

Abstract
Glycine-N-methyltransferase (GNMT) is essential to preserve liver homeostasis. Thus, cirrhotic patients show low expression of GNMT that is absent in HCC samples. Accordingly, GNMT deficiency in mice leads to steatohepatitis, fibrosis, cirrhosis and HCC. Lack of GNMT triggers NK cell activation in GNMT−/− mice and depletion of TRAIL significantly attenuates acute liver injury and inflammation in these animals. Chronic inflammation leads to fibrogenesis, further contributing to the progression of chronic liver injury regardless of the etiology. Taking all this together, the aim of our study is to elucidate the implication of TRAIL-producing NK cells in the progression of chronic liver injury and fibrogenesis. For this we generated double TRAIL−/−/GNMT−/− mice where we found that TRAIL deficiency efficiently protected the liver against chronic liver injury and fibrogenesis in the context of GNMT deficiency. Next, to better delineate the implication of TRAIL-producing NK cells during fibrogenesis we performed bile duct ligation (BDL) to GNMT−/− and TRAIL−/−/GNMT−/− mice. In GNMT−/− mice, exacerbated fibrogenic response after BDL concurred with NK1.1+ cell activation. Importantly, specific inhibition of TRAIL-producing NK cells efficiently protected GNMT−/− mice from BDL-induced liver injury and fibrogenesis. Finally, TRAIL−/−/GNMT−/− showed significantly less fibrosis after BDL than GNMT−/− mice further underlining the relevance of the TRAIL/DR5 axis in mediating liver injury and fibrogenesis in GNMT−/− mice. Finally, in vivo silencing of DR5 efficiently protected GNMT−/− mice from BDL-liver injury and fibrogenesis, overall underscoring the key role of the TRAIL/DR5 axis in promoting fibrogenesis in the context of absence of GNMT.
Conclusion
Overall, our work demonstrates that TRAIL-producing NK cells actively contribute to liver injury and further fibrogenesis in the pathological context of GNMT deficiency, a molecular scenario characteristic of chronic human liver disease.
Glycine-N-Methyltransferase (GNMT) is the most abundant methyltransferase in the liver. The relevance of GNMT to preserve liver homeostasis relies on its ability to tightly control the catabolism of SAMe, the main methyl donor of the body1. GNMT is down-regulated in cirrhotic patients (from HCV and ASH etiologies) and is absent in HCC samples2. In accordance, we described that mice lacking GNMT (GNMT−/−) develop spontaneous steatosis that progresses to steatohepatitis, cirrhosis and HCC3. More recently, we found that GNMT deficiency correlates with strong activation of NK cells, which mediate endotoxin-mediated inflammation and acute liver injury through TRAIL4. Moreover, we found that GNMT deficient livers and hepatocytes expressed more TRAILR2/DR5, further suggesting that the TRAIL/DR5 axis may play a key role in the progression of NASH that spontaneously develop GNMT−/− animals. Nevertheless, the implication of TRAIL/NK cells during chronic liver injury and fibrogenesis was not further explored.
Chronic liver injury leads to fibrogenesis and eventually to cirrhosis and hepatocellular carcinoma (HCC). Fibrosis is a common feature of the pathogenesis of chronic liver disease regardless of the etiology; NASH, HCV infection, alcohol abuse, primary biliary cirrhosis (PBC) and autoimmune hepatitis5. In the context of chronic liver injury, inflammation actively contributes to fibrogenesis, although the molecular mechanisms underlying this progression are poorly understood. It is commonly accepted that hepatocyte apoptotic cell death promotes an inflammatory response to remove cell debris, which in turn activates hepatic stellate cells (HSC) to deposit collagen, in a tissue remodeling/scarring process. Kupffer cells (KC) are the main cell compartment mediating this process, although HSC can also be directly activated by phagocytosis of apoptotic hepatocytes6, 7. Thus, the innate immune system and HSC are closely linked during fibrogenesis.
NKT/NK cells are part of the innate immune system, representing the first line of defense of the liver. NKT and NK cells seem to have differential roles during fibrogenesis. Thus, increased presence of NKT cells in cirrhotic livers contributes to fibrogenesis during NASH8, whereas NK cells are commonly described as anti-fibrogenic due to their ability to promote apoptosis of HSC through TRAIL/DR5 and NKG2D-RAE19, 10. Interestingly, a number of reports show NK cell activation during cholestatic liver diseases in patients. Thus, NK cells have a cytotoxic effect against autologous biliary cells/cholangiocytes in PBC and PSC patients11–13 and, in mice, TRAIL produced by NK cells contributes to cholestatic liver injury14. Also, in the context of NASH progression the presence of major-histocompatibility complex A and B proteins (MIC A/B), stress-ligands recognized by NK cells, directly correlate with the fibrosis stage in patients15. Overall, these studies suggest the potential implication of NK cells in mediating liver injury during chronic liver disease although its fibrogenic role remains uncertain.
Taking all this together, in the present work we aim to investigate the molecular mechanisms leading to fibrogenesis in the pathological context of GNMT deficiency, a scenario found in cirrhotic patients. Here, we provide evidence that TRAIL is a key player in the development of the spontaneous pathogenic phenotype observed in the liver of GNMT deficient animals. Also, we describe that NK cell activation concurs with exacerbated fibrogenesis in the context of GNMT deficiency after bile duct ligation (BDL). Finally, through different experimental strategies we show that selective inhibition of TRAIL-producing NK cells, knock down of TRAIL and in vivo silencing of DR5 in GNMT−/− mice efficiently protects the liver from BDL-induced liver injury, overall attenuating fibrogenesis in GNMT−/− mice.
MATERIALS AND METHODS
GNMT Knockout animals
GNMT−/− mice were generated as previously described16. TRAIL−/− mice were kindly provided by Amgen. Double KO mice were obtained crossing GNMT−/− with TRAIL−/− mice. Male 8–12 week old mice were used in our experiments. Bile duct ligation (BDL) was performed as previously described 17. Animals were treated according to the guidelines of the National Academy of Sciences (National Institutes of Health publication 86-23, revised 1985). Animal husbandry and procedures were approved by the Department of Environment, Planning, Agriculture and Fisheries of the Basque Country Government. All experiments shown in this MS were performed following the guidelines of the CICbioGUNE Facility’s guidelines with AALAC certificate.
Immunohistochemistry
Paraffin embedded liver samples were sectioned, dewaxed and hydrated. Immunohistochemistry (IHC) was performed using GNMT Ab, F4/80 (Serotec) and nuclei were counterstained with Hematoxylin. Fibrosis was determined with Sirius Red staining and IF using αSmooth Muscle Actin (αSMA) Ab (Sigma), labeled with Cy3 (red). Nuclei were counterstained with DAPI (blue). Quantification of F4/80, αSMA and collagen deposition in Sirius Red stained sections were analyzes using Frida software and represented as the % of the stained area relative to the total area (power field). 5–10 fields/per sample were pictured and analyzed.
Liver injury experimental model
Bile duct ligation was performed as previously described 17.
Human PBC samples
Glycine-N-methyltransferase (GNMT) was assessed by IHC in sections of deparaffinized liver samples obtained by percutaneous biopsy in 5 patients and samples from the explanted liver in 3 patients. The diagnosis of PBC was established by liver biopsy with characteristic features of the disease and presence of anti-mitochondrial antibodies. Biochemical and histological cholestasis were very prominent in the explanted samples, while features of chronic non-suppurative cholangitis, portal inflammation and ductopenia with fewer prominent biochemical cholestasis were present in the five patients in the initial stages of the disease. All the latter patients had Ludwig’s stage I, while 2 of the explanted livers had cirrhosis and the remaining livers had septal fibrosis (Ludwig’s stage 3) and severe ductopenia. The Ethical Committee of the institution approved the study and all participants provided written informed consent.
Characterization of liver damage and cell death
Alanine aminotransferase (ALT), aspartate aminotransferase (AST) and bilirubin were determined in serum samples. Histological analysis of the liver parenchyma was performed in liver sections that were stained with hematoxylin & eosin (H&E). Cell death was determined on frozen liver sections by TUNEL assay using the In Situ Cell Death Detection Kit (Roche diagnosis) following the manufacturer’s instructions. Caspase-3 activity was quantified in snap frozen livers as described 18. The presence of TNF was determined by ELISA (R&D systems) in whole liver extracts.
RNA Isolation and Quantitative Real-Time PCR
RNA was isolated with TriZol Reagent (Invitrogen) followed by first strand synthesis with Oligo dT primers and reverse transcription with M-MLV Reverse Transcriptase (Invitrogen). Quantitative real-time PCR (RT-PCR) was performed using SYBR Green reagent (Quantas biosciences) in a MyiQ single color Real-time PCR detection system (Biorad). Gene expression was normalized using Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and expressed in times versus control expression. Primers can be provided under request.
Western Blot Analysis
Whole liver protein extracts were resolved in sodium dodecyl sulfate–polyacrylamide gels and transferred to nitrocellulose membranes (Whatman). Membranes were probed with the following primary antibodies: DR5, p-JNK1/2 (Thr 183/Tyr185) from Cell signaling. FADD was obtained from Santa Cruz biotechnologies. As a loading control, we used GAPDH antibody (Abcam). As secondary antibodies, we used anti-rabbit-IgG–HRP-linked (Cell Signaling) and anti-mouse IgG–HRP-linked (Santa Cruz Biotechnology).
MNC isolation and flow cytometric analysis
Liver MNCs were obtained as previously described19. In brief, liver MNCs were isolated by enzymatic digestion with collagenase IV (Worthtington) during 30min at 37°. Extracts were passed through a 12-µm mesh (Becton Dickinson). MNCs were pelleted and washed by centrifugation at 300g for 10 minutes. Erythrocytes were removed using PharmLyse lysing buffer (BD). Cells were stained with CD45-APC-Cy7, CD3-APC, NK1.1-PE-Cy7, and TRAIL-PE (BD). Flow cytometry analysis was performed on a FACS Canto II (BD) and data was analyzed with FlowJo software (Tree Star). NK1.1+/CD3− (NK) and NK1.1+/CD3+ (NKT) cells were isolated in a FACSaria cell sorter.
NK/NKT and NK cell depletion
NK/NKT and specific NK cell depletion was achieved by i.p. administration of 250 µg/mouse of NK1.1+ antibody purified from hybridoma PK-136 (American Type Culture Collection) culture supernatants as previously described 19 and by ASIALO respectively. Both ASIALO and NK1.1+ Abs were injected 24h before BDL and every 72h after surgery. As control group, GNMT−/− mice were injected with IgG (250 µg/mouse from BD bioscience) at the same timepoints as ASIALO and NK1.1+ Ab.
In vivo DR5 overexpression
The TRAILR2/DR5 short hairpin (sh)RNA was cloned into the retroviral expression vector pSM2C (Open Biosystems) to induce sequence-specific silencing of DR5 in GNMT−/− mice. ShDR5 was administered by hydrodynamic i.v. injection at 24h before BDL and every 72h after surgery.
Statistical analysis
Data are expressed as mean ± standard deviation of the mean. Statistical significance was determined by two-way analysis of variance followed by a Student’s t test.
RESULTS
Reduced GNMT expression in patients with cholestatic liver disease
Previous work showed that glycine-N-methyltransferase (GNMT) expression is significantly downregulated in cirrhotic patients from alcohol and HCV etiologies2. Here we show that expression of GNMT was also decreased in the context of chronic cholestatic liver disease. Thus, livers from patients with primary biliary cirrhosis (PBC), both at initial stages and in patients with advanced disease that were transplanted, had significantly lower levels of GNMT than healthy livers as shown by IHC and further quantification (Fig. 1A, B). The finding that attenuation of GNMT was found at early stages of cholestatic liver disease, where fibrosis is not prominent, points to GNMT deficiency as a contributor/mediator of fibrogenesis rather than a consequence.
Figure 1. Low GNMT expression in human PBC livers.
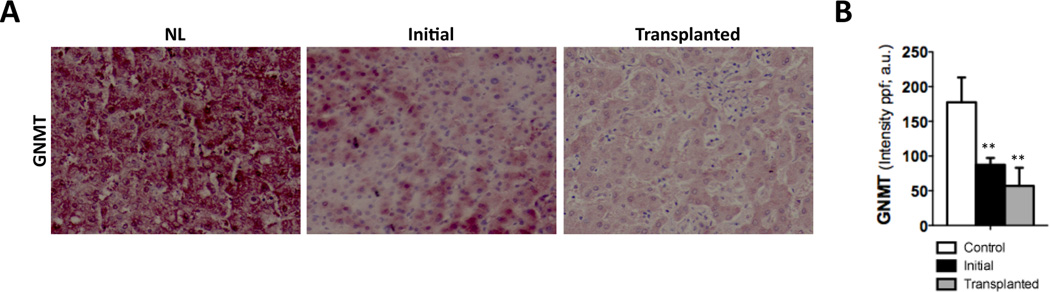
(A) IHC and (B) further quantification using FRIDA software showing lower GNMT expression in human samples from PBC patients at initial stages or after transplantation compared to healthy controls. n= 8–10. **p< 0.01 (Healthy vs early PBC and healthy vs late PBC). Error bars represent SD.
TRAIL deficiency reverts the spontaneous damaging phenotype found in GNMT−/− mice
Supporting the relevance of GNMT as a key player to preserve liver homeostasis, we previously described that GNMT deficient mice (GNMT−/−) show spontaneous steatohepatitis, cirrhosis and HCC development, mimicking the pathogenesis of NASH in humans3. Importantly, GNMT deficiency correlated with NK cell activation already at initial stages of liver disease, when mice show only a mild damaging phenotype4. Moreover, TRAIL producing NK cell depletion efficiently protected the liver against endotoxin-induced liver injury. However, the implication of TRAIL in the context of chronic liver disease was not further examined.
In order to investigate the contribution of TRAIL to chronic liver disease we generated TRAIL- and GNMT-deficient mice (TRAIL−/−/GNMT−/−). TRAIL−/−/GNMT−/− mice showed an apparent remission of the spontaneous damaging phenotype found in GNMT−/− mice3. H&E staining evidenced a clear attenuation of the liver parenchymal damage, especially in 9 month-old TRAIL−/−/GNMT−/− mice compared to GNMT−/− animals (Fig. 2A). Moreover, we observed a significant reduction of liver fibrogenesis in 3 and 9 month-old TRAIL−/−/GNMT−/− compared to GNMT−/− mice as evidenced by Sirius red staining (Fig. 2B) and further quantification using Frida software (Fig. 2C). Further analysis by qRTPCR confirmed the beneficial impact of TRAIL deficiency in the spontaneous fibrogenesis observed in GNMT−/− animals as evidenced by lower expression of collagen 1A1, αSMA and TGFβ found in 9 months-old TRAIL−/−/GNMT−/− mice (Fig. 2D). Macroscopical analysis further confirmed the important beneficial impact of TRAIL deficiency in GNMT deficient mice in the attenuation of the cirrhotic/tumorigenic phenotype found in GNMT−/− mice at 9m of age (Fig. 2E). Importantly, the improved phenotype found in TRAIL−/−/GNMT−/− mice correlated with a significantly lower activation of NK cells. Hence, we found a significant higher number of NK1.1+/CD3− (NK) cells in TRAIL−/−/GNMT−/− compared to GNMT−/− mice at 3 months of age (Fig. 2F). Restoration of the presence of NK cells to a percentage comparable to WT animals, correlated with a lower expression of NK cell activation markers such as IFNγ, Granzyme and CCL5 determined by qRTPCR analysis in FACSaria isolated NK1.1+ cells (Fig. 2G). Overall, TRAIL deficiency efficiently restored the number and activation status of NK cells; NK1.1+/CD3− cells in GNMT−/− mice that reached comparable levels to WT animals (Fig. 2F, G). These data support the implication of TRAIL-producing NK cells in mediating liver injury and fibrogenesis in the pathological context of reduced GNMT expression.
Figure 2. TRAIL deficiency reverts the spontaneous damaging phenotype in GNMT−/− mice, which show attenuated NK cell activation.

(A) H&E staining and (B) Sirius Red staining and further quantification using FRIDA software showing attenuation of liver injury and fibrogenesis in TRAIL−/−/GNMT−/− mice compared to GNMT−/− at 3 and 9 months of age. (D) qRTPCR analysis showing increased Collagen1A1, αSMA and TGFβ expression in 9 months old GNMT−/− mice compared to TRAIL−/−/GNMT−/− mice that show comparable levels than WT animals (E) Macroscopic view of livers from GNMT−/− and TRAIL−/−/GNMT−/− mice evidencing apparent regression of the damaging phenotype in the 9 months of age double KO animals (F) Dot plot of FACS analysis and bar-plot showing restoration of the number of NK1.1+ cells in TRAIL−/−/GNMT−/− compared to GNMT−/− animals, all 3 months old. (G) pPCR analysis on FACSaria isolated NK1.1+ cells confirming low activation in TRAIL−/−/GNMT−/− cells compared to GNMT−/−. n= 5–10. *p<0.05; **p< 0.01, ***<0.001 (GNMT−/− vs TRAIL−/−/GNMT−/−). Error bars represent SD.
Extensive fibrosis and liver damage in GNMT−/− mice after BDL concurs with activation of NK1.1+ cells
The significant attenuation of the progression of chronic liver disease and accordingly, the lower fibrosis/cirrhosis observed in TRAIL−/−/GNMT−/− mice (Fig 2) may seem conflicting with the anti-fibrogenic characteristic TRAIL-producing NK cells described by others9, 10. Thus, with the aim to elucidate the contribution of TRAIL-producing NK cells to fibrogenesis, we used a well-established animal model of fibrogenesis; bile duct ligation (BDL).
First, we found that only 25% of GNMT−/− mice survived the BDL up to 21 days, whereas survival was 60% in GNMT WT animals (GNMT+/+) (Suppl. Fig. 1A). The increased activation of hepatic stellate cells (HSC) and stronger collagen deposition found in GNMT−/− were evidenced by αSMA IF (Fig. 3A) and Sirius Red staining (Fig. 3B) in liver sections and further quantification at 14d and 21d after BDL (Suppl. Fig. 1B). qPCR analysis of αSMA and collagen1A1 expression confirmed the increased pro-fibrogenic response in GNMT−/− mice after BDL in comparison with WT animals (Fig. 3C). Interestingly, BDL led to significant increase of TLR9 in GNMT−/− mice (Fig. 3C), a receptor involved in the activation of HSC after engulfment of apoptotic hepatocyte-DNA7.
Figure 3. Exacerbated fibrogenesis in GNMT−/− mice concurs with activation of NK1.1+ cells.

(A) IF using an αSMA Ab and (B) Sirius Red staining showed increased fibrosis in GNMT−/− mice. (C) qPCR analysis of mRNA expression of αSMA, collagen1A1 and TLR9 confirmed HSC activation. (D) Dot plot of FACS analysis on isolated liver mononuclear cells (MNCs) showing lower number of CD45+/NK1.1.+ cells in GNMT−/− mice. (E) Histogram showing higher presence of TRAIL in the cell surface of NK1.1+ cells in GNMT−/− liver-MNCs. Bar-plot representing the data obtained by FACS analysis showing low presence of NK1.1+ cells correlating with high expression of TRAIL in GNMT−/− liver-MNCs. (F) qPCR analysis showing increased DR5 mRNA expression in GNMT−/− mice. (H) Western blot analysis confirmed that GNMT−/− mice have higher expression of DR5 after BDL correlating with increased phosphorylation of JNK after surgery. n = 5–7. *p< 0.05; **p< 0.01; ***P < 0.001 (GNMT+/+ vs GNMT−/−) Error bars represent SD.
Importantly, the increased fibrogenesis observed in GNMT−/− mice correlated with strong activation of NK1.1+ cells. Thus, FACS analysis revealed a lower presence of NK1.1+ cells in livers from GNMT−/− mice 14d after BDL (Fig. 3D), which denotes their activation4. This was supported by the higher expression of TRAIL found in liver NK1.1+ cells from GNMT−/− mice after BDL (Fig. 3E). Also, the elevated expression of granzyme and perforin, the mediators of NK1.1+ cell-mediated cell death, and RANTES/CCL5 found in GNMT−/− mice after BDL confirmed the activation of these cells (Suppl. Fig. 1C). We previously described that NK cells were cytotoxic against GNMT−/− hepatocytes and actively contribute to liver injury4. Accordingly, increased liver injury in GNMT−/− mice after BDL was evidenced by high serum transaminases and bilirubin levels (Suppl. Fig. 1D) and H&E staining on liver sections showing wide necrotic areas (Suppl. Fig. 1E, G). The increased apoptosis in GNMT−/− mice after BDL compared to WT mice was evidenced by TUNEL assay (Suppl. Fig. 1F) and quantification of caspase-3 activity (Suppl. Fig. 1G, lower panel). Importantly, increased NK1.1+ cell activation and liver injury in GNMT−/− mice correlated with higher mRNA and protein expression of DR5 observed in GNMT−/− mice, which may contribute to the cytotoxicity of TRAIL-producing NK1.1+ cells after BDL (Fig. 3F, G). In accordance, strong and sustained phosphorylation of JNK, the downstream kinase activated upon DR5 engagement, was found in GNMT−/− animals after BDL (Fig. 3G).
Tissue destruction triggers the inflammatory response in an attempt to remove cell debris, mainly mediated by Kupffer cells (KC). Previous work highlighted the implication of KC during fibrogenesis in different experimental models20, 21. Interestingly, despite the more severe liver injury and fibrosis observed in GNMT−/− mice we found lower presence of F4/80 positive cells after BDL compared to WT animals (Suppl. Fig. 1H). Finally, TNF serum levels were lower in GNMT−/− mice compared to WT after 14d supporting the low activation of KC observed (Suppl. Fig. 1I). These data are consistent with the lower KC-activation described in GNMT−/− mice in response to LPS, where NK cells but not KC mediated endotoxin-liver damage4.
Inhibition of NK1.1+ cells protects the liver against liver injury and fibrogenesis after BDL in the absence of GNMT
GNMT−/− mice show increased NK1.1+ cell activation that coincides with increased fibrogenesis after BDL (Fig.3). NK1.1 is a cell surface marker of both NK and NKT cells, which have shown opposing roles during fibrogenesis 8, 10. Our data point to a role of TRAIL-producing NK cells in mediating liver injury and fibrogenesis in the absence of GNMT (Fig. 2). Therefore, with the aim to better delineate the implication of NK cells during this process we performed two experimental approaches; i) we inhibited the whole NK1.1+ cell compartment using an NK1.1 depleting antibody and ii) we specifically depleted NK cells using the anti-ASIALO-GM1 antibody (ASIALO) before and after BDL.
The important beneficial impact of NK1.1+ cell depletion was evidenced by the 100% survival of NK1.1/GNMT−/− mice after BDL, in clear contrast to the high mortality observed in GNMT−/− mice (Fig. 4A). Lower serum transaminases levels, improved liver histology showing less necrotic areas, reduced presence of TUNEL positive cells and lower caspase-3 activity confirmed the beneficial impact of NK1.1+ cell-depletion in attenuating liver injury in GNMT−/− mice after BDL (Fig. 4B–D). In accordance with less cell death, liver fibrosis was significantly attenuated in NK1.1/GNMT−/− mice as evidenced by αSMA IF, Sirius Red staining, further quantification and qPCR of αSMA and collagen 1A1 expression (Fig. 4E–G). Also, the decreased expression of TLR9 found in NK1.1/GNMT−/− mice after BDL further supported the lower activation of HSC (Fig. 4G).
Figure 4. NK1.1+ cell depletion protects the liver of GNMT−/− mice against BDL-induced liver damage and fibrosis.

(A) Kaplan-Meier survival curve evidencing the beneficial impact of NK1.1+ cell depletion after BDL. (B) Serum transaminase and billirubin analysis, (C) H&E staining and TUNEL assay on liver sections showing lower liver injury after BDL in NK1.1/GNMT−/− mice. (D) Further quantification of necrotic areas on H&E sections and caspase-3 activity on whole liver extracts confirmed these data. (E) IHC using an αSMA Ab and Sirius Red staining and (F) further quantification evidenced reduced fibrosis in NK1.1/GNMT−/− mice. (G) qPCR analysis of αSMA, collagen1A1, TLR9 and DR5 expression. (H) WB analysis showing lower pJNK expression in NK1.1/GNMT−/− mice after BDL. n = 5–7. *p< 0.05; **p< 0.01; ***P < 0.001 (GNMT−/− vs NK1.1/GNMT−/−). Error bars represent SD.
KC-activation was partially restored in NK1.1/GNMT−/− mice as shown by F4/80 IHC and TNF serum levels (Suppl. Fig. 2A, B), suggesting an inhibitory cross-talk between KC and NK cells in the absence of GNMT. Finally, inhibition of NK1.1+ cells in GNMT−/− mice correlated with lower DR5 expression and less activation of JNK after BDL (Fig. 4G, H).
Overall, our data suggest that NK1.1+ cell activation leads to increased liver injury, strongly contributing to fibrogenesis after BDL in the context of GNMT absence.
Selective inhibition of TRAIL-producing NK cells has an anti-fibrogenic effect after BDL in the absence of GNMT
NK cells are described as the main producers of TRAIL in the liver 22, 23. Here we provide further evidence that NK cells isolated from GNMT−/− mice express significantly more TRAIL than NKT cells (Supplemental Fig. 3A). Moreover, we also show that NK cells from GNMT−/− mice express more TRAIL than GNMT+/+ both by FACS analysis and qRTPCR analysis in FASCaria isolated cells (Supplemental Fig. 3B, C), further supporting the potential implication of TRAIL producing NK cells in the pathogenesis of GNMT−/− mice.
Next, in order to better delineate the implication of TRAIL-producing NK cells during fibrogenesis we specifically depleted this cell compartment using the anti-ASIALO-GM1 antibody (ASIALO). Similarly to what we found in NK1.1/GNMT−/−, liver fibrosis was significantly attenuated in ASIALO/GNMT−/− mice as evidenced by αSMA, Sirius Red staining and further quantification (Fig. 5A, B). qPCR of αSMA and collagen 1A1 expression confirmed the reduced fibrosis found in ASIALO/GNMT−/− mice after BDL. Also, the decreased expression of TLR9 found in ASIALO/GNMT−/− mice after BDL pointed to a lower activation of HSC (Fig. 5B). The beneficial impact of specific inhibition of NK cells was supported by the high survival rate in ASIALO/GNMT−/− mice compared to GNMT−/− animals (Fig. 5C). Further analysis of serum transaminases (Fig. 5D), H&E staining and TUNEL assay (Fig. 5E, F) confirmed the significant improvement of the liver parenchyma status and the attenuation of cell death in NK cell-depleted mice after BDL. ASIALO/GNMT−/− mice showed lower DR5 expression and attenuated activation of JNK 14d after BDL (Fig. 5G, H). Finally, ASIALO/GNMT−/− mice showed partial recovery of KC activation evidenced by F4/80 IHC and TNF ELISA (Suppl. Fig. 4A, B), supporting the regulation between cell compartments in the context of cholestatic-liver damage in GNMT−/− animals. In parallel, a control group receiving IgG was performed showing no significant differences in fibrogenesis and liver injury when compared to the uninjected GNMT−/− mice, overall proving that the latter group can be considered as a proper control for both NK1.1+ and ASIALO treated groups (Suppl. Fig. 5).
Figure 5. Specific NK cell inhibition with ASIALO protects the liver of GNMT−/− mice from BDL-liver injury and fibrosis.

(A) αSMA IF (left panels) and Sirius red staining (right panels), further quantification using FRIDA software and (B) qPCR of αSMA, collagen1A1 and TLR9 expression evidenced the significantly lower fibrosis in ASIALO/GNMT−/− after BDL. (C) Kaplan-Meier survival curve showing improved animal survival in ASIALO/GNMT−/− compared to GNMT−/− after BDL. (D) Analysis of serum transaminases and bilirubin, (E) H&E staining (left panels) and TUNEL assay (right panels) showed an evident reduction of liver injury in ASIALO/GNMT−/− mice after BDL. (F) Quantification of necrotic areas in H&E stained liver sections after BDL showing less necrosis in ASIALO/GNMT−/− mice. (G) qPCR analysis of DR5 expression on liver samples and (H) lower phosphorylation of JNK detected by western blot analysis evidenced lower activation of the DR5-mediated cell death response in ASIALO/GNMT−/− mice after BDL. n = 5–7. **p< 0.01 (GNMT−/− vs ASIALO/GNMT−/−). Error bars represent SD.
Overall, our data points to the implication of TRAIL-producing NK cells in mediating BDL-induced liver injury and further fibrogenesis in the absence of GNMT.
TRAIL deficiency attenuates fibrogenesis in GNMT−/− mice but exacerbates the necrotic response after BDL
NK cells are the main source of TRAIL (Suppl. Fig 3), which mediates their antifibrogenic effect by promoting HSC death10. Conversely, previous work described that TRAIL has a main role in mediating cholestatic liver injury14. In this line, here we describe that TRAIL depletion notably attenuated the chronic damaging phenotype found in GNMT−/− mice (Fig. 2). Finally, inhibition of NK1.1+ cells and specifically of TRAIL-producing NK cells efficiently protected the liver against severe injury and fibrosis in GNMT−/− mice after BDL (Fig. 4, Fig. 5).
Overall, in order to better delineate the implication of TRAIL during fibrogenesis in the pathological context of GNMT deficiency we next performed BDL in TRAIL−/−/GNMT−/− mice. Liver fibrosis was significantly attenuated in TRAIL−/−/GNMT−/− when compared to GNMT−/− mice 21 days after BDL (Fig. 6A–C, Suppl. Fig. 6A). Also TLR9 was significantly less expressed in the double-KO mice, supporting lower activation of HSC after BDL (Suppl. Fig. 6B). Surprisingly, despite the obvious attenuation in liver fibrosis exerted by TRAIL depletion, BDL led to a similar lethality of TRAIL−/−/GNMT−/− mice, mainly occurring within the first days after surgery (Fig. 6D). Further analysis showed that mortality of TRAIL−/−/GNMT−/− mice correlated with severe liver necrosis, especially at 3 days after BDL (Fig. 6E, G upper panel). Interestingly, hepatocyte apoptosis was attenuated at 3, 7 and 14 days after BDL in TRAIL−/−/GNMT−/− mice (Fig. 6F, G lower panel). Accordingly with increased necrosis and lower apoptosis, we found less FADD expression in TRAIL−/−/GNMT−/− mice after BDL when compared to GNMT−/− and WT mice (Fig. 6H). Also, we observed reduced expression of DR5 in livers from the double-KO mice after BDL (Fig. 6I).
Figure 6. TRAIL deficiency in GNMT−/− mice attenuates fibrogenesis after BDL but shifts cell death from apoptosis to necrosis.

(A) TRAIL−/−/GNMT−/− mice show significantly lower fibrosis than GNMT−/− mice after BDL as shown by IF using an αSMA Ab, Sirius Red staining and further quantification. (B) Kaplan-Meier curve showing an overall comparable death rate after BDL in TRAIL−/−/GNMT−/− compared to GNMT−/− animals. (C) H&E staining and (G) further quantification (upper panel), (D) TUNEL assay and (G) Caspase3 activity (lower panel) of liver extracts showing lower apoptotic cell death but profuse necrosis in TRAIL−/−/GNMT−/− after BDL. (H) WB analysis showing lower FADD in TRAIL−/−/GNMT−/− mice after BDL. (I) qPCR showing lower DR5 expression in TRAIL−/−/GNMT−/− mice 14d after BDL. n = 5–7. *p< 0.05; **p< 0.01; ***P < 0.001 (TRAIL−/−/GNMT−/− vs GNMT−/−).
In vitro assays confirmed the higher susceptibility to necrosis conferred by TRAIL depletion in GNMT−/− mice. Thus, isolated hepatocytes from TRAIL−/−/GNMT−/− mice showed lower survival after deoxycholic acid (DCA) treatment, similar to GNMT−/− cells, when compared to cultured GNMT+/+ cells (Suppl. Fig. 7A). Interestingly, despite comparable low survival, caspase-3 activity in TRAIL−/−/GNMT−/− cells was significantly lower than in GNMT−/− hepatocytes after DCA and comparable to WT cells (Suppl. Fig. 7B). Finally, we found higher expression of RIP1 and RIP3 in TRAIL−/−/GNMT−/− cells exposed to DCA compared to GNMT−/− cells (Suppl. Fig. 7C).
Overall, our data show that TRAIL deficiency in GNMT−/− hepatocytes promotes a shift from apoptosis towards necrotic cell death. Moreover, our data suggest that TRAIL has a main role during fibrogenesis by mediating hepatocyte apoptosis in the pathogenic context of GNMT deficiency.
In vivo silencing of DR5 protects GNMT−/− mice against BDL-induced liver injury
Finally, silencing of DR5 in GNMT−/− mice confirmed the implication of the TRAIL/DR5 axis in mediating liver injury and further fibrogenesis. Thus, fibrosis was significantly reduced in ShDR5/GNMT−/− mice as we found lower αSMA levels and collagen deposition after BDL compared to GNMT−/− (Fig. 7A). Low fibrogenesis after DR5 silencing correlated with an overall significant improvement in the liver parenchyma of ShDR5/GNMT−/− mice that showed significantly lower AST, ALT and bilirubin serum levels after BDL (Fig. 7B). H&E staining, TUNEL assay, caspase-3 activity and a survival rate of 90% confirmed the liver protection exerted by silencing of DR5 in GNMT−/− mice (Fig. 7C–E).
Figure 7. In vivo silencing of DR5 in GNMT−/− mice attenuates fibrosis protecting the liver against BDL-induced injury.

(A) IF using αSMA Ab and Sirius Red staining and qPCR analysis of αSMA and Collagen 1A1 showed lower fibrosis in ShDR5/GNMT−/− mice. (B) Serum transaminases and bilirubin were lower in ShDR5/GNMT−/− mice compared to GNMT−/− mice after BDL. (C) H&E staining (left panels) and (D) further quantification (upper panel), (C) TUNEL assay (right panels) and (D) quantification of caspase-3 activity (lower panel) showed an obvious attenuation of both necrotic and apoptotic cell death in ShDR5/GNMT−/− mice. (E) Kaplan-Meier curve showing increased survival of ShDR5/GNMT−/− mice compared to GNMT−/− littermates. n = 5–7. *p< 0.05; **p< 0.01; ***P < 0.001 (GNMT−/− vs ShDR5/GNMT−/−). Error bars represent SD.
Overall, these data point to the hepatoprotective effect of DR5 deletion during BDL-induced liver injury in the pathogenic context of GNMT deficiency and suggest that attenuation of both DR5-mediated necrosis and apoptosis in GNMT−/− mice protects the liver against fibrogenesis after BDL.
DISCUSSION
Chronic liver injury triggers fibrogenesis, a tissue remodeling and scarring process that leads to cirrhosis and HCC regardless of the disease etiology5. GNMT, the enzyme that metabolizes S-adenosylmethionine, is downregulated in cirrhotic patients from diverse etiologies such as HCV and alcohol, and is absent in HCC2. Here, we show that GNMT is also downregulated during human cholestatic liver disease. Importantly, we observed that GNMT is reduced both at initial stages in patients with mild cholestasis and low degree of ductular alterations as well as in patients with more severe liver injury prior to transplantation. These data, together with previous work describing the lower expression of GNMT in NAFLD patients24, support the important role of GNMT during the pathogenesis of liver disease regardless of the etiology and suggest a causative role for the deficiency of GNMT rather than a consequence of altered liver homeostasis.
In accordance, GNMT-deficient mice develop fibrosis, cirrhosis and HCC3, which make them a valuable tool to investigate the molecular mechanisms underlying the pathogenesis of chronic liver disease. We previously described that in GNMT−/− mice, NK cells had a strong cytotoxic activity against autologous hepatocytes, which exhibited high expression of NK cells-activating ligands and TRAILR2/DR5, evidencing that GNMT deficiency promotes a ‘transformed-like’ phenotype in hepatocytes4. In GNMT−/− mice, NK cell activation led to increased apoptotic cell death, mainly mediated by TRAIL, which overall contributed to the inflammatory environment, distinctive of the progression of NASH4. Nonetheless, the role of TRAIL during chronic liver disease and fibrogenesis in the pathogenic context of GNMT deficiency was not further investigated. Here, we provide evidence that NK cells from GNMT−/− mice produce more TRAIL than WT cells and that depletion of TRAIL in GNMT−/− mice efficiently protects the liver against the progression of chronic liver disease characterized by profuse fibrogenesis. Importantly, the regression of the damaging phenotype in TRAIL−/−/GNMT−/− mice correlated with a lower activation of the NK1.1+ cell compartment and an overall restoration of the percentage of NK cells that reached comparable levels to WT animals.
NK and NKT cells have a differential role during fibrogenesis. Generally, while NKT cells are considered as pro-fibrogenic8, NK cells are commonly described as anti-fibrogenic since they induce HSC death through TRAIL and RAE1/NKG2D (reviewed in25). However, NK cell-mediated HSC death strongly depends on the activation status of the latter. Thus, NK cells kill early- but not fully-activated HSC, due to the loss of expression of RAE1 over time9. Similarly, HSC activated by phagocytosis of apoptotic bodies are resistant to TRAIL-mediated cell death26. Moreover, TRAIL has been described as a mediator of cholestatic liver injury in mice and a contributor to chronic cholestatic liver disease in humans through bile duct destruction11–14. Taking all these together, the concurrence of a damaging phenotype progressing to fibrosis with strong activation/cytotoxicity of NK cells found in the context of GNMT deficiency is somewhat intriguing. Thus, we aimed to elucidate the implication of NK cells during fibrogenesis in the pathogenic context of GNMT deficiency. We convincingly show that fibrosis progresses in the context of NK cell activation and that specific inhibition of NK cells efficiently protects the liver against BDL-injury/fibrosis in the absence of GNMT. Our data in TRAIL−/−/GNMT−/− mice further support the role of TRAIL during fibrogenesis in GNMT-deficient hepatocytes as mediators of hepatocyte apoptotic death. Interestingly, while transient amelioration of TRAIL in ASIALO/GNMT−/− mice efficiently protected the liver against BDL-acute injury, persistent blockade of apoptosis in TRAIL−/−/GNMT−/− mice promoted a high susceptibility to BDL-induced necrotic cell death. The shift from apoptosis to necrotic cell death is in accordance with previous work showing that persistent inhibition of apoptosis through genetic caspase-8 depletion sensitizes the liver to RIP1/3-mediated necrotic cell death27. The low expression of FADD; the endogenous inhibitor of RIP3-necrosis28 found in TRAIL−/−/GNMT−/− supports the increased incidence of BDL-induced necrosis. Accordingly TRAIL−/−/GNMT−/− hepatocytes were susceptible to necrotic rather than to apoptotic cell death in response to BA. Furthermore, our present data support the selective activation of HSC through phagocytosis of apoptotic hepatocytes (reviewed in29), as fibrogenesis was impaired in TRAIL−/−/GNMT−/− mice despite severe necrotic cell death and the potential pro-survival environment for HSC in a TRAIL-free context. Interestingly, protection against BDL-induced liver injury and fibrogenesis in GNMT−/− mice after depletion of NK cells and in TRAIL−/−/GNMT−/− mice all correlated with downregulation of DR5. These data support previous work highlighting the feedback-regulation of ligand and receptor30. Finally, in vivo DR5 silencing protected the GNMT deficient liver against BDL-injury, pointing to DR5 as a key mediator of fibrogenesis by means of hepatocyte death and further HSC activation, rather than acting as an anti-fibrotic agent causing HSC apoptosis. This anti-fibrogenic effect likely occurs in the pathological context of GNMT deficiency since, accordingly with the anti-fibrogenic role of DR5 as a mediator of HSC-death, in vivo silencing of DR5 in WT animals led to increased fibrogenesis after BDL in these healthy animals (data not shown).
Cell death triggers the inflammatory response in the liver, which is followed by tissue remodeling and fibrogenesis. Previous work highlighted the implication of Kupffer cells (KC) during fibrogenesis in different experimental models20. KC are recruited to the sites of inflammation and contribute to HSC activation, promoting liver fibrosis as part of the wound healing and regeneration process after injury21. Interestingly, despite increased fibrosis found in the context of GNMT deficiency, KC were poorly activated after BDL suggesting a negative regulatory feedback with NK cells. Accordingly, depletion of NK cells partially restored the activation of KC, which was, nevertheless, insufficient to trigger fibrogenesis in GNMT−/− livers likely due to the (transient) absence of TRAIL-cell death. The higher presence of KC in the liver of WT animals stimulated with poly I:C (which activate NK cells) would point to an attenuation of KC activation in the presence of activated NK cells is restricted to the context of GNMT absence (data not shown). Overall, it is tempting to speculate that due to the absence of KC activation found in GNMT−/− mice, removal of apoptotic cell debris by HSC may further contribute to their activation and fibrogenic activity. The increased expression of TLR9 consistently observed in GNMT−/− mice supports this mechanism, as DNA from mammalian apoptotic cells (DAMPs) promotes phagocytosis in HSC7, which readily become activated and resistant to TRAIL/DR5-mediated death26.
In summary, we here describe that NK cells mediate severe liver injury, further leading to fibrosis through the TRAIL/DR5 axis in the context of absence of GNMT. We propose that this undergoes through i) NK cell-mediated direct apoptotic killing of GNMT deficient hepatocytes by TRAIL, which further triggers an inflammatory response that ii) activates HSC-mediated fibrogenesis. In accordance, we here show that both NK cell inhibition and overall depletion of TRAIL significantly attenuates liver injury after BDL and further HSC activation and consequent fibrogenesis in GNMT−/− mice. Ultimately, silencing of DR5 also protected the liver against hepatocyte cell death and further fibrogenesis in the absence of GNMT. These data is of relevance since GNMT deficiency occurs in chronic human liver disease regardless of the etiology. Moreover, we here show that even at early stages of human liver (cholestatic) disease, when fibrosis is still not prominent, GNMT expression is low. This data, together with previous work showing attenuation of GNMT expression during NAFLD24 support the essential role of GNMT in maintaining liver homeostasis and preventing liver disease.
Overall, our present study provides new insights on the implication of TRAIL-producing NK cells in mediating liver injury and fibrogenesis when GNMT is absent. This knowledge may facilitate the development of potential therapeutic strategies based on NK cell inactivation to counteract hepatocyte injury in the pathogenic context of GNMT deficiency, a scenario that is critical for the progression from chronic to end-stage liver disease. Finally, our work underlines the importance of carefully assessing the molecular context of liver disease before proposing therapeutic strategies to counteract fibrogenesis in cirrhotic patients from different etiologies.
Supplementary Material
Acknowledgements
This work was supported by grants from the Instituto de Salud Carlos III; FIS, PS12/00402 (to N.B.), NIH AT-1576 (to S.C.L., M.L.M.-C., and J.M.M.), ETORTEK-2012 (to M.L.M.-C), Sanidad del Gobierno Vasco 2013 (to M.L.M.-C), PI11/01588 (to M.L.M.-C). Plan Nacional SAF2011-29851 (to JMM). N.B. is funded by the Program Ramon y Cajal (Ministry of Economy and Competitiveness, Spain). PI11/02942 (to A.L.). Ciberehd is funded by the Instituto de Salud Carlos III.
Footnotes
Conflict of Interest Statement: The authors declare that they have no competing financial interests.
REFERENCES
- 1.Martinez-Lopez N, Varela-Rey M, Ariz U, et al. S-adenosylmethionine and proliferation: new pathways, new targets. Biochemical Society transactions. 2008;36(Pt 5):848–852. doi: 10.1042/BST0360848. [DOI] [PubMed] [Google Scholar]
- 2.Avila MA, Berasain C, Torres L, et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol. 2000;33(6):907–914. doi: 10.1016/s0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Chantar ML, Vazquez-Chantada M, Ariz U, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47(4):1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomez-Santos L, Luka Z, Wagner C, et al. Inhibition of natural killer cells protects the liver against acute injury in the absence of glycine N-methyltransferase. Hepatology. 2012;56(2):747–759. doi: 10.1002/hep.25694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39(2):273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe A, Hashmi A, Gomes DA, et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology. 2007;46(5):1509–1518. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]
- 8.Syn WK, Agboola KM, Swiderska M, et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut. 2012;61(9):1323–1329. doi: 10.1136/gutjnl-2011-301857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Radaeva S, Wang L, Radaev S, et al. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am J Physiol Gastrointest Liver Physiol. 2007;293(4):G809–G816. doi: 10.1152/ajpgi.00212.2007. [DOI] [PubMed] [Google Scholar]
- 10.Radaeva S, Sun R, Jaruga B, et al. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D–dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130(2):435–452. doi: 10.1053/j.gastro.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 11.Shimoda S, Harada K, Niiro H, et al. Interaction between Toll-like receptors and natural killer cells in the destruction of bile ducts in primary biliary cirrhosis. Hepatology. 2011;53(4):1270–1281. doi: 10.1002/hep.24194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chuang YH, Lian ZX, Tsuneyama K, et al. Increased killing activity and decreased cytokine production in NK cells in patients with primary biliary cirrhosis. Journal of autoimmunity. 2006;26(4):232–240. doi: 10.1016/j.jaut.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Takeda K, Kojima Y, Ikejima K, et al. Death receptor 5 mediated-apoptosis contributes to cholestatic liver disease. Proc Natl Acad Sci U S A. 2008;105(31):10895–10900. doi: 10.1073/pnas.0802702105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kahraman A, Barreyro FJ, Bronk SF, et al. TRAIL mediates liver injury by the innate immune system in the bile duct-ligated mouse. Hepatology. 2008;47(4):1317–1330. doi: 10.1002/hep.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahraman A, Schlattjan M, Kocabayoglu P, et al. Major histocompatibility complex class I-related chains A and B (MIC A/B): a novel role in nonalcoholic steatohepatitis. Hepatology. 2010;51(1):92–102. doi: 10.1002/hep.23253. [DOI] [PubMed] [Google Scholar]
- 16.Luka Z, Capdevila A, Mato JM, et al. A glycine N-methyltransferase knockout mouse model for humans with deficiency of this enzyme. Transgenic Res. 2006;15(3):393–397. doi: 10.1007/s11248-006-0008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fickert P, Zollner G, Fuchsbichler A, et al. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology. 2002;123(4):1238–1251. doi: 10.1053/gast.2002.35948. [DOI] [PubMed] [Google Scholar]
- 18.Beraza N, Ludde T, Assmus U, et al. Hepatocyte-specific IKK gamma/NEMO expression determines the degree of liver injury. Gastroenterology. 2007;132(7):2504–2517. doi: 10.1053/j.gastro.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 19.Beraza N, Malato Y, Sander LE, et al. Hepatocyte-specific NEMO deletion promotes NK/NKT cell- and TRAIL-dependent liver damage. J Exp Med. 2009;206(8):1727–1737. doi: 10.1084/jem.20082152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13(11):1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 21.Seki E, De Minicis S, Gwak GY, et al. CCR1 and CCR5 promote hepatic fibrosis in mice. J Clin Invest. 2009;119(7):1858–1870. doi: 10.1172/JCI37444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smyth MJ, Cretney E, Takeda K, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J Exp Med. 2001;193(6):661–670. doi: 10.1084/jem.193.6.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeda K, Hayakawa Y, Smyth MJ, et al. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med. 2001;7(1):94–100. doi: 10.1038/83416. [DOI] [PubMed] [Google Scholar]
- 24.Chen CH, Huang MH, Yang JC, et al. Prevalence and risk factors of nonalcoholic fatty liver disease in an adult population of taiwan: metabolic significance of nonalcoholic fatty liver disease in nonobese adults. Journal of clinical gastroenterology. 2006;40(8):745–752. doi: 10.1097/00004836-200609000-00016. [DOI] [PubMed] [Google Scholar]
- 25.Gao B, Radaeva S. Natural killer and natural killer T cells in liver fibrosis. Biochimica et biophysica acta. 2013;1832(7):1061–1069. doi: 10.1016/j.bbadis.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang JX, Mikami K, Venugopal S, et al. Apoptotic body engulfment by hepatic stellate cells promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent pathways. J Hepatol. 2009;51(1):139–148. doi: 10.1016/j.jhep.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liedtke C, Bangen JM, Freimuth J, et al. Loss of caspase-8 protects mice against inflammation-related hepatocarcinogenesis but induces non-apoptotic liver injury. Gastroenterology. 2011;141(6):2176–2187. doi: 10.1053/j.gastro.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 28.Galluzzi L, Kepp O, Kroemer G. FADD: an endogenous inhibitor of RIP3-driven regulated necrosis. Cell research. 2011;21(10):1383–1385. doi: 10.1038/cr.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehal W, Imaeda A. Cell death and fibrogenesis. Seminars in liver disease. 2010;30(3):226–231. doi: 10.1055/s-0030-1255352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shetty S, Gladden JB, Henson ES, et al. Tumor necrosis factor-related apoptosis inducing ligand (TRAIL) up-regulates death receptor 5 (DR5) mediated by NFkappaB activation in epithelial derived cell lines. Apoptosis : an international journal on programmed cell death. 2002;7(5):413–420. doi: 10.1023/a:1020031023947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.