2014;340:86–90. doi: 10.1016/j.jns.2014.02.033. [DOI] [PubMed] [Google Scholar]
- Hendershot L, Bole D, Kohler G, Kearney JF. Assembly and secretion of heavy chains that do not associate posttranslationally with immunoglobulin heavy chain-binding protein. J Cell Biol. 1987;104:761–767. doi: 10.1083/jcb.104.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot LM, Wei J-Y, Gaut JR, Lawson B, Freiden PJ, Murti KG. In vivo expression of mammalian BiP ATPase mutants causes disruption of the endoplasmic reticulum. Mol Biol Cell. 1995;6:283–296. doi: 10.1091/mbc.6.3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot L, Wei J, Gaut J, Melnick J, Aviel S, Argon Y. Inhibition of immunoglobulin folding and secretion by dominant negative BiP ATPase mutants. Proc Natl Acad Sci USA. 1996;93:5269–5274. doi: 10.1073/pnas.93.11.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohfeld J, Jentsch S. GrpE-like regulation of the hsc70 chaperone by the anti-apoptotic protein BAG-1. EMBO J. 1997;16:6209–6216. doi: 10.1093/emboj/16.20.6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvers M, Anttonen AK, Lehesjoki AE, Morava E, Wortmann S, Vermeer S, van de Warrenburg BP, Willemsen MA. Marinesco-Sjogren syndrome due to SIL1 mutations with a comment on the clinical phenotype. Eur J Paediatr Neurol. 2013;17:199–203. doi: 10.1016/j.ejpn.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Howes J, Shimizu Y, Feige MJ, Hendershot LM. C-terminal mutations destabilize SIL1/BAP and can cause Marinesco-Sjogren syndrome. J Biol Chem. 2012;287:8552–8560. doi: 10.1074/jbc.M111.333286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner AA, Bertz J, Chan TY, van EJ, Polly P, Ittner LM. The nucleotide exchange factor SIL1 is required for glucose-stimulated insulin secretion from mouse pancreatic beta cells in vivo. Diabetologia. 2014;57:1410–1419. doi: 10.1007/s00125-014-3230-z. [DOI] [PubMed] [Google Scholar]
- Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- Kabani M, Beckerich JM, Gaillardin C. Sls1p stimulates Sec63p-mediated activation of Kar2p in a conformation-dependent manner in the yeast endoplasmic reticulum. Mol Cell Biol. 2000;20:6923–6934. doi: 10.1128/mcb.20.18.6923-6934.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger M, Roos A, Stendel C, Claeys KG, Sonmez FM, Baudis M, Bauer P, Bornemann A, de Goede C, Dufke A, et al. SIL1 mutations and clinical spectrum in patients with Marinesco-Sjogren syndrome. Brain. 2013;136:3634–3644. doi: 10.1093/brain/awt283. [DOI] [PubMed] [Google Scholar]
- Lawson B, Brewer JW, Hendershot LM. Geldanamycin, an HSP90/GRP94-binding drug, induces increased transcription of ER chaperones via the ER stress pathway. J Cell Physiol. 1998;174:170–178. doi: 10.1002/(SICI)1097-4652(199802)174:2<170::AID-JCP4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Lee Y-K, Brewer JW, Hellman R, Hendershot LM. BiP and Ig light chain cooperate to control the folding of heavy chain and ensure the fidelity of immunoglobulin assembly. Mol Biol Cell. 1999;10:2209–2219. doi: 10.1091/mbc.10.7.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HY, Masso-Welch P, Di YP, Cai JW, Shen JW, Subjeck JR. The 170-kDa glucose-regulated stress protein is an endoplasmic reticulum protein that binds immunoglobulin. Mol Biol Cell. 1993;4:1109–1119. doi: 10.1091/mbc.4.11.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinowski M, Holler M, Feige MJ, Baerend D, Lamb DC, Buchner J. Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat Struct Mol Biol. 2011;18:150–158. doi: 10.1038/nsmb.1970. [DOI] [PubMed] [Google Scholar]
- Marcinowski M, Rosam M, Seitz C, Elferich J, Behnke J, Bello C, Feige MJ, Becker CF, Antes I, Buchner J. Conformational selection in substrate recognition by Hsp70 chaperones. J Mol Biol. 2013;425:466–474. doi: 10.1016/j.jmb.2012.11.030. [DOI] [PubMed] [Google Scholar]
- Melnick J, Aviel S, Argon Y. The endoplasmic reticulum stress protein GRP94, in addition to BiP, associates with unassembled immunoglobulin chains. J Biol Chem. 1992;267:21303–21306. [PubMed] [Google Scholar]
- Moore GJ, Aldred P. Treatment of Pasteurella pneumotropica abscesses in nude mice (nu/nu) Lab Anim. 1978;12:227–228. doi: 10.1258/002367778781088413. [DOI] [PubMed] [Google Scholar]
- Okuda-Shimizu Y, Hendershot LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which requires Herp. Mol Cell. 2007;28:544–554. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero JH, Lizak B, Hendershot LM. Life and death of a BiP substrate. Semin Cell Dev Biol. 2010;21:472–478. doi: 10.1016/j.semcdb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Easton DP, Chen X, MacDonald IJ, Wang XY, Subjeck JR. The chaperoning properties of mouse grp170, a member of the third family of hsp70 related proteins. Biochemistry. 2003;42:14893–14902. doi: 10.1021/bi030122e. [DOI] [PubMed] [Google Scholar]
- Roos A, Buchkremer S, Kollipara L, Labisch T, Gatz C, Zitzelsberger M, Brauers E, Nolte K, Schroder JM, Kirschner J, et al. Myopathy in Marinesco-Sjogren syndrome links endoplasmic reticulum chaperone dysfunction to nuclear envelope pathology. Acta Neuropathol. 2014;127:761–777. doi: 10.1007/s00401-013-1224-4. [DOI] [PubMed] [Google Scholar]
- Senderek J, Krieger M, Stendel C, Bergmann C, Moser M, Breitbach-Faller N, Rudnik-Schoneborn S, Blaschek A, Wolf NI, Harting I, et al. Mutations in SIL1 cause Marinesco-Sjogren syndrome, a cerebellar ataxia with cataract and myopathy. Nat Genet. 2005;37:1312–1314. doi: 10.1038/ng1678. [DOI] [PubMed] [Google Scholar]
- Shen Y, Hendershot LM. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP's interactions with unfolded substrates. Mol Biol Cell. 2005;16:40–50. doi: 10.1091/mbc.E04-05-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren T. Hereditary congenital spinocerebellar ataxia accompanied by congenital cataract and oligophrenia; a genetic and clinical investigation. Confin Neurol. 1950;10:293–308. [PubMed] [Google Scholar]
- Spee P, Subjeck J, Neefjes J. Identification of novel peptide binding proteins in the endoplasmic reticulum: ERp72, calnexin, and grp170. Biochemistry. 1999;38:10559–10566. doi: 10.1021/bi990321r. [DOI] [PubMed] [Google Scholar]
- Steel GJ, Fullerton DM, Tyson JR, Stirling CJ. Coordinated activation of Hsp70 chaperones. Science. 2004;303:98–101. doi: 10.1126/science.1092287. [DOI] [PubMed] [Google Scholar]
- Superneau DW, Wertelecki W, Zellweger H, Bastian F. Myopathy in Marinesco-Sjogren syndrome. Eur Neurol. 1987;26:8–16. doi: 10.1159/000116305. [DOI] [PubMed] [Google Scholar]
- Torbergsen T, Aasly J, Borud O, Lindal S, Mellgren SI. Mitochondrial myopathy in Marinesco-Sjogren syndrome. J Ment Defic Res. 1991;35:154–159. doi: 10.1111/j.1365-2788.1991.tb01045.x. [DOI] [PubMed] [Google Scholar]
- van Anken E, Romijn EP, Maggioni C, Mezghrani A, Sitia R, Braakman I, Heck AJ. Sequential waves of functionally related proteins are expressed when B cells prepare for antibody secretion. Immunity. 2003;18:243–253. doi: 10.1016/s1074-7613(03)00024-4. [DOI] [PubMed] [Google Scholar]
- Vanhove M, Usherwood Y-K, Hendershot LM. Unassembled Ig heavy chains do not cycle from BiP in vivo, but require light chains to trigger their release. Immunity. 2001;15:105–114. doi: 10.1016/s1074-7613(01)00163-7. [DOI] [PubMed] [Google Scholar]
- Wei J-Y, Gaut JR, Hendershot LM. In vitro dissociation of BiP:peptide complexes requires a conformational change in BiP after ATP binding but does not require ATP hydrolysis. J Biol Chem. 1995;270:26677–26682. doi: 10.1074/jbc.270.44.26677. [DOI] [PubMed] [Google Scholar]
- Weitzmann A, Volkmer J, Zimmermann R. The nucleotide exchange factor activity of Grp170 may explain the non-lethal phenotype of loss of Sil1 function in man and mouse. FEBS Lett. 2006;580:5237–5240. doi: 10.1016/j.febslet.2006.08.055. [DOI] [PubMed] [Google Scholar]
- Wiest DL, Burkhardt JK, Hester S, Hortsch M, Meyer DI, Argon Y. Membrane biogenesis during B cell differentiation: most endoplasmic reticulum proteins are expressed coordinately. J Cell Biol. 1990;110:1501–1511. doi: 10.1083/jcb.110.5.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Li J, Sha B. Structural analysis of the Sil1-Bip complex reveals the mechanism for Sil1 to function as a nucleotide-exchange factor. Biochem J. 2011;438:447–455. doi: 10.1042/BJ20110500. [DOI] [PubMed] [Google Scholar]
- Zhao L, Longo-Guess C, Harris BS, Lee JW, Ackerman SL. Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nat Genet. 2005;37:974–979. doi: 10.1038/ng1620. [DOI] [PubMed] [Google Scholar]
- Zhao L, Rosales C, Seburn K, Ron D, Ackerman SL. Alteration of the unfolded protein response modifies neurodegeneration in a mouse model of Marinesco-Sjogren syndrome. Hum Mol Genet. 2010;19:25–35. doi: 10.1093/hmg/ddp464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.