

Abstract
We established a novel model of myocarditis induced with Theiler's murine encephalomyelitis virus (TMEV), which has been used as a viral model for multiple sclerosis and seizure/epilepsy. Following TMEV infection, C3H mice developed severe myocarditis with T cell infiltration, while C57BL/6 mice had mild lesions and SJL/J mice had no inflammation in the heart. In C3H mice, myocarditis was divided into three phases: acute viral, subacute immune, and chronic fibrotic phases. Using toll-like receptor (TLR) 4-deficient C3H mice, we found that interleukin (IL)-6, IL-17, TLR4, and anti-viral immune responses were associated with myocarditis susceptibility.
Keywords: Infectious disease, Tropism, Picornaviridae infection, CNS demyelinating disease, Fibrosis, Toll-like receptor, Autoimmunity, IL-6, Echocardiography, Troponin
1. Introduction
Theiler's murine encephalomyelitis virus (TMEV) is a non-enveloped, positive-sense, single-stranded RNA virus that belongs to the genus Cardiovirus, family Picornaviridae, and a natural pathogen of mice [1]. About 1 month after intracerebral TMEV injection, TMEV can persistently infect glial cells and macrophages in the central nervous system (CNS), leading to an inflammatory demyelinating disease in susceptible mouse strains, such as SJL/J mice [2]. TMEV-induced demyelinating disease (TMEV-IDD) resembles multiple sclerosis (MS) both clinically and histologically; TMEV-IDD has been widely used as a viral model of MS [3]. Although the precise pathomechanisms of TMEV-IDD are unclear, direct TMEV infection in the CNS as well as both cellular and humoral acquired immune responses against TMEV and CNS antigens have been shown to play pathogenic roles [4].
TMEV has also been used as a viral model of seizure/epilepsy, since TMEV can induce seizures in susceptible mouse strains, such as C57BL/6 (B6) mice, during the first week of infection [5]. The seizures induced with TMEV have been associated with innate immunity, such as interleukin (IL)-6 production from infiltrating macrophages [6]. Outside the CNS, although TMEV can cause viremia during the acute phase and has been isolated from the gastrointestinal tract, skeletal muscle, and cardiac muscle [7], there have been a few reports investigating the pathogenesis of TMEV infection in general organs [8].
In this study, we established a novel model of myocarditis induced with TMEV characterized by echocardiography, cardiac troponin (an indicator of cardiomyocyte damage), viral titration, histology (T cell infiltration and fibrosis), and virus-specific immune responses. Among SJL/J, B6, and C3H mice, interestingly, we found that only C3H mice, which were intermediately susceptible to TMEV-IDD and seizures, developed severe myocarditis, which was divided into three phases: acute viral (phase I), subacute immune (phase II), and chronic fibrotic (phase III) phases. To further characterize the myocarditis model, we infected wild-type and toll-like receptor (TLR) 4-deficient C3H mice and found decreased pro-inflammatory IL-6 and IL-17 production in TLR4-deficient C3H mice.
2. Materials and Methods
2.1. Comparative study among inbred mouse strains
Five-week-old B6 (Harlan Laboratories, Inc., Indianapolis, IN), SJL/J (Jackson Laboratory, Bar Harbor, ME), C3H/HeNTac (wild-type, Taconic Farms, Inc., Hudson, NY), and C3H/HeJ (TLR4-deficient, Jackson Laboratory) mice were infected intracerebrally with 2 × 105 plaque forming units (PFU) of the Daniels (DA) strain of TMEV, as described previously [9]. The severity of TMEV-induced seizures/epilepsy was graded using the Racine scale [5]. Mice were perfused with phosphate-buffered saline (PBS) followed by a 4% paraformaldehyde solution (Sigma-Aldrich, St. Louis, MO) in PBS. The spinal cord and heart tissues were harvested and fixed with 4% paraformaldehyde. The spinal cords and hearts were divided into 10 to 12 and four transverse sections, respectively, and embedded in paraffin. Four-μm-thick sections of the spinal cords and hearts were stained with Luxol fast blue (Solvent blue 38; Sigma-Aldrich) for myelin visualization, and hematoxylin (Electron Microscopy Sciences, Hatfield, PA) and eosin (Thermo Fisher Scientific, Inc., Rochester, NY) for myocarditis, respectively [9].
2.2. Characterization of viral myocarditis
Cardiac function and morphology were monitored by M- and B-mode of transthoracic echocardiography, respectively, using the Vevo 770 system (FUJIFILM VisualSonics, Inc., Ontario, Canada). Left ventricular end diastolic volume (LVEDV) and left ventricular end systolic volume (LVESV) were assessed in all mice. Left ventricular ejection fraction (LVEF) was calculated according to the following equation: LVEF = [(LVEDV -LVESV)/LVEDV] × 100. Blood was collected from TMEV-infected mice by submandibular bleeding. The levels of cardiac troponin I in sera (Life Diagnostic, Inc., West Chester, PA) were assessed by enzyme-linked immunosorbent assays (ELISA), according to the manufacturer's instructions. Viral genome was semi-quantified by real-time PCR of viral RNA using a pair of primers for a capsid protein VP2 of TMEV, as described previously [2]: forward; 5′-TGGTCGACTCTGTGGTTACG-3′ and reverse; 5′-GCCGGTCTTGCAAAGATAGT-3′. Glyceraldehyde-3-phosphate dehydrogenases (Gapd) expression was used as a housekeeping gene for normalization. CD3+ T cells and B220+ B cells were quantified by immunohistochemistry with anti-CD3ε antibody (Dako North America, Inc., Carpinteria, CA) and biotinylated anti-CD45R/B220 antibody (BD Biosciences, San Diego, CA), using the avidin-biotin-peroxidase complex technique (Vector, Burlingame, CA), as described previously [10; 11]. Fibrosis was quantified by Masson trichrome (Thermo Fisher Scientific, Inc.) or picrosirius red (ScyTek Laboratories, Inc., Logan, UT) staining. Infectious virus in the heart was titrated by plaque assays, as described previously [12].
2.3. Immune responses
The levels of serum anti-TMEV antibodies were assessed by ELISA, as described previously [12]. After blocking with 10% fetal bovine serum (FBS) and 0.2% Tween® 20 (Thermo Fisher Scientific), serial dilutions of sera were added to 96-well flat-bottom Nunc-Immuno plates (Thermo Fisher Scientific, Inc.) coated with 10 μg/ml of purified TMEV antigen. Following washing, a horseradish peroxidase-conjugated anti-mouse IgG (H + L) (Life Technologies, Gaithersburg, MD), anti-mouse IgG1 (Life Technologies), or anti-mouse IgG2a (Life Technologies) was added to the plates. Immunoreactive complexes were detected with o-phenylendiamine dihydrochloride (Sigma-Aldrich) and absorbance was read at 492 nm. Mononuclear cells (MNCs) were isolated from the spleens of TMEV-infected mice, stimulated with live TMEV at a multiplicity of infection (MOI) of 5, and cultured for 5 days. The levels of TMEV-specific lymphoproliferative responses were measured by [3H]thymidine (PerkinElmer, Inc., Waltham, MA) incorporation assays, as described previously [12]. All cultures were performed in triplicate and the data were expressed as stimulation indexes (experimental cpm/control cpm). For cytokine assays, MNCs isolated from the spleens of TMEV-infected mice were stimulated with 5 μg/ml of concanavalin A for 48 hours. The levels of IL-6 (BD Biosciences) and IL-17A (Biolegend, Inc., San Diego, CA) production in the culture supernatants were measured by ELISA, according to the manufacturer's instructions.
2.4. Principal component analysis (PCA)
To clarify and associate the potential effector mechanisms and pathology in TMEV infection, PCA was conducted on the histological and immunological data 2 months post infection (p.i.) from the two C3H mouse substrains, using a package ‘prcomp’ of R (http://www.R-project.org/), as described previously [13]. The proportion of variance and factor loading were also calculated.
2.5. Statistical analysis
Student t test was performed using OriginPro 8.1 (OriginLab Corporation, Northampton, MA) [9]. Results are mean ± standard error of the mean (SEM).
3. Results
3.1. Susceptibilities to myocarditis differ among mouse strains
After intracerebral TMEV injection, the susceptibilities to two TMEV-induced immune-mediated diseases in the CNS, TMEV-IDD and seizure/epilepsy, have been shown to be different among mouse strains [5], while the susceptibilities to TMEV-induced myocarditis remain unclear. In this study, we conducted comparative studies to determine the susceptibilities to myocarditis, using three mouse strains: SJL/J, B6, and C3H/HeNTac (wild-type) mice. We infected SJL/J, B6, and C3H mice intracerebrally with TMEV and compared the CNS and cardiac pathology during the chronic phase (2 months p.i.). As expected, SJL/J mice developed severe demyelination with meningitis and perivascular cuffing (inflammation) in the spinal cord, while no lesions were observed in the spinal cords of B6 mice (Figure 1a; Supplementary Table 1). Although all C3H mice developed demyelinating lesions in the spinal cord, the severity of TMEV-IDD was significantly less in C3H mice compared with SJL/J mice (mean demyelination scores ± SEM 2 months p.i.: SJL/J, 54.5 ± 4.1; C3H, 13.1 ± 3.9, P < 0.01, Student t test, Supplementary Figure 1). On the other hand, within 1 week p.i., during the acute phase of TMEV infection, 12 of 19 (63%) B6 mice had seizures, while no SJL/J mice (0 of 14 mice) developed seizures (Supplementary Table 1). TMEV-induced seizures were seen in 8% (2 of 24 mice) of C3H mice and the severity of seizures was lower in C3H mice than in B6 mice (mean maximum seizure grade ± SEM: B6, 5 ± 0; C3H, 3 ± 0).
Figure 1.
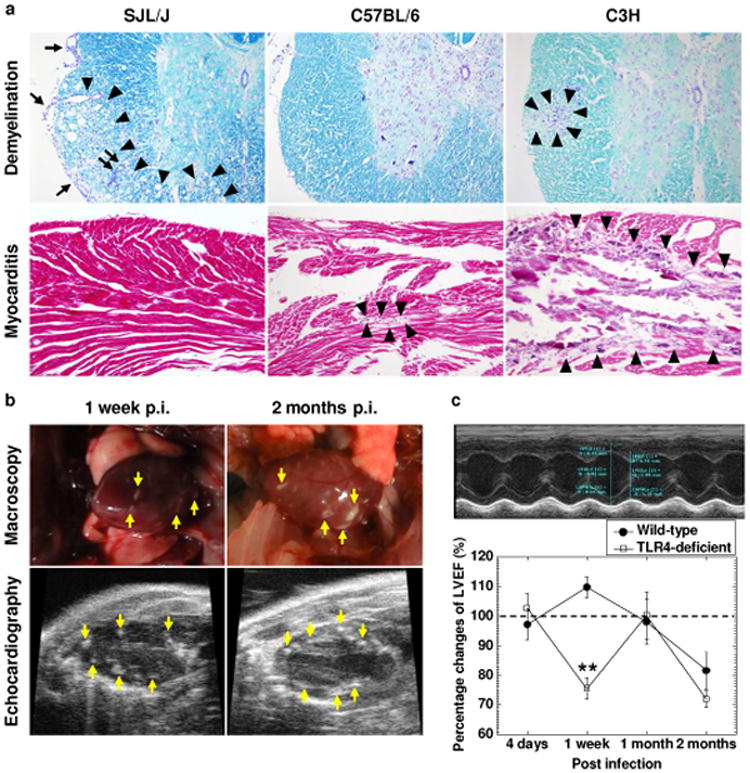
Contrasting spinal cord and cardiac pathology in the three inbred mouse strains following Theiler's murine encephalomyelitis virus (TMEV) infection. (a) Luxol fast blue staining of the spinal cord (upper panels). SJL/J mice had severe demyelinating lesions (arrowheads) with meningitis (arrows) and perivascular cuffing (paired arrows) in the spinal cord, while C57BL/6 mice did not develop TMEV-induced demyelinating disease (TMEV-IDD) and C3H mice had mild TMEV-IDD (arrowheads). Hematoxylin and eosin staining of the heart (lower panels). C3H mice developed severe myocarditis, including basophilic degeneration and calcification (arrowheads). C57BL/6 mice had only mild cardiac pathology (arrowheads), while no lesions were seen in SJL/J mice. SJL/J, C57BL/6, and C3H mice were infected with TMEV and killed 2 months post infection (p.i.). Magnification, ×46. The sections were representative of three to four independent experiments composed of 12 to 24 mice per mouse strain. (b) C3H mice had multiple macroscopic focal lesions (arrows, upper panel) in the heart 1 week and 2 months p.i. Using echocardiography, we also detected high intensity lesions (arrows, lower panels) in the left ventricle of TMEV-infected C3H mice. Results are representative of four experiments composed of five mice per time point. (c) Wild-type C3H/HeNTac mice had decreased left ventricular ejection fraction (LVEF) 2 months p.i., while TLR4-deficient C3H/HeJ mice showed a biphasic decrease in LVEF 1 week and 2 months p.i. TLR4-deficient mice had lower LVEF compared with wild-type mice 1 week p.i. (** P < 0.01, Student t test). LVEF was calculated by M-mode of transthoracic echocardiography (upper panel). The percentage changes of LVEF (LVEF of infected mice/mean LVEF of all age-matched uninfected control mice × 100) were compared between the two C3H mouse substrains at several time points (lower panel). Each time point was composed of five mice per mouse substrain.
During the course of the above experiments, we found that substantial numbers of infected C3H mice developed macroscopic lesions in the heart (Figure 1b). Microscopically, we found basophilic degeneration of cardiomyocytes and calcification in 19 of the 24 (79%) infected C3H mice (Figure 1a; Supplementary Table 1). The cardiac lesions were multifocal, but not diffuse; some focal lesions were transmural, but others were confined within the myocardium or extended to either the pericardium or endocardium. Then, we examined whether B6 and SJL/J mice also developed cardiac pathology. Five of the 19 (26%) infected B6 mice developed mild pathology in the heart, while no infected SJL/J mice had cardiac lesions. Since the route of viral infection may alter the incidence of myocarditis, we infected the three mouse strains with TMEV intraperitoneally. All C3H mice infected intraperitoneally with TMEV developed myocarditis, while the intraperitoneal inoculation did not alter the incidence of myocarditis in B6 and SJL/J mice [incidence: B6, 27% (4 of 15 mice); SJL/J, 0% (0 of 12 mice)]. Thus, in the following studies, we used C3H mice to characterize pathophysiological changes in the heart following TMEV infection.
3.2. Acute viral replication (4 days p.i.), subacute T cell infiltration (1-2 weeks p.i.), and chronic fibrosis (1-2 months p.i.) in TMEV-induced myocarditis
C3H mice are divided into two substrains, based on the presence or deficiency of TLR4. TLR4 has been reported to contribute to the pathogenesis of some picornavirus infections by enhancing viral entry and/or replication [14]. Using two C3H mouse substrains; C3H/HeNTac (wild-type) and C3H/HeJ (TLR4-deficient) mice, we conducted time course studies of TMEV-induced myocarditis. Using echocardiography, we longitudinally monitored the hearts of the two C3H mouse substrains and found high intensity lesions in the hearts of both C3H mouse substrains as early as 1 week p.i. (Figure 1b; Supplementary Video 1) but not 4 days p.i., while no lesions were seen in the hearts of age-matched uninfected control mice. Functionally, infected wild-type mice had a decreased cardiac function, LVEF, compared with age-matched uninfected control mice 2 months p.i.: LVEF of infected mice/mean LVEF of all age-matched uninfected control mice × 100 = 81.6 ± 6.3% (Figure 1c). Interestingly, TLR4-deficient mice showed lower LVEF not only 2 months p.i. but also 1 week p.i.
Using ELISA, we next biochemically quantified cardiomyocyte damage by measuring cardiac troponin I, a cardiac-specific protein, whose presence in sera indicates cardiomyocyte damage [15]. Cardiac troponin I in sera from infected mice was detectable 4 days p.i., peaked 1 week p.i., and became undetectable 2 weeks p.i. (Figure 2a). There were no statistical differences in the cardiac troponin I levels between the two C3H mouse substrains. The levels of cardiac troponin I in the sera and LVEF did not correlate at any time points (data not shown), suggesting that the cardiomyocyte damage during the acute phase was not severe enough to affect overall cardiac functions. On the other hand, decreased LVEF during the chronic phase could be explained by cardiac remodeling (see below Figure 3b), but not by ongoing active cardiomyocyte damage.
Figure 2.

Cardiomyocyte damage and viral replication in the hearts of TMEV-infected C3H mouse substrains. C3H/HeNTac (wild-type) and C3H/HeJ (TLR4-deficient) mice were infected with TMEV. (a) The concentrations of cardiac troponin I in sera peaked 1 week p.i. (* P < 0.05 compared with 4 days p.i., Student t test) and were comparable between the two C3H mouse substrains. Cardiac troponin I was quantified by enzyme-linked immunosorbent assays (ELISA). (b) Comparable high viral titers in the heart were detected in the two C3H mouse substrains 4 days p.i. by plaque assays. Infectious viral titers declined over the time course (* P < 0.05 and ** P < 0.01 compared with the preceding time point, Student t test) and were not detectable 1 months p.i. (c) The levels of viral RNA in the heart were highest 4 days p.i. in the two C3H mouse substrains, declined over the time course (* P < 0.05 and ** P < 0.01 compared with the preceding time point, Student t test), and were not detectable 2 months p.i. Viral genome was semi-quantified by real-time PCR of viral RNA using a pair of primers for a capsid protein VP2 of TMEV. Glyceraldehyde-3-phosphate dehydrogenases (Gapd) expression was used as a housekeeping gene for normalization. (d) The levels of cardiac troponin I in sera were statistically correlated with the levels of viral RNA in the hearts 4 days p.i. (◆, P < 0.01), but not 1 week p.i. (○). (a-c) Results are mean ± standard error of the mean (SEM). N.D., not detectable. Each group was composed of five to six mice per time pint.
Figure 3.
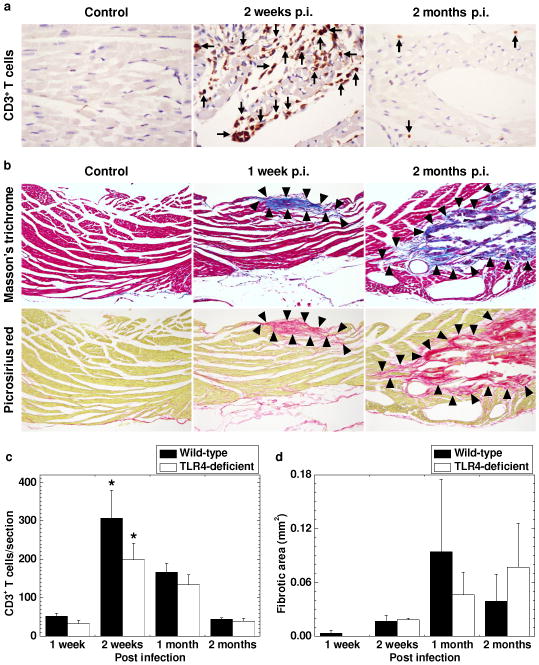
Time course studies of cardiac pathology in TMEV-infected C3H mice. C3H mice were infected with TMEV and killed 1 week, 2 weeks, 1 month, and 2 months p.i. (a) Immunohistochemistry against CD3 showed a larger number of CD3+ T cells (arrows) in the heart 2 weeks p.i. compared with 2 months p.i. No CD3+ T cells were seen in age-matched uninfected control mice. (b) Masson trichrome staining (upper panel) and picrosirius red staining (lower panel) visualized progressive cardiac fibrosis (arrowheads). (c) CD3+ T cell infiltration was detected 1 week p.i. and peaked 2 weeks p.i. (* P < 0.05 compared with the other time points, Student t test). There were no statistical differences in CD3+ T cell infiltration in the heart between wild-type and TLR4-deficeint mice. (d) Fibrotic areas were detected 1 week p.i., developed progressively, and peaked 1 and 2 months p.i. Using Image-Pro® Plus Version 6.3, the fibrotic areas were quantified by image analyses. (a, b) The heart was dissected into four transverse sections. Magnification, 185× (a) and 46× (b). The sections were representative of four independent experiments composed of 24 mice. (c, d) CD3+ T cells and fibrotic areas were quantified in the four sections per mouse. Results are mean ± SEM from six mice per time point.
At different time points following TMEV infection, we also titrated infectious virus and viral RNA in the heart by plaque assays (Figure 2b) and real-time PCR (Figure 2c), respectively. Infectious virus in the heart was detected 4 days p.i. and then the viral titers declined significantly by 2 weeks p.i. and were not detectable 1 month p.i. Similarly, high levels of viral RNA in the heart were detected 4 days p.i., while viral RNA levels decreased during the course of TMEV infection and became undetectable 2 months p.i. There were no differences in the levels of infectious virus or viral RNA between the two C3H mouse substrains. Since the time course kinetics of viral RNA and cardiac troponin I differed, we tested whether they correlated at each time point (Figure 2d). The levels of viral RNA in the hearts statistically correlated with the levels of cardiac troponin I in sera 4 days p.i. (r = 0.78, P < 0.01), but not 1 week p.i.
Cardiac pathology was examined for T cell infiltration (Figure 3a) with immunohistochemistry against CD3 (T-cell marker), and fibrosis (Figure 3b) by Masson trichrome staining (collagen) or picrosirius red staining (specific for collagen I and III). Few T cells were seen in the hearts of age-matched uninfected control mice (Figure 3a). T cell infiltration in the heart was detected 1 week p.i. and peaked 2 weeks p.i., while the number of T cells was declined 1 month p.i. and only a small number of T cells was sporadically detectable between muscle fibers 2 months p.i. (Figure 3c). The number of T cells was significantly higher at 2 weeks p.i. compared with the other time points in both C3H mouse substrains. Fibrosis was detectable as early as 1 week p.i., and the fibrotic areas increased 1 month and 2 months p.i. (Figure 3b and 3d). There were no statistical differences in T cell infiltration and fibrotic area in the heart between the two C3H mouse substrains during the 2 month observation period (Figure 3c and 3d). Since B cells and neutrophils can play a pathogenic role in myocarditis [16; 17], we quantified the number of B cells (Supplementary Figure 2a) and neutrophils (Supplementary Figure 2b) in the hearts of the two C3H mouse substrains with immunohistochemistry against B220 (B-cell marker) and hematoxylin and eosin staining. B cells and neutrophils were rarely seen in the hearts of either C3H mouse substrain at any time point.
3.3. TLR4 enhances production of pro-inflammatory cytokines in TMEV infection
We quantified TMEV-specific immune responses and pro-inflammatory cytokine production, both of which could play a protective and/or pathogenic role in viral infections. We found that both wild-type and TLR4-deficient C3H mouse substrains mounted substantial amounts of both TMEV-specific humoral and cellular immune responses as early as 1 week p.i., using ELISA and [3H]thymidine incorporation assays, respectively. The anti-TMEV antibody titers were higher in TLR4-deficient mice than in wild-type mice (2 weeks and 2 months p.i., P < 0.05, Figure 4a), while wild-type mice had higher levels of lymphoproliferative responses to TMEV, compared with TLR4-deficient mice, particularly 2 weeks p.i. (P < 0.05, Figure 4b). Since TLR4 has been shown to enhance IL-6 production, which regulates IL-17 production, in some viral infections [18; 19] as well as to enhance interferon (IFN)-γ production in an autoimmune model of myocarditis [20], we compared the amounts of the three cytokines between the two groups by ELISA. We detected substantial production of IL-6 (Figure 4c) and IL-17 (Figure 4d) from MNCs of both C3H mouse substrains. Wild-type mice had higher levels of IL-6 and IL-17 production than TLR4-deficient mice at all time points (IL-6, P < 0.05, at all time points; IL-17, 2 weeks and 1 month p.i., P < 0.05). There was no consistent trend in the production of IFN-γ between the two C3H mouse substrains (Supplementary Figure 3a).
Figure 4.
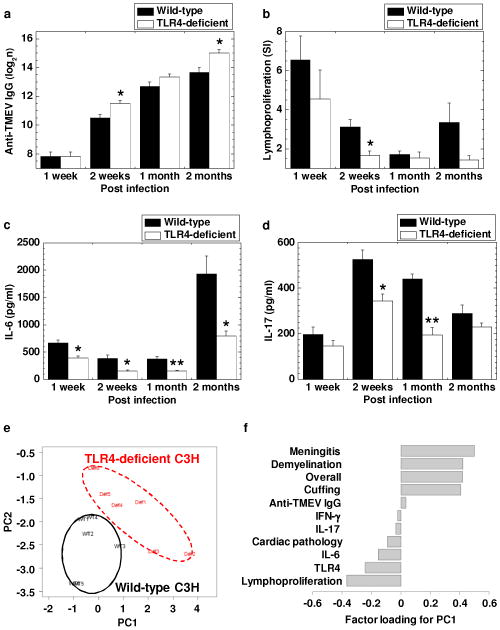
Immune responses and principal component analysis (PCA) between TMEV-infected C3H mouse substrains: C3H/HeNTac (wild-type) and C3H/HeJ (TLR4-deficient) mice. Overall, both C3H mouse substrains had substantial anti-viral antibody (a) and cellular immune responses (b), and pro-inflammatory cytokine production: interleukin (IL)-6 (c) and IL-17 (d). (a) Serum anti-TMEV IgG heavy and light chains antibody ELISA titers were higher in TLR4-deficient mice than in wild-type mice 2 weeks and 2 months p.i. (* P < 0.05, Student t test). (b) The levels of TMEV-specific lymphoproliferative responses were higher in wild-type mice than in TLR4-deficeint mice at all time points, particularly 2 weeks p.i. (* P < 0.05, Student t test). Mononuclear cells (MNCs) were isolated from the spleens and were stimulated with TMEV. TMEV-specific lymphoproliferative responses were quantified by [3H]thymidine incorporation assays and were expressed as stimulation indexes (SI, experimental cpm/control cpm). (c, d) Wild-type mice had significantly larger amounts of IL-6 (c) and IL-17 (d) production, compared with TLR4-deficient mice (* P < 0.05 and ** P < 0.01, Student t test). Splenic MNCs were stimulated with concanavalin A. The levels of IL-6 and IL-17 production in the culture supernatant were measured by ELISA. (e) PCA separated TMEV-infected wild-type (black circle) and TLR4-deficient (red dashed circle) mice as two distinct populations. PCA was conducted using 11 data ranked in (f): cardiac pathology, CNS pathology (meningitis, demyelination, cuffing, and overall, Supplementary Fig. 4d), TMEV-specific lymphoproliferation, anti-TMEV IgG, IL-6 and IL-17 levels, and the presence of TLR4 from the two C3H mouse substrains 2 months p.i. Each group was composed of six mice. (f) CNS pathology scores and anti-TMEV IgG antibody titers positively contributed to the principal component (PC) 1 value, while lymphoproliferative responses, TLR4 expression, pro-inflammatory cytokines, and cardiac pathology scores negatively contributed to the PC1 value. (a-d) Results are mean ± SEM from six mice per group per time point.
Lastly, we compared susceptibilities to TMEV-induced myocarditis, demyelination, and seizure/epilepsy between the two C3H mouse substrains. We found that the incidence of TMEV-induced myocarditis tended to be higher in wild-type mice (79%, 19 of 24 mice) than in TLR4-deficient mice (54%, 13 of 24 mice) during the 2 month observation period (P = 0.07, Supplementary Table 1). On the other hand, in the spinal cord, wild-type mice tended to have less severe chronic TMEV-IDD 2 weeks, 1 month, and 2 months p.i. compared with TLR4-deficient mice (Supplementary Figure 4a-d). In the brain, the two C3H mouse substrains showed similar brain pathology (Supplementary Figure 4e) and incidence of seizures (Supplementary Table 1) during the 2 month observation period. To further clarify and associate the potential effector mechanisms and pathology in TMEV infection, we conducted PCA using the above histological and immunological data 2 months p.i. from the two C3H mouse substrains. PCA clearly separated the two C3H mouse substrains as two distinct populations (Figure 4e). The proportions of variance of principal component (PC) 1 and PC2 were 60% and 18%, respectively. Factor loading for PC1 contributing to the separation of the two populations showed that TMEV-specific lymphoproliferation and TLR4 could be associated with the cardiac pathology positively, while these two factors may be associated with TMEV-IDD negatively (Figure 4f). The higher levels of TMEV-specific lymphoproliferation as well as IL-6 and IL-17 production were also associated with the higher incidence of myocarditis in wild-type mice. Thus, in TMEV infection, TLR4 may have detrimental effects in the heart by enhancing lymphoproliferative (cellular) responses, while TLR4 may have beneficial effects in the CNS.
4. Discussion
In CNS virus infections, neurovirulence (ability to cause disease in the CNS) is often not associated with levels of neurotropism (ability to infect neuronal cells in the CNS) or neuroinvasiveness (ability to gain access to the CNS tissue) [21]. During the acute phase of intracerebral TMEV infection, TMEV has been shown to infect and replicate in neurons in all mouse strains [4]. However, only B6 mice develop seizures with relatively high incidence, while other mouse strains are resistant or intermediately susceptible to seizures. Here, TMEV infection in the CNS is necessary but not sufficient to induce seizures; innate immunity has been proposed to play a key pathogenic role [22]. On the other hand, in TMEV-IDD, susceptible SJL/J mice develop demyelination, in which live virus persistence in glial cells and macrophages of the CNS is necessary, while demyelination has been associated with T cell infiltration rather than the amount of TMEV in the CNS [23]. Mouse strains resistant to TMEV-IDD, including B6 mice, can clear virus from the CNS during the acute phase of infection (there is no persistent virus infection). How about in viral myocarditis? Is the virulence associated with cardiac tropism or viral invasiveness to the heart? TMEV has been shown to induce viremia and infectious TMEV can be detected in the heart, even from SJL/J mice [24; 25]. Here, since TMEV can gain access to, and replicate in the heart in all mouse strains, the differences in innate and/or acquired immunities can be a key factor for subsequent viral clearance (and/or replication) and induction of inflammation in the heart.
TMEV is a natural pathogen of mice and has been shown to be able to infect the heart [1; 7]. Previously, Gómez et al. demonstrated that, after intraperitoneal TMEV injection, TMEV induced myocardial fibrosis without inflammation in severe combined immunodeficiency mice, suggesting that the immune system may not play an important role in TMEV-induced myocarditis, while no pathology data was shown in immunocompetent mice [8]. In this study, we found that, even after intracerebral TMEV injection, 79% of TMEV-infected C3H mice developed severe cardiac pathology, including macroscopic focal lesions, T cell infiltration, basophilic degeneration, and calcification, while B6 mice had only mild lesions and SJL/J mice had no lesions in the heart. We also found that intraperitoneal TMEV infection resulted in an increased incidence of myocarditis (100%) in C3H mice, but did not alter the incidence of myocarditis in B6 or SJL/J mice. The findings from the present study suggest that TMEV causes inflammation in the heart depending on the mouse strain; C3H mice are most susceptible.
In the time course studies of TMEV-induced myocarditis, we demonstrated that 1) viral RNA levels were correlated with cardiac troponin I in sera 4 days p.i. (phase I), 2) T cell infiltration was associated with decreased viral titers and high intensity lesions in echocardiograms 1-2 weeks p.i. (phase II), and 3) decreased LVEF with lower T cell infiltration, fibrosis, and calcification were observed 1-2 months p.i. (phase III). Furthermore, in phase III, there was progression of fibrotic lesions despite the decline in viral titers and T cell infiltration in the heart 1 month and 2 months p.i. These results suggest that cardiomyocyte damage could be caused by viral replication (viral pathology) in phase I, while T cell infiltration may not only suppress viral replication in the heart but also damage the heart (immunopathology) in phase II, leading to fibrosis and calcification in the heart in phase III. Our findings in TMEV infection are consistent with the pathophysiology that has been proposed in viral myocarditis in humans and other animal models, where myocarditis is initiated by viral entry/replication in the heart (phase I), followed by immunopathology caused by immune cell infiltration specific for the virus and/or cardiac antigens (phase II), and cardiac damage in phases I and II leads to cardiac remodeling and fibrosis (phase III) [26]. In phase III of human myocarditis in some cases, cardiac remodeling has been reported to develop progressively without viral persistence or ongoing cardiomyocyte damage [26; 27]. Cardiac remodeling can lead to decreased/depressed systolic function of the heart. The volume overload in the heart leads to further worsening of cardiac remodeling: enlargement of fibrotic lesions as well as cardiomyocyte hypertrophy. While there have been several existing myocarditis models, including coxsackievirus and reovirus infections and experimental autoimmune myocarditis [16], our TMEV-induced myocarditis model will be useful, since the pathogenesis of human myocarditis may be heterogeneous. For example, myocarditis can be caused by autoreactive CD4+ T cells, CD8+ T cells, autoantibodies, or the microbe itself. Some patients with myocarditis recover completely from acute myocarditis (phase I or II), while others develop chronic fibrosis in the heart (phase III) [16]. Experimentally, the reovirus model induces only phase I, while our TMEV model in C3H mice progresses to phase III. Thus, one model cannot represent all cases (which require differing models).
TLR4 has been shown to play a pathogenic role in some picornavirus infections. Poliovirus, which belongs to the family Picornaviridae, bound lipopolysaccharide (LPS) released from commensal microbiota in the gut. Poliovirus/LPS interactions were required for promoting viral replication in the gut, contributing to the pathogenesis, since mice treated with antibiotic for depleting microbiota had lower viral titers in the gut and a decreased mortality compared with untreated mice [14]. In infection with coxsackievirus B4, which also belongs to the family Picornaviridae, TLR4 signaling contributed to immunopathology by enhancing pro-inflammatory IL-6 production from pancreatic cells in vitro, although the mechanism of interaction between the virus and TLR4 remains unclear [18]. In an autoimmune model of myocarditis induced by injection of porcine cardiac myosin mixed with bacillus Calmette-Guérin (BCG) into two C3H mouse substrains, TLR4-deficient mice had mild cardiac pathology by decreasing pro-inflammatory IFN-γ production, while wild-type mice had large amounts of IFN-γ production, leading to severe inflammation in the heart [20]. In this study, PCA showed a positive association between TLR4 expression and cardiac pathology. Wild-type mice had higher IL-6 and IL-17 production compared with TLR4-deficient mice; this may be due to higher IL-6 production in wild-type mice, enhancing IL-17 production. TMEV might be recognized by TLR4 and activate TLR4 signaling, resulting in enhancement of pro-inflammatory cytokine production.
The roles of TLR4 in TMEV-induced pathology seemed to differ in the spinal cord depending on the time point. In the spinal cord, TLR4-deficient mice developed more severe chronic TMEV-IDD 2 weeks, 1 month, and 2 months p.i., while wild-type mice had more inflammation during the acute phase, 1 week p.i. Although the precise pathomechanisms of exacerbated chronic TMEV-IDD in TLR4-deficient mice are unclear, we found that the levels of anti-TMEV antibody responses correlated with the severity of demyelination in the spinal cord 2 months p.i. (r = 0.72, P < 0.01, Supplementary Figure 3b). Since anti-TMEV antibody has been shown to cross-react with myelin components, exacerbating demyelination [4], increased anti-TMEV antibody levels may contribute to exacerbated chronic TMEV-IDD in TLR4-deficient mice. The higher levels of anti-TMEV antibody responses in TLR4-deficeint mice may be due to Th2-biased immune responses that can help antibody production, since TLR4-deficeint mice have been shown to have Th2-biased immune responses compared with wild-type mice [20]. This is consistent with our findings that serum anti-TMEV IgG1 antibody titers, which is promoted by IL-4 (Th2 cytokine), were lower in wild-type mice (Supplementary Figure 3c), while serum anti-TMEV IgG2a antibody titers, which are regulated by IFN-γ (Th1 cytokine), were higher in wild-type mice (Supplementary Figure 3d) compared with TLR4-deficient mice. The roles of TLR4 in an autoimmune model of MS, experimental autoimmune encephalomyelitis (EAE), are controversial. Marta et al. reported that TLR4-deficient mice developed more severe EAE compared with wild-type mice [28], while Kerfoot et al. demonstrated that TLR4-deficient mice had less severe EAE or similar EAE, compared with wild-type mice [29]. Thus, further experiments are required for clarifying the roles of TLR4 in immune-mediated CNS diseases. The different effects of TLR4 on the spinal cord versus heart could be due to several factors, including the different expressions in endogenous TLR4 ligands, such as high-mobility group box 1 protein [30], since the levels of TLR4 expression have been reported to be similar between the spinal cord and heart [31].
In summary, we demonstrated different susceptibilities to myocarditis following TMEV infection among mouse strains, SJL/J, B6, and C3H mice. We found that the most susceptible strain for myocarditis was C3H mice. We further characterized the novel myocarditis model induced with TMEV, using wild-type and TLR4-deficient C3H mice, and found that TMEV-induced myocarditis was divided into three phases: acute viral, subacute immune, and chronic fibrotic phases. We also found decreased pro-inflammatory IL-6 and IL-17 production in TLR4-deficient C3H mice, which may contribute to a lower incidence of myocarditis. Since TMEV-induced myocarditis had three distinct phases and the susceptibilities differed depending on the genetic backgrounds of the host, TMEV infection can provide useful information as to how a natural pathogen (TMEV) can induce myocarditis in its natural host (mice).
Supplementary Material
Highlights.
We established a novel model of myocarditis induced with a cardiovirus
Myocarditis was divided into three phases: viral, immune, and fibrotic phases
Changes in B-mode and M-mode of echocardiography in viral myocarditis
Correlation between viral RNA in the heart and serum cardiac troponin levels
IL-6, IL-17, and TLR4 were associated with myocarditis susceptibility
Acknowledgments
This work was supported by the fellowships (F. Sato and S. Omura) from the Malcolm Feist Cardiovascular Research Endowment, Louisiana State University Health Sciences Center, Shreveport, and grants from the Department of Defence (PR100451, J.S. Alexander), the National Center for Research Resources of the National Institutes of Health (5P20RR018724, I. Tsunoda), and the National Institute of General Medical Sciences COBRE Grant (P30-GM110703, I. Tsunoda). We thank Drs. Noel G. Carlson, John W. Rose, Hong-Hua Mu, Masaaki Miyazawa, Fereidoon Shafiei, and Liam A. Morris for many helpful discussions and Sadie Faith Pearson, Lesya Ekshyyan, Elaine A. Cliburn, and Christi L. Eugene for excellent technical assistances.
Footnotes
Disclosure: The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hertzler S, Luo M, Lipton HL. Mutation of predicted virion pit residues alters binding of Theiler's murine encephalomyelitis virus to BHK-21 cells. J Virol. 2000;74:1994–2004. doi: 10.1128/jvi.74.4.1994-2004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martinez NE, Sato F, Kawai E, Omura S, Takahashi S, Yoh K, Tsunoda I. Th17-biased RORγt transgenic mice become susceptible to a viral model for multiple sclerosis. Brain Behav Immun. 2014 doi: 10.1016/j.bbi.2014.07.008. Epubahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez NE, Sato F, Omura S, Minagar A, Alexander JS, Tsunoda I. Immunopathological patterns from EAE and Theiler's virus infection: Is multiple sclerosis a homogenous 1-stage or heterogenous 2-stage disease? Pathophysiology. 2013;20:71–84. doi: 10.1016/j.pathophys.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sato F, Tanaka H, Hasanovic F, Tsunoda I. Theiler's virus infection: Pathophysiology of demyelination and neurodegeneration. Pathophysiology. 2011;18:31–41. doi: 10.1016/j.pathophys.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Libbey JE, Kirkman NJ, Smith MCP, Tanaka T, Wilcox KS, White HS, Fujinami RS. Seizures following picornavirus infection. Epilepsia. 2008;49:1066–74. doi: 10.1111/j.1528-1167.2008.01535.x. [DOI] [PubMed] [Google Scholar]
- 6.Cusick MF, Libbey JE, Patel DC, Doty DJ, Fujinami RS. Infiltrating macrophages are key to the development of seizures following virus infection. J Virol. 2013;87:1849–60. doi: 10.1128/JVI.02747-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mi W, Young CR, Storts RW, Steelman AJ, Meagher MW, Welsh CJR. Restraint stress facilitates systemic dissemination of Theiler's virus and alters its pathogenecity. Microb Pathog. 2006;41:133–43. doi: 10.1016/j.micpath.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Gómez RM, Rinehart JE, Wollmann R, Roos RP. Theiler's murine encephalomyelitis virus-induced cardiac and skeletal muscle disease. J Virol. 1996;70:8926–33. doi: 10.1128/jvi.70.12.8926-8933.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sato F, Martinez NE, Shahid M, Rose JW, Carlson NG, Tsunoda I. Resveratrol exacerbates both autoimmune and viral models of multiple sclerosis. Am J Pathol. 2013;183:1390–6. doi: 10.1016/j.ajpath.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsunoda I, Libbey JE, Fujinami RS. TGF-β1 suppresses T cell infiltration and VP2 puff B mutation enhances apoptosis in acute polioencephalitis induced by Theiler's virus. J Neuroimmunol. 2007;190:80–9. doi: 10.1016/j.jneuroim.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsunoda I, Libbey JE, Kuang LQ, Terry EJ, Fujinami RS. Massive apoptosis in lymphoid organs in animal models for primary and secondary progressive multiple sclerosis. Am J Pathol. 2005;167:1631–46. doi: 10.1016/S0002-9440(10)61247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez NE, Karlsson F, Sato F, Kawai E, Omura S, Minagar A, Grisham MB, Tsunoda I. Protective and Detrimental Roles for Regulatory T Cells in a Viral Model for Multiple Sclerosis. Brain Pathol. 2014;24:436–51. doi: 10.1111/bpa.12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Omura S, Kawai E, Sato F, Martinez NE, Chaitanya GV, Rollyson PA, Cvek U, Trutschl M, Alexander JS, Tsunoda I. Bioinformatics multivariate analysis determined a set of phase-specific biomarker candidates in a novel mouse model for viral myocarditis. Circ Cardiovasc Genet. 2014;7:444–54. doi: 10.1161/CIRCGENETICS.114.000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, Dermody TS, Pfeiffer JK. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;334:249–52. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan J, Yu M, Lin QW, Cao AL, Yu X, Dong JH, Wang JP, Zhang JH, Wang M, Guo HP, Cheng X, Liao YH. Th17 cells contribute to viral replication in coxsackievirus B3-induced acute viral myocarditis. J Immunol. 2010;185:4004–10. doi: 10.4049/jimmunol.1001718. [DOI] [PubMed] [Google Scholar]
- 16.Whitton JL, Feuer R. Myocarditis, microbes and autoimmunity. Autoimmunity. 2004;37:375–86. doi: 10.1080/08916930410001713089. [DOI] [PubMed] [Google Scholar]
- 17.Goland S, Luthringer D, Shirvani V, Trento A, Czer LSC. An unusual case of allograft neutrophilic myocarditis mimicking acute myocardial infarction in a post-heart transplant patient. J Heart Lung Transplant. 2009;28:843–6. doi: 10.1016/j.healun.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 18.Triantafilou K, Triantafilou M. Coxsackievirus B4-induced cytokine production in pancreatic cells is mediated through toll-like receptor 4. J Virol. 2004;78:11313–20. doi: 10.1128/JVI.78.20.11313-11320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 20.Nishikubo K, Imanaka-Yoshida K, Tamaki S, Hiroe M, Yoshida T, Adachi Y, Yasutomi Y. Th1-type immune responses by Toll-like receptor 4 signaling are required for the development of myocarditis in mice with BCG-induced myocarditis. J Autoimmun. 2007;29:146–53. doi: 10.1016/j.jaut.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Flint SJ, Enquist LW, Racaniello VR, Skalka AM. Infection of a susceptible host. In: Flint SJ, Enquist LW, Racaniello VR, Skalka AM, editors. Principals of virology. ASM Press; Washington, DC: 2009. pp. 2–27. [Google Scholar]
- 22.Kirkman NJ, Libbey JE, Wilcox KS, White HS, Fujinami RS. Innate but not adaptive immune responses contribute to behavioral seizures following viral infection. Epilepsia. 2010;51:454–64. doi: 10.1111/j.1528-1167.2009.02390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato F, Omura S, Martinez NE, Tsunoda I. Animal models of multiple sclerosis. In: Minagar A, editor. Neuroinflammation. Elsevier; London, UK: 2011. pp. 55–79. [Google Scholar]
- 24.Tsunoda I, McCright IJ, Kuang LQ, Zurbriggen A, Fujinami RS. Hydrocephalus in mice infected with a Theiler's murine encephalomyelitis virus variant. J Neuropathol Exp Neurol. 1997;56:1302–13. doi: 10.1097/00005072-199712000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Tsunoda I, Iwasaki Y, Terunuma H, Sako K, Ohara Y. A comparative study of acute and chronic diseases induced by two subgroups of Theiler's murine encephalomyelitis virus. Acta Neuropathol. 1996;91:595–602. doi: 10.1007/s004010050472. [DOI] [PubMed] [Google Scholar]
- 26.Liu PP, Mason JW. Advances in the understanding of myocarditis. Circulation. 2001;104:1076–82. doi: 10.1161/hc3401.095198. [DOI] [PubMed] [Google Scholar]
- 27.Smith SC, Ladenson JH, Mason JW, Jaffe AS. Elevations of cardiac troponin I associated with myocarditis. Experimental and clinical correlates. Circulation. 1997;95:163–8. [PubMed] [Google Scholar]
- 28.Marta M, Andersson Å, Isaksson M, Kämpe O, Lobell A. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur J Immunol. 2008;38:565–75. doi: 10.1002/eji.200737187. [DOI] [PubMed] [Google Scholar]
- 29.Kerfoot SM, Long EM, Hickey MJ, Andonegui G, Lapointe BM, Zanardo RCO, Bonder C, James WG, Robbins SM, Kubes P. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J Immunol. 2004;173:7070–7. doi: 10.4049/jimmunol.173.11.7070. [DOI] [PubMed] [Google Scholar]
- 30.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–5. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol Pharm Bull. 2005;28:886–92. doi: 10.1248/bpb.28.886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.