

Abstract
Rationale
Low-efficacy mu opioid receptor agonists may be useful for some clinical indications, but clinically available low-efficacy mu agonists also have low selectivity for mu vs. kappa opioid receptors. NAQ is a novel opioid receptor ligand with low-efficacy at mu receptors and greater mu-receptor selectivity than existing low-efficacy agonists.
Objectives
This study examined behavioral effects of NAQ in rats using an intracranial self-stimulation (ICSS) procedure that has been used previously to examine other opioids. NAQ effects were examined before, during and after chronic morphine treatment, and effects of NAQ were compared to effects of nalbuphine and naltrexone.
Methods
Adult male Sprague-Dawley rats were trained to respond for electrical brain stimulation delivered via electrodes implanted in the medial forebrain bundle. A range of brain stimulation frequencies maintained a wide range of baseline ICSS rates. Effects of NAQ (0.32-10 mg/kg), nalbuphine (1.0 mg/kg) and naltrexone (0.1 mg/kg) were determined before morphine treatment and during treatment with 3.2 and 18 mg/kg/day morphine. NAQ effects were also redetermined beginning two weeks after termination of morphine treatment.
Results
NAQ produced weak ICSS facilitation in morphine-naïve rats but more robust ICSS facilitation during and after morphine treatment and also reversed morphine withdrawal-associated depression of ICSS. These effects were similar to effects of nalbuphine.
Conclusions
These results agree with the in vitro characterization of NAQ as a low-efficacy mu agonist. Opioid exposure may enhance abuse-related effects of NAQ, but NAQ may also serve as a low-efficacy and relatively safe option for treatment of opioid withdrawal or dependence.
Keywords: NAQ, intracranial self-stimulation, rat, morphine, opioid, abuse liability
Introduction
Mu opioid receptor agonists are used for a range of clinical applications that include treatment of pain, diarrhea and cough, and they are also used as maintenance medications to treat opioid addiction (Gutstein and Akil 2006; Negus and Banks 2013). Two important determinants of opioid action are selectivity for, and efficacy at, mu opioid receptors. Regarding selectivity, drugs may bind not only to mu receptors, but also to kappa or delta opioid receptors, or to non-opioid receptors (Emmerson et al. 1994; Raynor et al. 1994). Regarding efficacy, drugs vary along a continuum from high to low efficacy at mu receptors, and mu receptor ligands may also function as antagonists or inverse agonists (Emmerson et al. 1996; Selley et al. 1998). For mu agonists that are currently available clinically, the most selective mu agonists also have high efficacy at mu receptors (e.g. methadone and fentanyl) (Emmerson et al. 1996; Raynor et al. 1994; Selley et al. 1998). Conversely, mu agonists with lower efficacy at mu receptors (e.g. nalbuphine, pentazocine and butorphanol) also have low selectivity for mu receptors, especially for mu vs. kappa opioid receptors (Emmerson et al. 1996; Emmerson et al. 1994; Peng et al. 2007; Raynor et al. 1994; Selley et al. 1998). Low-efficacy mu agonists may have clinical value for some indications because they might retain sufficient efficacy to produce therapeutic effects such as analgesia against modest pain or alleviation of opioid withdrawal, but they might lack sufficient efficacy to produce some side effects such as significant respiratory depression or lethality (Gutstein and Akil 2006; Hoskin and Hanks 1991; Negus and Banks 2013). However, kappa opioid receptors mediate dysphoric subjective effects and some other undesirable effects (Pfeiffer et al. 1986; Walsh et al. 2001), and kappa-mediated effects may be one factor that limits clinical utility of existing low-efficacy opioids. Identification of more selective low-efficacy mu agonists might therefore be useful for some clinical indications.
NAQ (17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-[(3′-isoquinolyl) acetamido]morphinan) has recently been synthesized as a novel opioid receptor ligand, and in vitro studies of receptor binding and functional efficacy suggest that it functions as a low-efficacy mu agonist with greater mu selectivity than existing clinically available low-efficacy mu agonists (Li et al. 2009). In binding studies, NAQ displays nearly 50-fold selectivity for mu vs. kappa receptors, and even greater selectivity for mu vs. delta receptors (Li et al. 2009). Conversely, nalbuphine, pentazocine and butorphanol all display little or no selectivity for mu vs. kappa receptors, and similar or less selectivity than NAQ for mu vs. delta receptors (Emmerson et al. 1994; Li et al. 2009; Peng et al. 2007; Raynor et al. 1994). In functional studies of agonist-stimulated GTPγS binding, NAQ displays low efficacy similar to that of nalbuphine, pentazocine and butorphanol at mu receptors (Emmerson et al. 1996; Li et al. 2009; Selley et al. 1998), but weaker efficacy than these compounds at kappa receptors (Peng et al. 2007; Remmers et al. 1999; Yuan et al. 2011). The mu selectivity and very low kappa efficacy of NAQ both contribute to its potential for in vivo selectivity as a low-efficacy mu agonist.
This study evaluated behavioral effects of NAQ in rats using intracranial self-stimulation (ICSS), a behavioral procedure that has been used previously to examine other opioids that vary in selectivity for, and efficacy at, opioid receptors (Altarifi et al. 2012; Altarifi and Negus 2011; Altarifi et al. 2013; Kornetsky and Esposito 1979; Negus et al. 2010; Pereira Do Carmo et al. 2009; Todtenkopf et al. 2004). This procedure has been useful to dissociate both efficacy and receptor selectivity of opioid agonists, and also because drug effects on ICSS can be interpreted in terms of abuse potential, a key side effect of opioid analgesics (Kornetsky et al. 1979; Negus and Miller 2014; Vlachou and Markou 2011; Wise 1996). NAQ effects were examined before, during and after a regimen of repeated morphine treatment, because previous studies found that a history of opioid exposure can enhance abuse-related effects of opioids on ICSS (Altarifi and Negus 2011; Altarifi et al. 2013). Also, relief of morphine withdrawal by NAQ may suggest consideration of NAQ to treat opioid dependence (Negus and Banks 2013). Lastly, effects of NAQ were compared to effects of nalbuphine and naltrexone, two clinically available mu opioids with low mu vs. kappa selectivity and low or inverse efficacy at mu receptors, respectively (Emmerson et al. 1996; Raynor et al. 1994).
Methods
Subjects
Six male Sprague-Dawley rats (Harlan, Fredrick, MD, USA) weighing 310-350 g at the time of surgery were individually housed and maintained on a 12-h light/dark cycle with lights on from 6:00 a.m. to 6:00 p.m. Rats had free access to food and water except during testing. Animal maintenance and research were in compliance with National Institutes of Health guidelines on care and use of animal subjects in research, and all animal use protocols were approved by the Virginia Commonwealth University Institutional Care and Use Committee.
Apparatus and Training
Training and testing procedures were identical to those described previously for other opioid agonists (Altarifi et al. 2013). Briefly, experiments were conducted in six operant conditioning chambers (Model ENV-007-VP-CT, Med Associates) equipped with a single response lever, stimulus lights, house light, and an ICSS stimulator (Model SG-510, Med Associates) that delivered brain stimulation via an electrode implanted in the left medial forebrain bundle at the level of the lateral hypothalamus (2.8 mm posterior and 1.7 mm lateral from the bregma, and 8.8 mm below the skull). The house light was illuminated during behavioral sessions, and responding under a FR 1 schedule produced a 0.5-s train of 0.1-ms square-wave cathodal pulses (0.1-ms pulse duration) together with 0.5 s illumination of stimulus lights over the response lever. The terminal schedule consisted of sequential 10-min components. During each component, one of a descending series of 10 current frequencies was presented (158-56 Hz in 0.05-log increments), with a 60-s trial at each frequency. Each frequency trial consisted of a 10-s timeout, during which five noncontingent stimulations were delivered at the frequency available during that trial, followed by a 50-s “response” period, during which responding produced electrical stimulation. Training was considered to be complete when two criteria were met. First, rats had to respond at rates greater than 50% MCR (see Data Analysis) during the first three frequency trials of all components for three consecutive days. Second, rats were then tested with saline until these injections had no effect on frequency-rate curves as determined by two-way analysis of variance (see Data Analysis).
Testing
Once training was complete, “pre-drug baseline” sessions were conducted over a period of three consecutive days to establish baseline ICSS performance before administration of any drug. Each pre-drug baseline session consisted of three components as described above. After collecting pre-drug baseline data, testing proceeded in four phases to evaluate NAQ effects before chronic morphine (phase 1), during daily treatment with 3.2 mg/kg/day morphine (phase 2) and 18 mg/kg/day morphine (phase 3), and beginning two weeks after termination of morphine treatment (phase 4). During each phase, each rat received vehicle and NAQ (0.32-10 mg/kg) in a mixed dose order followed by testing with 1.0 mg/kg nalbuphine and 0.1 mg/kg naltrexone in that order. These reference doses of nalbuphine and naltrexone were selected based on results from previous studies (Altarifi et al., 2012; Altarifi et al., 2013).
The first phase of testing started immediately after the pre-drug baseline sessions and lasted for 15 days. Daily ICSS sessions in this and all subsequent phases consisted of three consecutive daily-baseline components followed first by a 30 min time out and then by two ICSS test components. Saline or drug was administered at the beginning of the time out. Thus, ICSS was assessed twice each day: once during the daily-baseline components before that day's injection (and approximately 23 hr after the previous day's injection), and once during the test components that began 30 min after that day's injection. After the last test component, subjects were removed from the test chambers and returned to their home cages. Test sessions with each dose of NAQ, nalbuphine or naltrexone were separated by at least three days. Saline was administered on intervening days. ICSS sessions were sometimes omitted on weekends, but injections were delivered daily.
The second phase started with a 7-day maintenance period, during which 3.2 mg/kg/day morphine was administered during the time out of each daily ICSS session. On day 8, test sessions resumed, and drug effects were redetermined using the same dose order and intervals as the first phase. In addition, two additional test days were also added during which rats were tested with 0.1 mg/kg naltrexone + 1.0 mg/kg NAQ or 0.1 naltrexone + 1.0 mg/kg nalbuphine. For these tests, naltrexone was administered 5 minutes before NAQ or nalbuphine, which was administered 30 min before onset of test ICSS components. On days that subjects did not receive test drug, they received 3.2 mg/kg morphine. As in phase 1, test sessions were separated by at least three days, so that each test session was preceded by at least two days of treatment with the chronic morphine dose. Also, when subjects were tested with vehicle, they received a supplemental dose of 3.2 mg/kg morphine after the ICSS test session on that day, before returning to their home cages. Phase two lasted 25 days.
The third phase began with a gradual increase in the morphine dose administered during the time out of consecutive daily ICSS sessions. Subjects received 5.6 mg/kg/day morphine for 2 days, followed by 10 mg/kg/day morphine for 4 days and 18 mg/kg/day for the remainder of the third phase. Once the terminal dose of 18 mg/kg/day morphine was achieved, it was maintained for seven days. Subsequently, test sessions resumed, and drug effects were redetermined using the same dose order and intervals as the first phase. Subjects in this phase received 18 mg/kg/day morphine on the days that they did not receive test drug, and 18 mg/kg morphine was also administered at the end of test sessions during which vehicle or low NAQ doses (0.32-1.0 mg/kg) were tested. Phase three lasted 26 days.
At the end of the third phase, daily morphine injections were terminated, and ICSS sessions and drug treatments were suspended for two weeks. Training was then resumed for three days, after which NAQ effects were redetermined using the same dose order and intervals as the first phase. Nalbuphine and naltrexone were not tested during this phase.
Fecal Bolus Production
During repeated 18 mg/kg/day morphine, operant chambers were scored for the presence of fecal boli at the end of each test session. The number of subjects with two or more fecal boli was counted for each treatment as a sign of enhanced gastrointestinal transit often associated with morphine withdrawal.
Data Analysis
The primary dependent variable was the reinforcement rate in stimulations/trial during each frequency trial. To normalize these raw data, reinforcement rates from each trial in each rat were converted to Percent Maximum Control Rate (%MCR) for that rat. The maximum control rate was determined for each rat during the pre-drug baseline sessions at the beginning of the experiment. The first component from these sessions (and from all other sessions) was considered to be an acclimation component, and data were discarded. The maximum control rate was defined as the mean of the maximal rates observed during any frequency trial of the second and third components of the three pre-drug baseline sessions (six total pre-drug baseline components). Subsequently, %MCR for each trial was calculated as (Reinforcement Rate During a Frequency Trial ÷ Maximum Control Rate) × 100. Graphs show mean frequency-rate curves, with brain stimulation frequency on the abscissa, and ICSS rate expressed as %MCR on the ordinate.
Frequency-rate curves from test sessions during each phase of the study were submitted for statistical analysis. As noted above, these frequency-rate curves were assessed twice on each test day: once during daily baseline components before that day's injection (and approximately 23 hr after the previous day's injection), and again during test components that began 30 min after that day's injection. Daily baseline data and test data from test sessions were analyzed separately. The daily baseline data provided information on changes in baseline ICSS produced by the chronic treatment (saline, 3.2 mg/kg/day morphine, 18 mg/kg/day morphine, or protracted morphine withdrawal). During chronic morphine treatment, daily baseline components were conducted approximately 23 hr after the most recent morphine injection, and as a result, these results provided data on changes in ICSS produced by 23 hr withdrawal from the morphine maintenance dose. Daily baseline data from test days within each phase were averaged across all rats to yield mean baseline ICSS data during chronic treatment with saline, 3.2 mg/kg/day morphine, 18/mg/kg/day morphine, and protracted morphine withdrawal. Mean daily-baseline data during each phase were compared to the pre-drug baseline data using two-way ANOVA, with phase of chronic treatment as one factor and ICSS frequency as the other factor. A significant ANOVA was followed by a Holm-Sidak post hoc test, and the criterion for significance was set at p < 0.05. To permit within-subject analysis, data were included only for those rats that completed all four phases of chronic morphine treatment (N=5; one rat lost its head cap and electrode at the end of phase 2, and was not tested in phases 3 and 4).
Test data from each test session were averaged across rats within each condition and analyzed to assess drug effects on ICSS frequency-rate curves during each phase of chronic morphine/vehicle treatment. Within each phase, ICSS test data for vehicle and NAQ were compared by two-way ANOVA, with NAQ dose as one factor and ICSS frequency as the other factor. A similar approach was used to compare effects of vehicle, nalbuphine and naltrexone. A significant ANOVA was followed by a Holm-Sidak post hoc test, and the criterion for significance was set at p < 0.05. Data were included for all rats that completed a given phase. (N=6 for saline and 3.2 mg/kg/day morphine, N=5 for 18 mg/kg/day morphine and post-morphine).
To provide an additional summary measure of baseline and test ICSS performance, the total number of stimulations per component was calculated as the average of the total stimulations delivered across all 10 frequency trials of each component. Baseline and test data were expressed as a percentage of the total stimulations per component earned during the “pre-drug baseline” components (% Control). Thus, % Control was calculated as (Mean Total Stimulations During Daily Baseline or Test Components ÷ Mean Total Stimulations During Pre-Drug Baseline Components) × 100.
Drugs
Morphine sulfate and naltrexone HCl were provided by the National Institute on Drug Abuse Drug Supply Program (Bethesda, MD). Nalbuphine HCl was provided by Dr. Kenner Rice (Chemical Biology Branch, National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD). NAQ ((17-Cyclopropyl-methyl-3,14β-dihydroxy-4,5α-epoxy-6α-[(3′-isoquinolyl)acetamido]-morphinan)) was synthesized in Dr. Zhang's laboratory as described previously (Li et al. 2009). Morphine, nalbuphine and naltrexone were dissolved in saline, while NAQ was dissolved in water. All doses are expressed in the salt forms above, and all injections were delivered subcutaneously in a volume of 1 ml/kg body weight.
Results
Baseline ICSS before, during and after chronic morphine
During pre-drug baseline sessions, the mean ± SEM maximum control rate (MCR) for all rats in the study was 64.1±7.4 stimulations per trial, and the mean ± SEM control number of total stimulations delivered across all frequencies was 344.0±55.0. Figure 1 shows baseline frequency-rate ICSS curves before treatment and during each phase of chronic morphine treatment. There were significant main effects of frequency [F(9,36)=95.8; P<0.001] and treatment phase [F(4,16)=9.1; P<0.001], and a significant phase X frequency interaction [F(36,144)=4.0; P<0.001]. Electrical brain stimulation maintained a frequency-dependent increase in ICSS rates before initiation of any drug treatments (Pre-drug baseline), and chronic vehicle did not significantly alter ICSS. Chronic treatment with 3.2 mg/kg/day morphine slightly decreased ICSS relative to the pre-drug baseline (significant at 89 Hz), and 18 mg/kg/day morphine produced a further decrease in ICSS (significant at 89-126 Hz). These baseline ICSS frequency-rate curves were determined approximately 23hr after the most recent morphine injection on the previous day, and they show the effects of spontaneous 23hr withdrawal from that morphine maintenance dose. ICSS partially recovered at two weeks after termination of morphine treatment, although ICSS rates were still significantly decreased at three frequencies (89-112 Hz).
Figure 1.
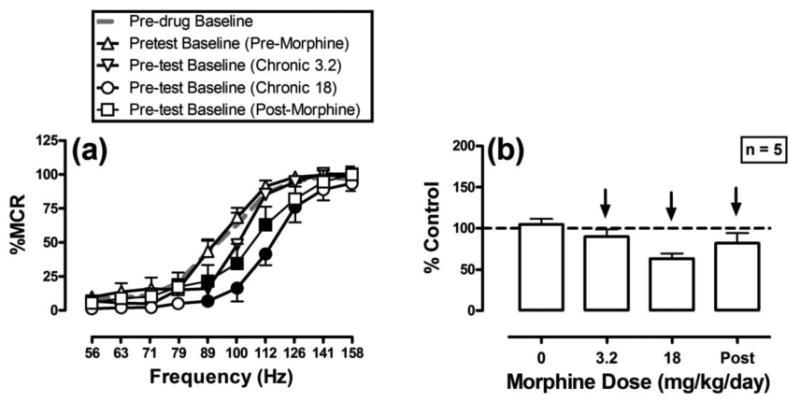
Baseline ICSS performance before, during and after chronic morphine treatment. ICSS curves were analyzed during pre-drug baseline sessions (grey dashed line), and before test drug administration during each phase of morphine exposure. The left panel shows ICSS frequency-rate curves. Abscissae: frequency of electrical brain stimulation in hertz (log scale). Ordinates: ICSS rate expressed as percent maximum control rate (%MCR). Filled symbols indicate frequencies at which ICSS rates were lower than those observed during the pre-drug baseline components as determined by the Holm-Sidak post-hoc test following a significant two-way ANOVA. Right panel shows summary data for the total number of stimulations per test component expressed as a percentage of total pre-drug baseline control stimulations. Abscissa: daily morphine dose in mg/kg/day during each phase of testing. Ordinate: percent control stimulations per test component. Arrows indicate the presence and direction of significant differences from pre-drug baseline as determined by analyses of frequency-rate data in the left panel, such that downward arrows indicate significant depression of ICSS at ≥1 frequency of the frequency-rate curve. All points show mean ± SEM for the 5 rats that completed all phases of the study; one rat lost its head cap during treatment with 18 mg/kg/day morphine and is not included in this figure.
Effects of NAQ on ICSS before, during and after chronic morphine
Figure 2 shows effects of NAQ on ICSS. During vehicle treatment, NAQ produced weak effects that were not monotonically related to dose (significant main effect of frequency [F(9,45)=66.7; P<0.001], no significant main effect of dose [F(4,20)=1.8; P=0.178], and a significant interaction [F(36,180)=2.0; P=0.001]). Thus, exclusive increases in ICSS were produced by NAQ doses of 0.32 mg/kg (71 Hz) and 3.2 (63-79 Hz) mg/kg, whereas the highest dose of 10 mg/kg produced biphasic effects (increased ICSS at 71 Hz and decreased ICSS at 158 Hz). A dose of 1.0 mg/kg NAQ did not significantly alter ICSS at any frequency. Repeated treatment with 3.2 mg/kg/day morphine eliminated expression of the rate-decreasing effects of NAQ and enhanced the dose-dependence and magnitude of NAQ's rate-increasing effects (significant main effects of frequency [F(9,45)=32.5; P<0.001] and dose [F(4,20)=17.7; P<0.001], and a significant interaction [F(36,180)=2.5; P<0.001]). For example, 10 mg/kg NAQ increased ICSS at frequencies of 63-100 Hz and did not decrease ICSS at any frequency (figure 2 c,d). During treatment with 18 mg/kg/day morphine, NAQ produced similar effects consisting of exclusive increases in ICSS across all NAQ doses (significant main effects of frequency [F(9,36)=43.7; P<0.001] and dose [F(4,16)=3.4; P=0.033], and a significant interaction [F(36,144)=1.8; P=0.007]). However, the magnitude of NAQ effects was generally smaller during treatment with 18 than 3.2 mg/kg/day morphine. For example, during treatment with 3.2 mg/kg/morphine, 10 mg/kg NAQ increased ICSS at six frequencies (63-100 Hz), but during treatment with 18 mg/kg morphine, the same NAQ dose only increased ICSS at two frequencies (100-112 Hz). Two weeks after termination of morphine treatment, all NAQ doses continued to produce a significant increase in ICSS (significant main effects of frequency [F(9,36)=19.4; P<0.001] and dose [F(4,16)=8.2; P<0.001], but no significant interaction [F(36,144)=1.3; P=0.151].
Figure 2.
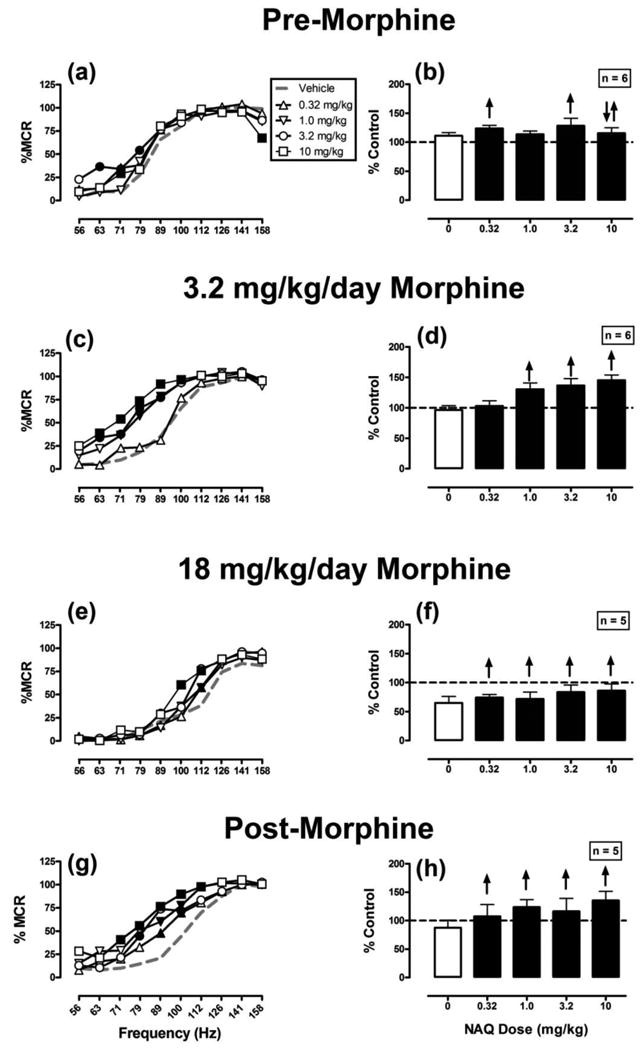
Effects of NAQ on ICSS before, during and after chronic morphine treatment. NAQ doses (or vehicle) were administered during treatment with repeated vehicle (a,b), repeated 3.2 mg/kg/day morphine (c,d), repeated 18 mg/kg/day morphine (e,f), or beginning two weeks after termination of morphine treatment (g,h). Left panels show full frequency-rate curves. Left abscissae: frequency of electrical brain stimulation in hertz (log scale). Left ordinates: ICSS rate expressed as percent maximum control rate (%MCR). Filled symbols indicate frequencies at which ICSS rates after NAQ were different than those observed after vehicle, as determined by the Holm-Sidak post-hoc test following a significant two-way ANOVA. Summary data in the right panels show the total number of stimulations per test component expressed as a percentage of total pre-drug baseline control stimulations. Abscissae: dose of NAQ in mg/kg. Ordinates: percent control stimulations per test component. Upward and/or downward arrows indicate the presence and direction of significant differences from vehicle as determined by analyses of frequency-rate data in the left panel, such that upward arrows indicate significant facilitation of ICSS at ≥1 frequency, and downward arrows indicate significant depression of ICSS at ≥1 frequency of the frequency-rate curve. All points show mean ± SEM for 6 rats (panels a-d) or 5 rats (panels e-h).
To evaluate the role of opioid receptors in NAQ-induced facilitation of ICSS, 0.1 mg/kg naltrexone was administered as a pretreatment to 1.0 mg/kg NAQ during treatment with 3.2 mg/kg/day morphine, and results are shown in Figure 3. Naltrexone blocked NAQ-induced facilitation of ICSS at a naltrexone dose that had no effect alone (significant main effect of frequency [F(9,45)=57.4; P<0.001] and treatment [F(3,15)=16.3; P<0.001], and a significant interaction [F(27,135)=2.7; P<0.001].
Figure 3.
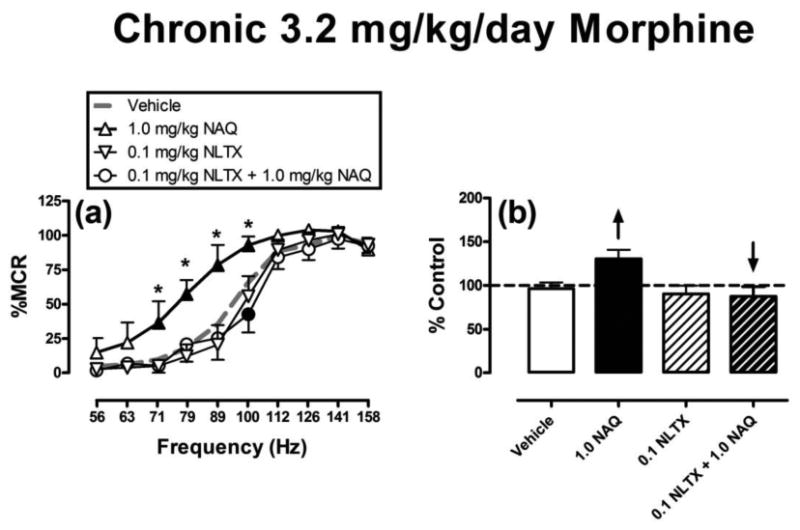
Naltrexone antagonism of NAQ effects during chronic treatment with 3.2 mg/kg/day morphine. All points show mean ± SEM for 6 rats. For description of axes and symbols, please refer to figures 1 and 2. Filled points indicate significantly different from “Vehicle.” Asterisks indicate significantly different from “0.1 NLTX + 1.0 NAQ”.
Effect of nalbuphine and naltrexone on ICSS
Figure 4 compares the effects of 1.0 mg/kg nalbuphine and 0.1 mg/kg naltrexone on ICSS before and during chronic morphine. In all phases, 1.0 mg/kg nalbuphine produced significant facilitation of ICSS compared to vehicle, whereas 0.1 mg/kg naltrexone failed to produce any significant changes. As observed with NAQ, 0.1 mg/kg naltrexone blocked nalbuphine-induced facilitation of ICSS during chronic treatment with 3.2 mg/kg/day morphine. ANOVA results for each panel of Figure 4 were as follows: Chronic vehicle: Significant main effects of frequency [F(9,45)=37.7; P<0.001] and treatment [F(2,10)=1.8; P=0.007], and a significant interaction [F(18,90)=2.4; P=0.004]. Repeated 3.2 mg/kg/day morphine: Significant main effects of frequency [F(9,45)=47.9; P<0.001] and treatment [F(3,15)=13.5; P<0.001], and a significant interaction [F(27,135)=2.6; P<0.001]. Repeated 18 mg/kg/day morphine: Significant main effects of frequency [F(9,36)=46.0; P<0.001] and treatment [F(2,8)=17.2; P=0.001], and significant interaction [F(18,72)=2.1; P=0.016].
Figure 4.
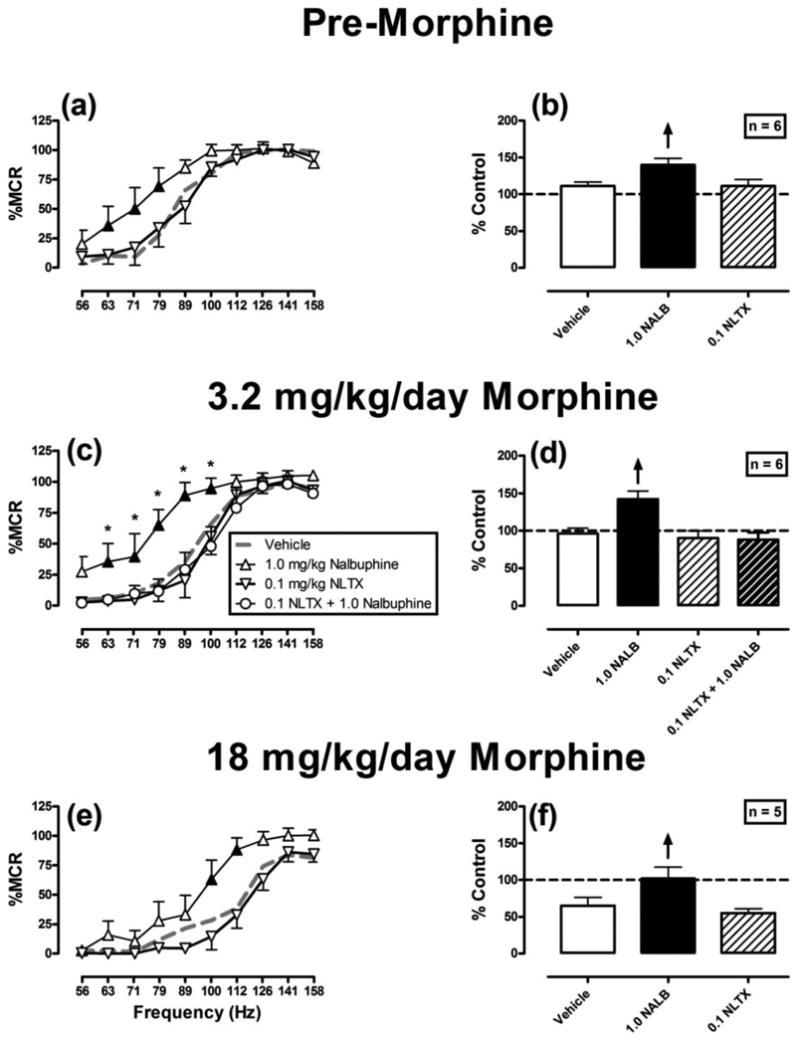
Effects of nalbuphine (1.0 mg/kg) and naltrexone (0.1 mg/kg) on ICSS before and during chronic morphine treatment. All points show mean ± SEM for 6 rats (panels a-d) or 5 rats (panels e,f). For description of axes and symbols, please refer to figures 1 and 2. Filled points indicate significantly different from “Vehicle.” Asterisks indicate significantly different from “0.1 NLTX + 1.0 nalbuphine”.
Fecal bolus production
Fecal boli were counted after test sessions during treatment with 18 mg/kg/day morphine, and the number of rats with two or more fecal boli was counted for each treatment. No rats met this criterion after vehicle treatment, and all rats met this criterion after naltrexone treatment. No more than two rats met this criterion during treatment with any NAQ dose, and similarly, only two rats met this criterion after 1.0 mg/kg nalbuphine.
Discussion
This study evaluated effects of the novel opioid receptor ligand NAQ on ICSS in rats before, during and after a regimen of chronic morphine treatment. There were three main findings. First, the profile of NAQ effects throughout testing was consistent with its in vitro profile as a selective and low-efficacy mu agonist. Second, NAQ produced weak and inconsistent ICSS facilitation in opioid-naïve rats, but it produced dose-dependent and more robust ICSS facilitation in opioid-experienced rats both during and after morphine treatment. Insofar as ICSS facilitation is predictive of abuse potential, these results suggest that NAQ might have weak abuse liability in opioid-naïve subjects and higher abuse liability in subjects with histories of opioid exposure. Lastly, repeated daily morphine treatment was associated with a withdrawal-associated depression of ICSS, and NAQ had sufficient mu-agonist efficacy to reverse this withdrawal effect. This finding suggests that NAQ may be useful as a low-efficacy and relatively safe strategy for treatment of opioid withdrawal and addiction.
NAQ as a low-efficacy mu agonist
Results of this study are consistent with the characterization of NAQ as a low-efficacy mu agonist. We have reported previously that other mu opioid receptor ligands produce efficacy-dependent effects on ICSS in opioid-naive rats (Altarifi et al. 2012; Altarifi and Negus 2011; Altarifi et al. 2013). Specifically, mu antagonists such as naltrexone and β-funaltrexamine have little effect on ICSS but block effects of mu agonists. Low-efficacy mu agonists such as nalbuphine produce weak facilitation of low ICSS rates maintained by low frequencies of brain stimulation, and weak depression of high ICSS rates maintained by high frequencies of brain stimulation. Finally, high-efficacy mu agonists such as morphine, fentanyl or methadone also produce weak facilitation of low ICSS rates maintained by low frequencies of brain stimulation; however, in comparison to low-efficacy agonists, high-efficacy mu agonists produce more robust decreases in high ICSS rates maintained by high frequencies of brain stimulation. In the present study, NAQ effects observed prior to initiation of morphine treatment resembled nalbuphine effects in opioid-naive rats reported here with 1.0 mg/kg nalbuphine or previously with a wider nalbuphine dose range (Altarifi et al. 2012; Altarifi et al. 2013).
Mu agonists also display a characteristic change in effects on ICSS after regimens of opioid exposure (Koob et al. 1975; Kornetsky and Esposito 1979). For example, we reported previously that a regimen of morphine exposure identical to that used here produced tolerance to ICSS rate-decreasing effects and enhanced expression of ICSS rate-increasing effects of both low- and high-efficacy mu agonists (Altarifi and Negus 2011; Altarifi et al. 2013). Consistent with these previous results with other mu agonists, repeated morphine also produced tolerance to any rate-decreasing effects and enhanced expression of rate-increasing effects of NAQ in this study. The enhanced expression of rate-increasing effects was especially evident during treatment with the intermediate morphine dose of 3.2 mg/kg/day morphine and appeared weaker during treatment with the higher dose of 18 mg/kg/day morphine. These results suggest that treatment with high morphine doses may produce tolerance not only to rate-decreasing effects of NAQ, but also to its abuse-related rate-increasing effects.
The characterization of NAQ as a low-efficacy mu-selective agonist is also supported by two other sources of data. First, NAQ-induced facilitation of ICSS was blocked by a naltrexone dose shown previously to block ICSS effects of other mu agonists (Altarifi et al. 2012). Second, the profile of NAQ effects observed here did not resemble the profile of ICSS effects produced by agonists at either kappa opioid receptors (Beguin et al. 2008; Negus et al. 2010; Negus et al. 2012a; Todtenkopf et al. 2004) or delta opioid receptors (Negus et al. 2012b; Pereira Do Carmo et al. 2009). In contrast to mu agonists, systemically administered kappa and delta agonists produce only depression of ICSS, and although repeated treatment may produce tolerance to these rate-decreasing effects, it does not unmask expression of rate-increasing effects (Negus and Altarifi 2013).
In vitro studies have suggested that NAQ differs from other low-efficacy mu agonists such as nalbuphine in the degree of its selectivity for mu vs. kappa and delta opioid receptors (Li et al. 2009). Previous studies have not found evidence for a role of non-mu opioid receptors in mediating nalbuphine effects on ICSS, and NAQ and nalbuphine effects on ICSS are similar in studies conducted so far. However, nalbuphine has been reported to produce effects mediated by kappa opioid receptors (Pick et al. 1992), or other opioid receptor types (e.g. nociceptin/orphanin FQ receptors; Gear et al. 2014) under other conditions in rodents. Future studies will be required to investigate the degree to which NAQ might lack these non-mu effects.
Implications for expression and treatment of abuse
ICSS is one preclinical procedure that has been used to assess abuse potential of drugs in laboratory animals, and drug-induced facilitation of low ICSS rates maintained by low brain stimulation frequencies or intensities is often interpreted as evidence for abuse potential (Kornetsky and Esposito 1979; Negus and Miller 2014; Vlachou and Markou 2011; Wise 1996). From this perspective, the present results suggest that NAQ has sufficient efficacy at mu opioid receptors to produce abuse-related effects in other preclinical procedures such as drug self-administration and to have some abuse liability in humans. For example, NAQ effects on ICSS were similar to those of nalbuphine, and nalbuphine maintains self-administration by laboratory animals (Balster and Lukas 1985; Steinfels et al. 1981) and is occasionally abused by humans (Preston and Jasinski 1991; Wines et al. 1999). However, behavioral economic studies in rhesus monkeys suggest that nalbuphine functions as a weaker reinforcer than higher efficacy mu agonists (Hursh and Winger 1995), and the present ICSS results suggest that abuse-related effects of NAQ are similar to those of nalbuphine.
The present study also found that NAQ reversed the depression of ICSS produced by spontaneous morphine withdrawal. Specifically, in agreement with previous studies (Altarifi and Negus 2011; Altarifi et al. 2013), once-per-day treatment with escalating morphine doses produced a dose-dependent depression in ICSS measured 23hr after each daily morphine injection, and this ICSS depression partially recovered within two weeks after termination of morphine treatment. Depression of ICSS associated with withdrawal from opioids or other abused drugs has been interpreted as a potential sign of withdrawal-associated anhedonia, which may contribute to continued drug use and addiction (Kenny et al. 2006; Liu and Schulteis 2004; Stoker and Markou 2011). Accordingly, NAQ reversal of withdrawal-induced depression of ICSS suggests that NAQ may be useful as a tool in treatment of opioid dependence to prevent or reverse anhedonic signs of opioid withdrawal. Existing treatments for opioid withdrawal include the antagonist naltrexone, the intermediate-efficacy agonist buprenorphine, and the high-efficacy agonist methadone (Gutstein and Akil 2006; Kreek et al. 2002; Negus and Banks 2013). One advantage of NAQ as a complement to these existing medications is its relative efficacy between naltrexone and buprenorphine. Thus, unlike naltrexone, NAQ has sufficient mu receptor efficacy to reverse withdrawal-induced depression of ICSS (Altarifi et al. 2013). Perhaps because of its weak but measurable efficacy at mu receptors, NAQ also produced weaker somatic withdrawal signs than naltrexone in morphine-dependent mice (Yuan et al. 2011), and weaker withdrawal-associated activation of gastrointestinal transit than naltrexone in morphine-dependent rats in the present study. However, NAQ has lower efficacy at mu receptors than either buprenorphine or methadone (Emmerson et al. 1996; Li et al. 2009; Selley et al. 1998) and might therefore be less likely than these higher efficacy mu agonists to produce undesirable side effects such as sedation or respiratory depression. The low efficacy of NAQ to produce ICSS rate-decreasing effects in this study in both opioid-naive and opioid-experienced subjects is consistent with this potential for NAQ to be safer than higher efficacy mu agonists.
Acknowledgments
This research was supported by NIH grants R01-NS070715, R01-DA026946 and R01-DA024022.
Footnotes
Disclosure: The authors declare no conflict of interest.
References
- Altarifi AA, Miller LL, Negus SS. Role of mu-opioid receptor reserve and mu-agonist efficacy as determinants of the effects of micro-agonists on intracranial self-stimulation in rats. Behav Pharmacol. 2012;23:678–9. doi: 10.1097/FBP.0b013e328358593c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarifi AA, Negus SS. Some determinants of morphine effects on intracranial self-stimulation in rats: dose, pretreatment time, repeated treatment, and rate dependence. Behav Pharmacol. 2011;22:663–73. doi: 10.1097/FBP.0b013e32834aff54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarifi AA, Rice KC, Negus SS. Abuse-related effects of mu-opioid analgesics in an assay of intracranial self-stimulation in rats: modulation by chronic morphine exposure. Behav Pharmacol. 2013;24:459–70. doi: 10.1097/FBP.0b013e328364c0bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balster RL, Lukas SE. Review of self-administration. Drug Alcohol Depend. 1985;14:249–61. doi: 10.1016/0376-8716(85)90060-2. [DOI] [PubMed] [Google Scholar]
- Beguin C, Potter DN, Dinieri JA, Munro TA, Richards MR, Paine TA, Berry L, Zhao Z, Roth BL, Xu W, Liu-Chen LY, Carlezon WA, Jr, Cohen BM. N-methylacetamide analog of salvinorin A: a highly potent and selective kappa-opioid receptor agonist with oral efficacy. J Pharmacol Exp Ther. 2008;324:188–95. doi: 10.1124/jpet.107.129023. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Chartoff EH. Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nat Protoc. 2007;2:2987–95. doi: 10.1038/nprot.2007.441. [DOI] [PubMed] [Google Scholar]
- Emmerson PJ, Clark MJ, Mansour A, Akil H, Woods JH, Medzihradsky F. Characterization of opioid agonist efficacy in a C6 glioma cell line expressing the mu opioid receptor. J Pharmacol Exp Ther. 1996;278:1121–7. [PubMed] [Google Scholar]
- Emmerson PJ, Liu MR, Woods JH, Medzihradsky F. Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J Pharmacol Exp Ther. 1994;271:1630–7. [PubMed] [Google Scholar]
- Gear RW, Bogen O, Ferrari LF, Green PG, Levine JD. NOP receptor mediates anti-analgesia induced by agonist-antagonist opioids. Neuroscience. 2014;257:139–48. doi: 10.1016/j.neuroscience.2013.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutstein H, Akil H. Opioid analgesics. In: Brunton L, Lazo J, Parker K, editors. Goodman and Gilman's The Pharmacological Basis of Therapeutics. 11th. McGraw-Hill; New York: 2006. pp. 547–590. [Google Scholar]
- Hoskin PJ, Hanks GW. Opioid agonist-antagonist drugs in acute and chronic pain states. Drugs. 1991;41:326–44. doi: 10.2165/00003495-199141030-00002. [DOI] [PubMed] [Google Scholar]
- Hursh SR, Winger G. Normalized demand for drugs and other reinforcers. J Exp Anal Behav. 1995;64:373–84. doi: 10.1901/jeab.1995.64-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ, Chen SA, Kitamura O, Markou A, Koob GF. Conditioned withdrawal drives heroin consumption and decreases reward sensitivity. J Neurosci. 2006;26:5894–900. doi: 10.1523/JNEUROSCI.0740-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Spector NH, Meyerhoff JL. Effects of heroin on lever pressing for intracranial self-stimulation, food and water in the rat. Psychopharmacologia. 1975;42:231–4. doi: 10.1007/BF00421261. [DOI] [PubMed] [Google Scholar]
- Kornetsky C, Esposito RU. Euphorigenic drugs: effects on the reward pathways of the brain. Fed Proc. 1979;38:2473–6. [PubMed] [Google Scholar]
- Kornetsky C, Esposito RU, McLean S, Jacobson JO. Intracranial self-stimulation thresholds: a model for the hedonic effects of drugs of abuse. Arch Gen Psychiatry. 1979;36:289–92. doi: 10.1001/archpsyc.1979.01780030055004. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, LaForge KS, Butelman E. Pharmacotherapy of addictions. Nat Rev Drug Discov. 2002;1:710–26. doi: 10.1038/nrd897. [DOI] [PubMed] [Google Scholar]
- Li G, Aschenbach LC, Chen J, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, Zhang Y. Design, synthesis, and biological evaluation of 6alpha- and 6beta-N-heterocyclic substituted naltrexamine derivatives as mu opioid receptor selective antagonists. J Med Chem. 2009;52:1416–27. doi: 10.1021/jm801272c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Schulteis G. Brain reward deficits accompany naloxone-precipitated withdrawal from acute opioid dependence. Pharmacol Biochem Behav. 2004;79(1):101–108. doi: 10.1016/j.pbb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Negus SS, Altarifi A. Mu, delta and kappa opioid agonist effects in novel assays of pain-depressed behavior. In: Ko H, Husbands SM, editors. Research and Development of Opioid Releated Ligands. American Chemical Society; Washington, DC: 2013. pp. 163–176. [Google Scholar]
- Negus SS, Banks ML. Medications development for opioid abuse. Cold Spring Harb Perspect Med. 2013;3:a012104. doi: 10.1101/cshperspect.a012104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Miller LL. Intracranial self-stimulation to evaluate abuse potential of drugs. Pharmacol Rev. 2014;66(3):869–917. doi: 10.1124/pr.112.007419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Morrissey EM, Rosenberg M, Cheng K, Rice KC. Effects of kappa opioids in an assay of pain-depressed intracranial self-stimulation in rats. Psychopharmacology (Berl) 2010;210:149–59. doi: 10.1007/s00213-009-1770-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, O'Connell R, Morrissey E, Cheng K, Rice KC. Effects of peripherally restricted kappa opioid receptor agonists on pain-related stimulation and depression of behavior in rats. J Pharmacol Exp Ther. 2012a;340:501–9. doi: 10.1124/jpet.111.186783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Rosenberg MB, Altarifi AA, O'Connell RH, Folk JE, Rice KC. Effects of the delta opioid receptor agonist SNC80 on pain-related depression of intracranial self-stimulation (ICSS) in rats. J Pain. 2012b;13:317–27. doi: 10.1016/j.jpain.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Knapp BI, Bidlack JM, Neumeyer JL. Pharmacological properties of bivalent ligands containing butorphan linked to nalbuphine, naltrexone, and naloxone at mu, delta, and kappa opioid receptors. J Med Chem. 2007;50:2254–8. doi: 10.1021/jm061327z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira Do Carmo G, Folk JE, Rice KC, Chartoff E, Carlezon WA, Jr, Negus SS. The selective non-peptidic delta opioid agonist SNC80 does not facilitate intracranial self-stimulation in rats. Eur J Pharmacol. 2009;604:58–65. doi: 10.1016/j.ejphar.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233:774–6. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- Pick CG, Paul D, Pasternak GW. Nalbuphine, a mixed kappa 1 and kappa 3 analgesic in mice. J Pharmacol Exp Ther. 1992;262:1044–50. [PubMed] [Google Scholar]
- Preston KL, Jasinski DR. Abuse liability studies of opioid agonist-antagonists in humans. Drug Alcohol Depend. 1991;28:49–82. doi: 10.1016/0376-8716(91)90053-2. [DOI] [PubMed] [Google Scholar]
- Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol. 1994;45:330–4. [PubMed] [Google Scholar]
- Remmers AE, Clark MJ, Mansour A, Akil H, Woods JH, Medzihradsky F. Opioid efficacy in a C6 glioma cell line stably expressing the human kappa opioid receptor. J Pharmacol Exp Ther. 1999;288:827–33. [PubMed] [Google Scholar]
- Selley DE, Liu Q, Childers SR. Signal transduction correlates of mu opioid agonist intrinsic efficacy: receptor-stimulated [35S]GTP gamma S binding in mMOR-CHO cells and rat thalamus. J Pharmacol Exp Ther. 1998;285:496–505. [PubMed] [Google Scholar]
- Steinfels GF, Young GA, Khazan N. The new mixed agonist-antagonist analgesics, nalbuphine and butorphanol, vs. pentazocine: relapse and substitution in morphine-addict rats. NIDA Res Monogr. 1981;34:138–44. [PubMed] [Google Scholar]
- Stoker AK, Markou A. The intracranial self-stimulation procedure provides quantitative measures of brain reward function. In: Gould TD, editor. Mood and Anxiety Related Phenotypes in MIce Characterization Using Behavioral Tests, Volume II Neuromethods Volume 63. Humana Press; Totowa, NJ: 2011. pp. 307–331. [Google Scholar]
- Todtenkopf MS, Marcus JF, Portoghese PS, Carlezon WA., Jr Effects of kappa-opioid receptor ligands on intracranial self-stimulation in rats. Psychopharmacology (Berl) 2004;172:463–70. doi: 10.1007/s00213-003-1680-y. [DOI] [PubMed] [Google Scholar]
- Vlachou S, Markou A. Intracranial self-stimulation. In: Olmstead MC, editor. Animal Models of Drug Addiction. Humana Press; New York: 2011. pp. 3–56. [Google Scholar]
- Walsh SL, Strain EC, Abreu ME, Bigelow GE. Enadoline, a selective kappa opioid agonist: comparison with butorphanol and hydromorphone in humans. Psychopharmacology (Berl) 2001;157:151–62. doi: 10.1007/s002130100788. [DOI] [PubMed] [Google Scholar]
- Wines JD, Jr, Gruber AJ, Pope HG, Jr, Lukas SE. Nalbuphine hydrochloride dependence in anabolic steroid users. Am J Addict. 1999;8:161–4. doi: 10.1080/105504999305965. [DOI] [PubMed] [Google Scholar]
- Wise RA. Addictive drugs and brain stimulation reward. Annu Rev Neurosci. 1996;19:319–40. doi: 10.1146/annurev.ne.19.030196.001535. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Li G, He H, Stevens DL, Kozak P, Scoggins KL, Mitra P, Gerk PM, Selley DE, Dewey WL, Zhang Y. Characterization of 6alpha- and 6beta-N-heterocyclic substituted naltrexamine derivatives as novel leads to development of mu opioid receptor selective antagonists. ACS Chem Neurosci. 2011;2:346–51. doi: 10.1021/cn2000348. [DOI] [PMC free article] [PubMed] [Google Scholar]