

Midkine (MDK) expression is associated with the proliferation of many cancers, including glioma. SP1 directly up-regulates the expression of MDK, and the SP1-MDK axis cooperates in glioma tumorigenesis.
Abstract
Midkine (MDK) expression is associated with the proliferation of many cancers, including glioma. However, the upstream signaling that leads to MDK accumulation remains elusive. This study investigates the molecular mechanism that induces MDK overexpression in human glioma. The Repository for Molecular Brain Neoplasia Data was analyzed to identify potential MDK regulators. Expression of MDK and specificity protein 1 (SP1) was compared in glioma specimens. Chromatin immunoprecipitation assay was used to confirm the transcriptional regulation. MDK-force–expressed, SP1-silenced glioma cells were used to test rescue effects in vitro and in vivo. MDK and SP1 expression in gliomas was significantly higher than in adjacent tissues and was positively correlated in glioma clinical samples and cell lines. The promoter of the human MDK gene has a putative SP1 binding site. SP1 binds to the promoter of the MDK gene and directly regulates MDK expression. MDK or SP1 gene silencing inhibited the proliferation of glioma cells and reduced the tumor volume in nude mice. Overexpression of MDK in SP1-silenced cells could partially rescue the SP1 inhibition effects in vivo and in vitro. SP1 directly up-regulated the expression of MDK, and the SP1-MDK axis cooperated in glioma tumorigenesis.
INTRODUCTION
As the most common brain cancer, glioma account for >60% of primary brain tumors in adults (Stupp et al., 2009; Jemal et al., 2010). Surgery followed by radiotherapy, with concomitant and adjuvant chemotherapy with temozolomide, is the standard treatment of glioma; however, the prognosis remains poor (Minniti et al., 2009; Stupp et al., 2009). Most malignant glioma patients die within 1 yr of the diagnosis, and only 5% survive >5 yr despite aggressive therapies (Wen and Kesari, 2008; Mrugala, 2013). Early diagnosis is critically important to the prognosis of glioma but is highly challenging due to the lack of sensitive and specific biological markers and molecular targets for treatment. The pathogenesis of glioma is largely unknown, although it has been shown that genetic alterations in a number of genes are associated with the development of glioma (Nagane et al., 1997; Yan et al., 2009; Melin, 2011; Nakada et al., 2011). Unclear pathogenesis prevents the early diagnosis and efficient treatment of glioma. Thus investigating the pathogenesis of glioma and identifying potential targets that play key roles in its development are critically important for the diagnosis and treatment of this fatal disease.
Midkine (MDK) was originally reported to be the product of a retinoic acid–responsive gene during embryogenesis (Kaname et al., 1993; Muramatsu, 2011). By binding to oversulfated structures in heparan sulfate and chondroitin sulfate, MDK interacts with downstream proteins to activate several signaling pathways and contributes to various cellular process (Muramatsu, 2011; Weckbach et al., 2011). The expression of MDK was high during embryogenesis but was not detectable in healthy adults (Kadomatsu and Muramatsu, 2004). Identification of MDK in serum and tissues is usually associated with diseases, including inflammation and cancers (Ikematsu et al., 2000; Muramatsu, 2010; Weckbach et al., 2011). MDK starts downstream signaling systems such as Src family kinases and tyrosine phosphorylation (Maeda et al., 1999; Muramatsu et al., 2000). Increased tyrosine phosphorylation of paxillin leads to migration at osteoblast-like cells and is followed by the suppression of caspases and the activation of phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK; Maeda et al., 1999; Owada et al., 1999; Muramatsu et al., 2000; Ohuchida et al., 2004). Overexpression of MDK has been observed in many cancers, including lung cancer (Hao et al., 2013; Ma et al., 2013), gastric cancer (Xu et al., 2012; Zhu et al., 2013a), and breast cancer (Miyashiro et al., 1997; Ibusuki et al., 2009) as well as CNS tumors (Mishima et al., 1997; Lorente et al., 2011; Kishida and Kadomatsu, 2014), and also may contribute to cancer resistance to chemotherapy (Kang et al., 2007; Xu et al., 2012; Zhu et al., 2013a). This results suggest that MDK plays an important role in the human carcinogenesis process and also that it might be an ideal target for cancer treatment.
Although the region downstream of MDK has been widely investigated and interference with the pathway has been applied in glioma therapy (Kohno et al., 2004; Lorente et al., 2011; Nakada et al., 2011), the upstream features of the MDK pathway remain largely unknown. Identification of proteins controlling the expression of MDK may provide a novel approach to the treatment of glioma. In this study, we observe a correlation between MDK and SP1 expression in glioma clinical samples and cell lines and identify a novel regulatory mechanism of MDK by SP1, using a combination of bioinformatics and molecular biology approaches. The identified regulatory mechanism might be applicable in the future treatment of glioma.
RESULTS
MDK overexpression correlates with up-regulation of SP1 expression in human glioma specimens and glioma cell lines
Search in the Repository for Molecular Brain Neoplasia Data (REMBRANDT) database at the National Cancer Institute demonstrated that the expression of both MDK and SP1 in glioma cells was significantly higher than in adjacent tissues (Supplemental Figures S1–S4). To validate this result, we determined MDK and SP1 expression in serial sections of 80 human glioma specimens by immunohistochemical analyses. Both MDK and SP1 were present in the glioma mass but absent from the “normal” brain tissues surrounding the tumor (Figure 1A). We then quantified the immunohistochemical staining of MDK and SP1 in glioma specimens on scale from 0 to 8. We analyzed the scores and found significant correlation between MDK and SP1 expression levels (Figure 1B; r = 0.79, p < 0.001). Next we performed Western blot analyses using total protein extracts from three low-grade astrocytoma (grade 2), three anaplastic astrocytoma (grade 3), and three glioblastoma (grade 4) frozen human glioma tissues and their paired adjacent “normal” tissues. As shown in Figure 1C, both MDK and SP1 are overexpressed in glioma tissues compared with the “normal” brain, and MDK expression correlated with SP1 expression with regard to protein level. To exclude the possibility that this correlation was due to the tissues other than glioma cells, we next determined the expression of MDK and SP1 mRNA and protein in glioma cell lines A172, LN18, U118, U251, U87, LN382, and LN444 by using reverse transcription (RT) PCR and Western blot and set normal human astrocytes (NHAs) as control. As shown in Figure 1D, the levels of mRNA and protein of MDK in these glioma cell lines were significantly higher than with NHAs, and the increased level of MDK mRNA is positively correlated with SP1 protein. Therefore our results indicate that both MDK and SP1 are markedly overexpressed in human glioma, and expression of MDK mRNA and protein is correlated with SP1 protein level.
FIGURE 1:

SP1 overexpression correlates with up-regulation of MDK expression in human glioma samples and cell lines. (A) Immunohistochemical staining with specific anti-SP1 and anti-MDK antibodies on glioma tissues. Normal, brain tissues surrounding the tumor; Tumor, human glioma tissues. (B) We quantitatively scored the tissue sections according to the percentage of positive cells and staining intensity as described in Materials and Methods. We then combined the percentage and intensity scores to obtain a total score (range, 0–8). SP1 expression levels correlated positively with MDK expression levels in glioma samples (Pearson's r = 0.79; p < 0.001). (C) SP1 and MDK protein expression levels for three paired low-grade astrocytoma (II), three paired anaplastic astrocytoma (III), and three paired glioblastoma (IV) frozen tissues as determined by Western blot analysis. (D) The SP1 and MDK protein levels in NHA and various glioma cell lines as determined by Western blot.
Altered SP1 expression affects MDK expression in glioma cells
To determine the effect of decreased SP1 expression on MDK expression, we studied two human glioma cell lines, U87 and U251, with high levels of SP1 expression. By establishing stable SP1- knockdown cell lines using short hairpin RNA (shRNA) lentivirus, we found that SP1 knockdown in U87 and U251 cells significantly reduced both MDK mRNA and protein expression (Figure 2A). These results indicate that preventing SP1 expression suppressed MDK expression in glioma cells.
FIGURE 2:
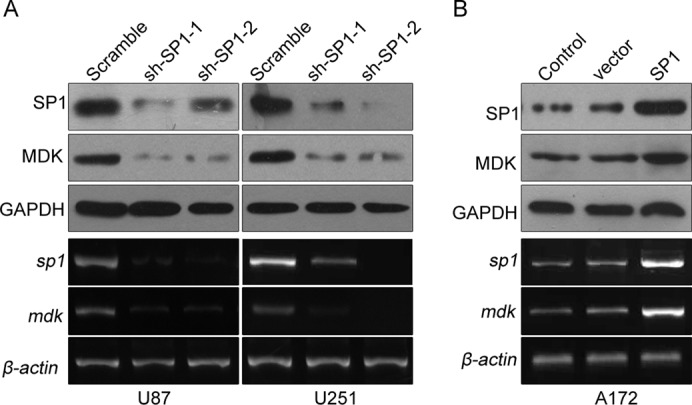
Effects of altered SP1 expression on MDK expression in human glioma cell lines. (A) SP1 gene silencing of glioma cell lines U87 and U251 was conducted by lentivirus-delivered shRNA. Cell lysates were subjected to Western blot and RT-PCR. (B) SP1 overexpression and control A172 cell lines were established using lentivirus. MDK and SP1 expression were determined by Western blot and RT-PCR. Data are representative of three separate experiments.
Conversely, to determine increased SP1 expression on MDK expression, we stably transfected A172 glioma cell lines, which have low levels of SP1 and MDK expression, with the SP1 expression lentivirus. Forced expression of SP1 significantly up-regulated the SP1 mRNA and protein levels in A172 cell lines. These cells also exhibit significantly increased MDK mRNA and protein expression after the infection (Figure 2B). These results indicate that SP1 overexpression in glioma cells increases MDK expression.
SP1 directly interacts with and regulates the activity of the MDK promoter
To determine whether MDK could be a direct transcriptional target of SP1, we analyzed MDK promoter reporter constructs by measuring the activities of firefly and Renilla luciferase. As shown in Figure 3A, compared with the control group, SP1 stably knocked down U87 and U251 cells showed dramatically lower luciferase activity in the −562 to +33 region of the MDK promoter, whereas overexpression of SP1 in A172 cells could activate the same region of the MDK promoter (Figure 3B), indicating that a putative SP1 binding site located on MDK promoter encompasses the −562 to +33 fragment. Next we generated a set of sequentially truncated MDK promoter constructions and compared their activity in U87 cells. As shown in Figure 3C, deletion mutants from −562 to −125 were as active as the original construct, whereas further deletion to −85 nearly abolished the activation, indicating that the putative SP1 binding site could be located in the region from −125 to −85 of the MDK promoter. Searching this spectrum using promoter analysis tools Patch and MatInspector, we identified a putative SP1 binding site, GGCGGG, at positions −108 to −102, suggesting that this site interacts with SP1.
FIGURE 3:

An SP1 site located at the MDK promoter is necessary for MDK promoter activation in glioma cell lines. (A) SP1-knockdown stable U87 and U251 cell lines were transfected with MDK (-562/+33)-luc or pGL-3 Bacis vector and pRL-TK. 24 h after transfection, the levels of luciferase activity were measured and normalized to Renilla luciferase activity. (B) SP1 overexpression and control A172 cell lines were transfected with MDK (−562/+33)-luc or pGL-3 Bacis vector and pRL-TK. At 24 h after transfection, the levels of luciferase activity were measured. (C) Structures of MDK 5′ sequential deletion constructs. U87 cells were cotransfected with reporter plasmids and pRL-TK for 24 h. Promoter activity was determined by luciferase assay. (D) DNA isolated and purified from ChIP immunoprecipitated material was amplified by PCR with primers to amplify an MDK promoter fragment spanning the SP1 site; equal amounts of total genomic DNA were used for input. Data are representative of three separate experiments.
To provide direct proof that SP1 is recruited to the endogenous MDK promoter during transcription in vivo, we performed standard chromatin immunoprecipitation (ChIP) assays with U87, U251, and A172 cells. We found that the SP1-binding regions of the MDK promoter bound to SP1 protein in all three cell types (Figure 3D). Furthermore, in U87 and U251 control cells, which expressed higher SP1, five to six times more DNA promoter was bound to SP1 than in the A172 cells, which expressed low SP1 levels. Together our findings show that SP1 bound specifically to SP1 binding sites in the MDK promoter in glioma cells.
SP1 binding sites are critical for the activation of the MDK promoter in glioma cells
To assess the functional role of the SP1 binding site in MDK gene regulation, we performed site-specific mutagenesis within the SP1 binding site of the MDK promoter. As shown in Figure 4A, a point mutation (GGCGGG → TTCGGG) was generated from the wild-type MDK promoter construct. We transfected the mutant luciferase reporter into U87 and U251 cells and compared the activity with that of the wild-type MDK promoter. Disrupting the SP1-specific binding site on the MDK promoter significantly attenuated MDK promoter transactivation, suggesting that the SP1 binding site is critical for MDK promoter activation in glioma cells (Figure 4B). To further confirm our finding, we used an SP1-DNA binding inhibitor, mithramycin A, to test the transactivation effect of SP1 on MDK promoter. As shown in Figure 4C, the expression of MDK mRNA and protein was significantly reduced in U87 cells when mithramycin A was introduced. In addition, with an increase in the concentration of mithramycin A, the activity of reporter gene luciferase reduced significantly (Figure 4D). Taken together, these results demonstrate that SP1 directly regulates MDK expression by activating MDK promoter in glioma cells.
FIGURE 4:

Mutation of the SP1 site abolished activation of the MDK promoter. (A) The core regions of MDK promoter were analyzed by Patch and MatInspector, and the putative conserved SP1 binding site was located at −108/−102. The wild-type SP1 site, GGCGGG (WT), was mutated to TTCGGG (Mut). (B) NHA, U87, and U251 were cotransfected with MDK-Luc wt or mdk-Luc mut, together with pRL-TK for 24 h, respectively. Luciferase activity was measured. (C) Mithramycin A attenuated the up-regulation of MDK in a dose-dependent manner. U87 cells were placed in medium in the absence or presence of mithramycin A at the indicated concentrations for 12 h. MDK expression was assessed by Western blot and RT-PCR (left). (D) Mithramycin A attenuated the activity of mdk promoter. mdk promoter activities were measured by luciferase assay. The experiments were repeated three times, with duplicates for each treatment.
Down-regulation of MDK or SP1 significantly inhibited the proliferation of glioma cells and the growth of glioma tumor in nude mice
Next we examined the functional consequence of the SP1/MDK interaction by assessing their roles in glioma proliferation. Western blot indicated that the key proliferation-related proteins, including p-Akt, CDK4, and CDK6, were at significantly lower levels in MDK stable knockdown glioma cells (Figure 5A). By comparing the MDK stable knockdown U87 and U251 cells and their control cells, we found that the growth of glioma cell lines with MDK knockdown was significantly inhibited (Figure 5B). In addition, the volume of solid tumor in nude mice planted with glioma cells with MDK knockdown is significantly smaller than that planted with glioma cells of wild type (Figure 5C). Further, stably knocking down SP1 in U87 and U251 also decreased the expression of p-Akt, p-ERK, CDK4, and CDK6 and inhibited tumor cell growth in vivo and in vitro (Figure 5, D–F). In the converse case, forced expression of SP1 or MDK in A172 cells increased cell proliferation, up-regulated p-Akt, CDK4, and CDK6 expression, and induced tumor growth in vivo and in vitro (Figure 5, G–I). These results indicate that the SP1/MDK axis plays a crucial role in the proliferation of glioma cells.
FIGURE 5:

Effects of altered MDK or SP1 expression on proliferation capacity and tumorigenetic capability of glioma cells. (A) MDK was stably knocked down by two independent shRNAs in U87 and U251 cells, and the expression levels of total AKT, p-AKT, CDK-6, and CDK-6 were measured by Western blot. (B) MDK was stably knocked down by two independent shRNAs in U87 and U251 cells, and the proliferation rate was measured for 96 h. The data represent ± SEM of four experiments. (C) MDK was stably knocked down by two independent shRNAs in U87 and U251 cells before the cells were injected into the nude mice. The control group and MDK-knockdown group each included five mice. The tumor volume was measured for 4 wk. (D) SP1 was stably knocked down by two independent shRNAs in U87 and U251 cells, and the expression levels of total AKT, p-AKT, CDK-6, and CDK-6 were measured by Western blot. (E) SP1 was stably knocked down by two independent shRNAs in U87 and U251 cells, and the proliferation rate was measured for 96 h. The data represent ± SEM of four experiments. (F) SP1 was stably knocked down by two independent shRNAs in U87 and U251 cells before the cells were injected into the nude mice. The control group and SP1-knockdown group each included five mice. The tumor volume was measured for 4 wk. (G) MDK and SP1 expression plasmids were stably transfected into A172 glioma cells, respectively. The expression levels of total AKT, p-AKT, CDK-6, and CDK-6 were measured by Western blot. (H) The cell proliferation rate was measured for 96 h in SP1 or MDK stably overexpressed A172 cells and control cells. The data represent ± SEM of four experiments. (I) SP1 or MDK stably overexpressed A172 cells and control cells were injected into nude mice. The tumor volume was measured for 4 wk. The data represent ± SEM of five mice in each group.
Overexpression of MDK antagonized the effects of SP1 silencing
To further investigate the interaction of SP1 and MDK in the development of glioma, we overexpressed MDK in U87 and U251 cells, which we transfected with lentiviral vectors expressing SP1 siRNA. With SP1 silencing, the expression of MDK and phosphorylation of related proteins, including p-Akt, p-ERK, CDK4, and CDK6, were significantly reduced. However, overexpression of MDK partially antagonized the effects of SP1 silencing, indicated by the partial rescue of p-Akt, p-ERK, CDK4, and CDK6 expression (Figure 6A) and the fact that the cell proliferation rate was significantly faster after MDK was reexpressed in SP1-silenced U87 and U251 cells (Figure 6B). In addition, reexpression of MDK in SP1 stably silenced U87 and U251 cells reactivated their capacity for tumorigenesis in nude mice (Figure 6C). Together our results indicate that SP1 regulated glioma cell proliferation and tumorigenesis, as least in part, through direct regulation of MDK expression.
FIGURE 6:

Overexpression of MDK antagonized the effects of SP1 silencing. (A) Western blot was conducted to evaluate the expression of MDK, AKT, and various related proteins in U87 and U251 cells with SP1 stably silenced or SP1 stably silenced plus MDK overexpression. SP1 silencing significantly down-regulated the expression of MDK and phosphorylated AKT, ERK, CDK4, and CDK6, but MDK overexpression antagonized the effects of SP1 silencing on the expression of these proteins. (B) The proliferation rate of the control group, SP1 stably silenced group, and SP1 stably silenced plus MDK overexpression group were measured in U87 and U251 cells for 96 h. The data represent ± SEM of four experiments. (C) The in vivo tumorigenicity of the control group, SP1 stably silenced group, and SP1 stably silenced plus MDK overexpression group was measured in U87 and U251 cells for 4 wk. The data represent ± SEM of five mice in each group.
DISCUSSION
Tumorigenesis is a multistep process in which cells accumulate multiple genetic alterations as they progress to a more malignant phenotype. Although a number of genetic factors, such as alternation of genes p53, PTEN, and EGFR, are associated with the pathogenesis of glioma (Nagane et al., 1997; Melin, 2011), the molecular biology of glioma is still largely unknown. In this study, we demonstrated that the transcriptional factor SP1, which has been shown to be involved in many cancers (Bernsen et al., 1999; Li and Davie, 2010; Hsu et al., 2012; Zhao et al., 2013), is involved in the development of glioma cells by directly up-regulating the expression of MDK, a heparin-binding growth factor associated with the development of glioma (Kang et al., 2007; Lorente et al., 2011; Xu et al., 2012; Hao et al., 2013). We found that human glioma cells constitutively express MDK and that the level strongly correlates with the levels of SP1 expression. Moreover, we found that SP1 transactivates MDK through direct binding to the MDK gene promoter. Overexpression of SP1 significantly increases the expression of MDK and the ability of glioma cells to proliferate, whereas inhibition of SP1 decreases MDK expression and glioma proliferation. Forced overexpression of MDK in SP1-silenced gliomas partially rescues the ability of gliomas to proliferate, both in vitro and in vivo. Thus it appears that overexpression of SP1 results in MDK overexpression and is critical for proliferation in human glioma cells.
SP1 family transcription factors play a vital role in various cellular processes and have been shown to be associated with tumorigenesis in many types of cancer (Li and Davie, 2010; Fulciniti et al., 2011; Hsu et al., 2012; Zhao et al., 2013; Lee et al., 2014), including glioma (Ishibashi et al., 2000; Lin et al., 2011; Castro-Gamero et al., 2013). SP1 typically binds to the GC-rich promoter element through three Cys2His2-type zinc fingers and regulates the expression of targeted genes that are involved in several cellular processes, including the growth and proliferation of tumor cells (Black et al., 2001; Wang et al., 2003; Mansilla and Portugal, 2008; Li and Davie, 2010). In this study, by decreasing sequence analysis of the promoter of MDK, we found that the region between −125 and −85 of the human MDK gene was the binding region of transcriptional factor and has a putative SP1 binding site, GGCGGG, suggesting both that MDK can be regulated by SP1 and that it is involved in the proliferation of glioma. There have been several studies addressing how SP1 influences the outcome of glioma. For instance, Guan et al. (2012) demonstrated that SP1 controls glioma invasion by directly regulating MMP-2 expression. Seznec et al. (2011) reported that SP1-specific inhibitor mithramycin A is a potential therapeutic drug for glioblastoma. SP1 also seems to influence the self-renewal ability of glioma initiating/stem cells by regulating the CD133 promoter (Gopisetty et al., 2013). In a more recent report, SP1 expression was confirmed as a contributor to Bcl-w–induced aggressiveness in glioblastoma (Lee et al., 2014). All of these studies indicate that SP1 is crucial to glioma tumorigenesis in multiple ways.
Several studies have demonstrated the importance of MDK in cell transformation, growth, survival, migration, and angiogenesis (Choudhuri et al., 1997; Owada et al., 1999; Stoica et al., 2002; Nobata et al., 2005). MDK has been shown to inhibit apoptosis through activation of ERK1/2 and PI3K/AKT pathways in neurons, cardiomyocytes, and cancer cells (Owada et al., 1999; Horiba et al., 2006; Tong et al., 2007; Jin et al., 2008). MDK is known to activate not only the PI3K/AKT pathway, but also the MAPK pathway in primary neuronal culture (Owada et al., 1999). Zhu et al. (2013b) found that serum MDK was significantly elevated in most hepatocellular carcinomas, including those with negative AFP and at an early stage, which may serve as a novel diagnostic marker in the early diagnosis and postoperative monitoring of hepatocellular carcinomas (Zhu et al., 2013b). Lorente et al. (2011) showed that MDK is directly involved in the resistance of glioma cells to cannabinoid treatment through the MDK/ALK axis. Given all of this evidence and the fact that MDK overexpression is widely seen in multiple human malignancies, targeting MDK is potentially a novel therapeutic strategy in future treatment. For example, Hao et al. (2013) found that inhibition of MDK by a novel small-molecule compound (iMDK) efficiently suppressed the growth of H441 lung adenocarcinoma cells by inhibiting the PI3K pathway and inducing apoptosis. However, little is known about the upstream regulation and mechanisms of MDK expression in glioma. To our knowledge, this is the first report to show that MDK is a direct target of SP1. This study furthers our understanding of the mechanism of action of SP1 in glioma oncogenesis.
In summary, we identified a putative transcriptional factor, SP1, and showed its role in the development of glioma by binding to the promoter of MDK and up-regulating the expression of MDK, which is aberrantly activated in glioma cells. Our study points to potentially rich new treatment strategies in glioma that target MDK and SP1.
MATERIALS AND METHODS
Primary human glioma specimens and immunohistochemical analysis
We stained tissue sections from paraffin-embedded glioma specimens with an antibody against human MDK or an anti–human SP1 antibody (Santa Cruz Biotechnology, Dallas, TX). We quantitatively scored the tissue sections according to the percentage of positive cells and staining intensity, using the following criteria. We assigned a score of 0 if 0% of the tumor cells showed positive staining; 1 if 0–1% of cells were stained; 2 if 1–10% were stained; 3 if 11–30% were stained; 4 if 31–70% were stained; and 5 if 71–100% were stained. We rated the intensity of staining on a scale of 0 to 3: 0, negative; 1, weak; 2, moderate; and 3, strong. We then combined the proportion and intensity scores to obtain a total score (range, 0–8). The use of human glioma specimens was approved by the institutional review board at Sun Yat-sen University.
Cell lines
Primary NHAs were purchased from Sciencell Research Laboratories (Carlsbad, CA) and cultured according to the manufacturer's instructions. Glioma cell lines, including U118, A172, U87, U251, LN18, LN464, LN382, and LN444, were kindly provided by Shiyuan Chen, Northwestern University, and grown in DMEM supplemented with 10% fetal bovine serum (Life Technologies, Carlsbad, CA).
Western blot
Western blot was performed according to standard methods as described previously (Li et al., 2008), using anti-MDK and anti-SP1 (Santa Cruz Biotechnology), anti-AKT, anti-p-AKT, anti-ERK, anti–CDK-4, and anti–CDK-6 antibodies (Cell Signaling Technology, Boston, MA). The membranes were stripped and reprobed with an anti–α-tubulin antibody (Sigma-Aldrich, St. Louis, MO) or glyceraldehyde-3-phosphate dehydrogenase antibody (Cell Signaling Technology) as a loading control. After washing, the membranes were incubated with horseradish peroxidase–conjugated goat anti–mouse or anti–rabbit secondary antibodies (Jackson ImmunoResearch, West Grove, PA) and visualized using enhanced chemiluminescence reagents (Forevergen Biosciences, Guangzhou, China).
Isolation of total RNA and RT-PCR
Total RNA was isolated from cultured glioma cells and fresh glioma tissues using TRIzol RNA isolation reagent (Life Technologies) according to the manufacturer's instructions. RNA integrity and concentration were determined using Agilent RNA 6000 Nano Kit and Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA). Reverse transcription was performed using SuperscriptIII reverse transcriptase (Life Technologies) and oligo-dT primers. The primers used were mdk forward, 5′-CGCGGTCGCCAAAAAGAAAG-3′; mdk reverse, 5′-TACTTGCAGTCGGCTCCAAAC-3′; sp1 forward, 5′-TGGCAGCAGTACCAATGGC-3′; sp1 reverse, 5′-CCAGGTAGTCCTGTCAGAACTT-3′; β-actin forward, 5′-TGGATCAGCAAGCAGGAGTA-3′; and β-actin reverse, 5′-TCGGCCACATTGTGAACTTT-3′.
Luciferase assay
Glioma cells were seeded in triplicate in 48-well plates and allowed to settle for 24 h. A 100-ng amount of luciferase reporter plasmid or the control-luciferase plasmid, plus 1 ng of pRL-TK Renilla plasmid (Promega, Madison, WI), was transfected into glioma cells using Lipofectamine 2000 reagent (Life Technologies) according to the manufacturer's recommendation. Luciferase and Renilla signals were measured 24 h after transfection using the Dual Luciferase Reporter Assay Kit (Promega, Madison, WI) according to a protocol provided by the manufacturer. Three independent experiments were performed, and the data are presented as means ± SD.
Determination of the MDK promoter region
Human MDK gene sequence (GenBank accession number 4192) was retrieved from the National Center for Biotechnology Information GenBank database. Different regions of MDK upstream (−562/+33, −422/+33, −158/+33, −125/+33, −85/+33) were amplified from NHA cells and inserted into luciferase reporter gene vector pGL3-Basic to generate a set of MDK promoter-reporter constructs. Promoter activities of all of the MDK promoter constructs were analyzed by luciferase assay. U251 and U87 were cotransfected with MDK promoter plasmid and pRL-TK for 24 h. Activities of firefly and Renilla luciferase were measured by the Dual-Luciferase Reporter assay system. Firefly luciferase activity was normalized with Renilla luciferase activity. The experiments were repeated four times, with triplicates for each condition. The data represent ± SEM of four experiments.
Determination of the SP1 binding site on MDK promoter by site-directed mutagenesis
To investigate the interaction of SP1 and MDK and understand the regulation of SP1 in the expression of MDK, the predicted binding region of SP1 was mutated (GGCGGG → TTCGGG) and tested by luciferase assay. Site-directed mutagenesis was conducted using a QuikChange II site-directed mutagenesis kit according to the manufacturer's instructions (Stratagene, La Jolla, CA), as well as the protocol described by Kriwacki et al. (1992). Primers used for introduction of point mutations were forward primer, 5′-CTGGGGGCTGGATTCGGGGGTGGGGGTC-3′, and reverse primer, 5′-ACCCCCACCCCCGAATCCAGCCCCCAGC-3′. Mutations of the binding region of MDK promoter were confirmed by DNA sequencing. The influence of mutation of the promoter was analyzed by comparing the luciferase activity between mutation plasmid (pmdk -Luc-mut) and wild-type plasmid (pmdk -Luc-wt).
Chromatin immunoprecipitation assay
The ChIP assay was performed using the EZ-ChIP Chromatin Immunoprecipitation Kit (Millipore, Billerica, MA) according to the manufacturer's instructions. Briefly, glioma cells were grown in 100-mm Petri dishes (∼80–90% confluent) and washed with phosphate-buffered saline, pH 7.4, before ChIP assay using the appropriate antibody. Approximately 5 × 107 cells were cross-linked using formaldehyde and broken down with ultrasound. SP1/DNA complex was immunoprecipitated using anti-SP1 antibody and treated with proteinase K and RNase A at 55°C for 15 min and incubated at 65°C overnight to release cross-links. PCR was used to detect mdk DNA in the SP1-precipitated protein/DNA complex using primers 5′-GGCGGCCGGAGCGGGACGGG-3′ (mdk forward primer) and 5′-GGGGCGGCCCCTCGCCGCTA-3′ (mdk reverse primer).
Generation of lentiviruses
SP1- and MDK-expressing plasmids were constructed by cloning the cDNA encoding SP1 and MDK into the pCDH-CMV-MCS-EF1-GFP-Puro vector (System Biosciences, Mountain View, CA). RNA interference sequences were SP1-1, 5′-GCAGTACCAATGGCAGCAATG-3′; SP1-2, 5′-GCAGACCTTTACAACTCAA-3′; MDK-1, 5′-GTTTGGAGCCGACTGCAAG-3′; and MDK-2, 5′-CCGACTGCAAGTACAAGTT-3′; and the scramble sequence for negative control was 5′-TTCTCCGAACGTGTCACGT-3′. The shRNA expression cassettes containing sense-loop (TTCAAGAGA)-antisense-termination signal T6 were inserted downstream of the U6 promoter of pLL3.7-Puro vector. The expression vectors were mixed with plasmids pGag/Pol, pRev, and pVSV-G and transfected into 293T cells using Lipofectamine 2000. Supernatant was collected, and infections were carried out in the presence of 5–10 μg/ml Polybrene. Cells were selected with 2 μg/ml puromycin after transduction.
Proliferation of glioma cells (MTS assay)
Cell viability was quantitated by 3-(4,5-dimethylthiazol-2-yl)-5- (3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assays in 96-well, flat-bottom plates. Glioma cells were plated at a density of 1 × 103/well in 96-well plates. After 24 h, test drugs were added, and the cultures were incubated for different time periods (24, 48, 72, and 96 h) at 37°C. The treated cells were then stained with MTS reagent in complete medium for 4 h. The absorbance values of each well were measured using a microplate reader (Diatek, San Diego, CA) at a wavelength of 490 nm. Each experiment was performed in triplicate and repeated three times. The mean value was calculated, and the proliferation curves were constructed.
Animal experiments
Female BALB/c nude mice were purchased from the Sun Yat-sen University Laboratory Animal Center. U87 and U251 glioma cells were removed from culture flasks by trypsinization and then washed and resuspended in phosphate-buffered saline. The cells (2.5 × 106 cells/50 μl) were transplanted subcutaneously into the right flanks of 4-wk-old mice. The grown tumors were measured daily with a Vernier caliper. The length (L) and width (W; in millimeters) of the tumors were taken and used to calculate the volume (V) by using the formula V = (W2L)/2.
Statistical analysis
Data are shown as mean ± SD. The significance of differences in the human glioblastoma multiforme data was determined using Pearson's correlation test, for in vitro data using Student's t test (two-tailed), and for in vivo data using the Mann–Whitney U test. p < 0.05 was considered significant.
Human and animal studies
All human and animal studies were approved by the Institute Research Medical Ethics Committee of Sun Yat-sen University. All human studies were performed in accordance with the ethical standards laid down in the 1975 Declaration of Helsinki and its later amendments.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81370072, 81072082), the Fundamental Research Funds for the Central Universities (11ykzd06), the Natural Science Foundation of Guangdong Province for Distinguished Young Scholars (S2013050014535), the Key Project Supported by the Science and Technology Planning Project of Guangdong Province (S2012020006147), Pearl River New Star Science, the technology program of Guangzhou City (2013J2200022), the Innovative Funds for Small and Medium-Sized Enterprises of Guangzhou (1210000302), the PhD Start-up Fund of the Natural Science Foundation of Guangdong Province (S2013040014989), and the Medical Scientific Research Foundation of Guangdong Province (B2013144).
Abbreviations used:
- ChIP
chromatin immunoprecipitation
- MDK
midkine
- NHA
normal human astrocytes
- REMBRANDT
Repository for Molecular Brain Neoplasia Data
- RT
reverse transcription
- SP1
specificity protein 1.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-10-1443) on November 26, 2014.
*These authors contributed equally to this work.
The authors declare that they have no competing interests.
REFERENCES
- Bernsen HJ, Rijken PF, Hagemeier NE, van der Kogel AJ. A quantitative analysis of vascularization and perfusion of human glioma xenografts at different implantation sites. Microvasc Res. 1999;57:244–257. doi: 10.1006/mvre.1999.2143. [DOI] [PubMed] [Google Scholar]
- Black AR, Black JD, Azizkhan-Clifford J. Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001;188:143–160. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- Castro-Gamero AM, Borges KS, Moreno DA, Suazo VK, Fujinami MM, de Paula GQ R, de Oliveira HF, Carlotti CJ, Scrideli CA, Tone LG. Tetra-O-methyl nordihydroguaiaretic acid, an inhibitor of Sp1-mediated survivin transcription, induces apoptosis and acts synergistically with chemo-radiotherapy in glioblastoma cells. Invest New Drugs. 2013;31:858–870. doi: 10.1007/s10637-012-9917-4. [DOI] [PubMed] [Google Scholar]
- Choudhuri R, Zhang HT, Donnini S, Ziche M, Bicknell R. An angiogenic role for the neurokines midkine and pleiotrophin in tumorigenesis. Cancer Res. 1997;57:1814–1819. [PubMed] [Google Scholar]
- Fulciniti M, Amin S, Nanjappa P, Rodig S, Prabhala R, Li C, Minvielle S, Tai YT, Tassone P, Avet-Loiseau H, et al. Significant biological role of sp1 transactivation in multiple myeloma. Clin Cancer Res. 2011;17:6500–6509. doi: 10.1158/1078-0432.CCR-11-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopisetty G, Xu J, Sampath D, Colman H, Puduvalli VK. Epigenetic regulation of CD133/PROM1 expression in glioma stem cells by Sp1/myc and promoter methylation. Oncogene. 2013;32:3119–3129. doi: 10.1038/onc.2012.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan H, Cai J, Zhang N, Wu J, Yuan J, Li J, Li M. Sp1 is upregulated in human glioma, promotes MMP-2-mediated cell invasion and predicts poor clinical outcome. Int J Cancer. 2012;130:593–601. doi: 10.1002/ijc.26049. [DOI] [PubMed] [Google Scholar]
- Hao H, Maeda Y, Fukazawa T, Yamatsuji T, Takaoka M, Bao XH, Matsuoka J, Okui T, Shimo T, Takigawa N, et al. Inhibition of the growth factor MDK/midkine by a novel small molecule compound to treat non-small cell lung cancer. PLoS One. 2013;8:e71093. doi: 10.1371/journal.pone.0071093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiba M, Kadomatsu K, Yasui K, Lee JK, Takenaka H, Sumida A, Kamiya K, Chen S, Sakuma S, Muramatsu T, et al. Midkine plays a protective role against cardiac ischemia/reperfusion injury through a reduction of apoptotic reaction. Circulation. 2006;114:1713–1720. doi: 10.1161/CIRCULATIONAHA.106.632273. [DOI] [PubMed] [Google Scholar]
- Hsu TI, Wang MC, Chen SY, Yeh YM, Su WC, Chang WC, Hung JJ. Sp1 expression regulates lung tumor progression. Oncogene. 2012;31:3973–3988. doi: 10.1038/onc.2011.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibusuki M, Fujimori H, Yamamoto Y, Ota K, Ueda M, Shinriki S, Taketomi M, Sakuma S, Shinohara M, Iwase H, et al. Midkine in plasma as a novel breast cancer marker. Cancer Sci. 2009;100:1735–1739. doi: 10.1111/j.1349-7006.2009.01233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikematsu S, Yano A, Aridome K, Kikuchi M, Kumai H, Nagano H, Okamoto K, Oda M, Sakuma S, Aikou T, et al. Serum midkine levels are increased in patients with various types of carcinomas. Br J Cancer. 2000;83:701–706. doi: 10.1054/bjoc.2000.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi H, Nakagawa K, Onimaru M, Castellanous EJ, Kaneda Y, Nakashima Y, Shirasuna K, Sueishi K. Sp1 decoy transfected to carcinoma cells suppresses the expression of vascular endothelial growth factor, transforming growth factor beta1, and tissue factor and also cell growth and invasion activities. Cancer Res. 2000;60:6531–6536. [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- Jin Z, Lahat G, Korchin B, Nguyen T, Zhu QS, Wang X, Lazar AJ, Trent J, Pollock RE, Lev D. Midkine enhances soft-tissue sarcoma growth: a possible novel therapeutic target. Clin Cancer Res. 2008;14:5033–5042. doi: 10.1158/1078-0432.CCR-08-0092. [DOI] [PubMed] [Google Scholar]
- Kadomatsu K, Muramatsu T. Midkine and pleiotrophin in neural development and cancer. Cancer Lett. 2004;204:127–143. doi: 10.1016/S0304-3835(03)00450-6. [DOI] [PubMed] [Google Scholar]
- Kaname T, Kuwano A, Murano I, Uehara K, Muramatsu T, Kajii T. Midkine gene (MDK), a gene for prenatal differentiation and neuroregulation, maps to band 11p11.2 by fluorescence in situ hybridization. Genomics. 1993;17:514–515. doi: 10.1006/geno.1993.1359. [DOI] [PubMed] [Google Scholar]
- Kang HC, Kim IJ, Park HW, Jang SG, Ahn SA, Yoon SN, Chang HJ, Yoo BC, Park JG. Regulation of MDK expression in human cancer cells modulates sensitivities to various anticancer drugs: MDK overexpression confers to a multi-drug resistance. Cancer Lett. 2007;247:40–47. doi: 10.1016/j.canlet.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Kishida S, Kadomatsu K. Involvement of midkine in neuroblastoma tumourigenesis. Br J Pharmacol. 2014;171:896–904. doi: 10.1111/bph.12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno S, Nakagawa K, Hamada K, Harada H, Yamasaki K, Hashimoto K, Tagawa M, Nagato S, Furukawa K, Ohnishi T. Midkine promoter-based conditionally replicative adenovirus for malignant glioma therapy. Oncol Rep. 2004;12:73–78. [PubMed] [Google Scholar]
- Kriwacki RW, Schultz SC, Steitz TA, Caradonna JP. Sequence-specific recognition of DNA by zinc-finger peptides derived from the transcription factor Sp1. Proc Natl Acad Sci USA. 1992;89:9759–9763. doi: 10.1073/pnas.89.20.9759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WS, Kwon J, Yun DH, Lee YN, Woo EY, Park MJ, Lee JS, Han YH, Bae IH. Specificity protein 1 expression contributes to Bcl-w-induced aggressiveness in glioblastoma multiforme. Mol Cells. 2014;37:17–23. doi: 10.14348/molcells.2014.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang N, Song LB, Liao WT, Jiang LL, Gong LY, Wu J, Yuan J, Zhang HZ, Zeng MS, et al. Astrocyte elevated gene-1 is a novel prognostic marker for breast cancer progression and overall patient survival. Clin Cancer Res. 2008;14:3319–3326. doi: 10.1158/1078-0432.CCR-07-4054. [DOI] [PubMed] [Google Scholar]
- Li L, Davie JR. The role of Sp1 and Sp3 in normal and cancer cell biology. Ann Anat. 2010;192:275–283. doi: 10.1016/j.aanat.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Lin PC, Lin SZ, Chen YL, Chang JS, Ho LI, Liu PY, Chang LF, Harn YC, Chen SP, Sun LY, et al. Butylidenephthalide suppresses human telomerase reverse transcriptase (TERT) in human glioblastomas. Ann Surg Oncol. 2011;18:3514–3527. doi: 10.1245/s10434-011-1644-0. [DOI] [PubMed] [Google Scholar]
- Lorente M, Torres S, Salazar M, Carracedo A, Hernandez-Tiedra S, Rodriguez-Fornes F, Garcia-Taboada E, Melendez B, Mollejo M, Campos-Martin Y, et al. Stimulation of the midkine/ALK axis renders glioma cells resistant to cannabinoid antitumoral action. Cell Death Differ. 2011;18:959–973. doi: 10.1038/cdd.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Li H, Wang B, Shen Q, Cui E, Min L, Qian F, Ping J, Dai L. Midkine mRNA level in peripheral blood mononuclear cells is a novel biomarker for primary non-small cell lung cancer: a prospective study. J Cancer Res Clin Oncol. 2013;139:557–562. doi: 10.1007/s00432-012-1357-1. [DOI] [PubMed] [Google Scholar]
- Maeda N, Ichihara-Tanaka K, Kimura T, Kadomatsu K, Muramatsu T, Noda M. A receptor-like protein-tyrosine phosphatase PTPzeta/RPTPbeta binds a heparin-binding growth factor midkine. Involvement of arginine 78 of midkine in the high affinity binding to PTPzeta. J Biol Chem. 1999;274:12474–12479. doi: 10.1074/jbc.274.18.12474. [DOI] [PubMed] [Google Scholar]
- Mansilla S, Portugal J. Sp1 transcription factor as a target for anthracyclines: effects on gene transcription. Biochimie. 2008;90:976–987. doi: 10.1016/j.biochi.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Melin B. Genetic causes of glioma: new leads in the labyrinth. Curr Opin Oncol. 2011;23:643–647. doi: 10.1097/CCO.0b013e32834a6f61. [DOI] [PubMed] [Google Scholar]
- Minniti G, Muni R, Lanzetta G, Marchetti P, Enrici RM. Chemotherapy for glioblastoma: current treatment and future perspectives for cytotoxic and targeted agents. Anticancer Res. 2009;29:5171–5184. [PubMed] [Google Scholar]
- Mishima K, Asai A, Kadomatsu K, Ino Y, Nomura K, Narita Y, Muramatsu T, Kirino T. Increased expression of midkine during the progression of human astrocytomas. Neurosci Lett. 1997;233:29–32. doi: 10.1016/s0304-3940(97)00619-8. [DOI] [PubMed] [Google Scholar]
- Miyashiro I, Kaname T, Shin E, Wakasugi E, Monden T, Takatsuka Y, Kikkawa N, Muramatsu T, Monden M, Akiyama T. Midkine expression in human breast cancers: expression of truncated form. Breast Cancer Res Treat. 1997;43:1–6. doi: 10.1023/a:1005748728351. [DOI] [PubMed] [Google Scholar]
- Mrugala MM. Advances and challenges in the treatment of glioblastoma: a clinician's perspective. Discov Med. 2013;15:221–230. [PubMed] [Google Scholar]
- Muramatsu H, Zou K, Sakaguchi N, Ikematsu S, Sakuma S, Muramatsu T. LDL receptor-related protein as a component of the midkine receptor. Biochem Biophys Res Commun. 2000;270:936–941. doi: 10.1006/bbrc.2000.2549. [DOI] [PubMed] [Google Scholar]
- Muramatsu T. Midkine, a heparin-binding cytokine with multiple roles in development, repair and diseases. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:410–425. doi: 10.2183/pjab.86.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu T. Midkine: a promising molecule for drug development to treat diseases of the central nervous system. Curr Pharm Des. 2011;17:410–423. doi: 10.2174/138161211795164167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagane M, Huang HJ, Cavenee WK. Advances in the molecular genetics of gliomas. Curr Opin Oncol. 1997;9:215–222. doi: 10.1097/00001622-199709030-00001. [DOI] [PubMed] [Google Scholar]
- Nakada M, Kita D, Watanabe T, Hayashi Y, Teng L, Pyko IV, Hamada J. Aberrant signaling pathways in glioma. Cancers (Basel) 2011;3:3242–3278. doi: 10.3390/cancers3033242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobata S, Shinozawa T, Sakanishi A. Truncated midkine induces transformation of cultured cells and short latency of tumorigenesis in nude mice. Cancer Lett. 2005;219:83–89. doi: 10.1016/j.canlet.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Ohuchida T, Okamoto K, Akahane K, Higure A, Todoroki H, Abe Y, Kikuchi M, Ikematsu S, Muramatsu T, Itoh H. Midkine protects hepatocellular carcinoma cells against TRAIL-mediated apoptosis through down-regulation of caspase-3 activity. Cancer. 2004;100:2430–2436. doi: 10.1002/cncr.20266. [DOI] [PubMed] [Google Scholar]
- Owada K, Sanjo N, Kobayashi T, Mizusawa H, Muramatsu H, Muramatsu T, Michikawa M. Midkine inhibits caspase-dependent apoptosis via the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase in cultured neurons. J Neurochem. 1999;73:2084–2092. [PubMed] [Google Scholar]
- Seznec J, Silkenstedt B, Naumann U. Therapeutic effects of the Sp1 inhibitor mithramycin A in glioblastoma. J Neurooncol. 2011;101:365–377. doi: 10.1007/s11060-010-0266-x. [DOI] [PubMed] [Google Scholar]
- Stoica GE, Kuo A, Powers C, Bowden ET, Sale EB, Riegel AT, Wellstein A. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J Biol Chem. 2002;277:35990–35998. doi: 10.1074/jbc.M205749200. [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- Tong Y, Mentlein R, Buhl R, Hugo HH, Krause J, Mehdorn HM, Held-Feindt J. Overexpression of midkine contributes to anti-apoptotic effects in human meningiomas. J Neurochem. 2007;100:1097–1107. doi: 10.1111/j.1471-4159.2006.04276.x. [DOI] [PubMed] [Google Scholar]
- Wang L, Wei D, Huang S, Peng Z, Le X, Wu TT, Yao J, Ajani J, Xie K. Transcription factor Sp1 expression is a significant predictor of survival in human gastric cancer. Clin Cancer Res. 2003;9:6371–6380. [PubMed] [Google Scholar]
- Weckbach LT, Muramatsu T, Walzog B. Midkine in inflammation. ScientificWorldJournal. 2011;11:2491–2505. doi: 10.1100/2011/517152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- Xu YY, Mao XY, Song YX, Zhao F, Wang ZN, Zhang WX, Xu HM, Jin F. Midkine confers Adriamycin resistance in human gastric cancer cells. Tumour Biol. 2012;33:1543–1548. doi: 10.1007/s13277-012-0406-3. [DOI] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zhang W, Guo Z, Ma F, Wu Y, Bai Y, Gong W, Chen Y, Cheng T, Zhi F, et al. Inhibition of the transcription factor Sp1 suppresses colon cancer stem cell growth and induces apoptosis in vitro and in nude mouse xenografts. Oncol Rep. 2013;30:1782–1792. doi: 10.3892/or.2013.2627. [DOI] [PubMed] [Google Scholar]
- Zhu CY, Lv YP, Yan DF, Gao FL. Knockdown of MDR1 increases the sensitivity to adriamycin in drug resistant gastric cancer cells. Asian Pac J Cancer Prev. 2013a;14:6757–6760. doi: 10.7314/apjcp.2013.14.11.6757. [DOI] [PubMed] [Google Scholar]
- Zhu WW, Guo JJ, Guo L, Jia HL, Zhu M, Zhang JB, Loffredo CA, Forgues M, Huang H, Xing XJ, et al. Evaluation of midkine as a diagnostic serum biomarker in hepatocellular carcinoma. Clin Cancer Res. 2013b;19:3944–3954. doi: 10.1158/1078-0432.CCR-12-3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.