

Abstract
Background
Cleft palate occurs in up to 1:1000 live births and is associated with mutations in multiple genes. Palatogenesis involves a complex choreography of palatal shelf elongation, elevation, and fusion. Transforming growth factor β (TGFβ) and bone morphogenetic protein 2 (BMP2) canonical signaling is required during each stage of palate development. The type III TGFβ receptor (TGFβR3) binds all three TGFβ ligands and BMP2, but its contribution to palatogenesis is unknown.
Results
The role of TGFβR3 during palate formation was found to be during palatal shelf elongation and elevation. Tgfbr3-/- embryos displayed reduced palatal shelf width and height, changes in proliferation and apoptosis, and reduced vascular and osteoblast differentiation. Abnormal vascular plexus organization as well as aberrant expression of arterial (Notch1, Alk1), venous (EphB4), and lymphatic (Lyve1) markers was also observed. Decreased osteoblast differentiation factors (Runx2, alk phos, osteocalcin, col1A1, and col1A2) demonstrated poor mesenchymal cell commitment to the osteoblast lineage within the maxilla and palatal shelves in Tgfbr3-/- embryos. Additionally, in vitro bone mineralization induced by osteogenic medium (OM+BMP2) was insufficient in Tgfbr3-/- palatal mesenchyme, but mineralization was rescued by overexpression of TGFβR3.
Conclusions
These data reveal a critical, previously unrecognized role for TGFβR3 in vascular and osteoblast development during palatogenesis.
Keywords: cleft palate, osteogenesis, vascularization, palatogenesis, TGFβ, BMP
2.1 Introduction
The palatal shelves appear around E11.5 in the mouse (week 6 in humans) and grow vertically along the lateral sides of the tongue. As the jaw continues to grow, the tongue descends, allowing space for the palatal shelves to elongate (E13.5) and elevate (E14.5) into a horizontal position. This is followed by adhesion and dissolution of the medial edge epithelial (MEE) seam at E15.5 giving rise to the single confluent palatal structure (Ferguson, 1988; Xu et al., 2006). All of these steps are dependent on coordinated proliferation, extracellular matrix production, and apoptosis. The palate is comprised predominantly of mesenchymal cells of neural crest origin and endothelial cells of mesodermal origin which are covered by epithelium of ectodermal origin (Yoshida et al., 2008). Neural crest migration into the frontonasal and maxillary prominences brings the lip and palatal processes into apposition to allow lip and palate fusion to occur (Murray and Schutte, 2004). Once the palatal shelves are fused, the anterior portion undergoes intramembranous ossification to form the hard palate and the posterior palate is invested by skeletal muscle to form the soft palate. Cleft palate occurs when: 1) palatal shelf elevation is impeded 2) palatal shelf elongation stops and/or 3) epithelial dissolution of the apposed palatal shelves does not occur (Chai and Maxson, 2006).
Transforming growth factor beta (TGFβ) and bone morphogenetic protein (BMP) signaling is involved in cell growth and differentiation during embryonic development and has been shown to be essential for palate development. The type I (TGFβR1 or ALK 5) and the type II (TGFβR2) receptors bind TGFβ ligands and function as serine-threonine kinases that phosphorylate SMADs 2/3, intracellular proteins that conduct TGFβ signaling by inducing transcription of downstream gene targets. BMPs also signal in a similar complex of type I (BMPR1 or ALK2/3) and type II (BMPR2) receptors that phosphorylate SMADs 1/5/8. TGFβR3 binds all 3 TGFβ ligands as well as BMP2, and is specifically required for high affinity binding of TGFβ2 to its receptor (Lopez-Casillas et al., 1991). TGFβR3 has no known enzymatic activity and can act indirectly by presenting ligand to the receptor complex to augment TGFβ or BMP canonical signaling. However, TGFβR3 is required for the activation of other pathways via G Interacting Protein C (GIPC), β-arrestin2, and Par6/Smurf/RhoA effectors (Hill et al., 2012; Sanchez and Barnett, 2012; Sanchez et al., 2011; You et al., 2009).
Although, previous studies have demonstrated that alterations in the TGFβ pathway during palate formation lead to cleft palate (Baek et al., 2011; Cui et al., 2003; Doetschman et al., 2012; Dudas et al., 2006; Dudas et al., 2004a; Ito et al., 2003; Kaartinen et al., 1995; Levi et al., 2006; Li et al., 2013; Loeys et al., 2005; Proetzel et al., 1995; Sanford et al., 1997; Shiomi et al., 2006; Taya et al., 1999; Van Laer et al., 2014; Xu et al., 2006), none have revealed the function of TGFβR3 during palate formation. Previous studies showed that TGFβR3 expression during palatogenesis occurs throughout the epithelium and is specifically localized to the medial edge epithelium (MEE) during palatal shelf fusion in mice (Cui and Shuler, 2000). Knockdown of Tgfbr3 with siRNA in a palatal shelf culture model inhibited in vitro palatal shelf fusion due to persistence of the palatal epithelium (Nakajima et al., 2007). Recent studies demonstrated a partial rescue of the cleft palate phenotype in Wnt1-Cre;Tgfbr2F/F;Tgfbr3+/- mice suggesting that TGFβR3 plays a pivotal role in maintaining homeostasis of TGFβ signaling in the palate (Iwata et al., 2012).
Here we have identified the TGFβR3-dependent processes during palate development. Cleft palate occurred at E14.5 in Tgfbr3-/- mice concomitant with alterations in proliferation and apoptosis, aberrant vascular remodeling and specification, as well as reduced mesenchymal cell commitment to osteoblast fate. In vitro cultures of mouse embryonic palatal mesenchymal (MEPM) cells in osteogenic medium (OM+BMP2) revealed that TGFβR3 was necessary and sufficient to induce mineralization and transcription of key genes expressed early during pre-osteoblast differentiation and later throughout osteoblast development. Furthermore, the loss of TGFβR3 resulted in atypical expression of several components of both TGFβ and BMP signaling pathways within the palatal shelves in vivo. Taken together, our results demonstrated that global deletion of TGFβR3 perturbed the balance of TGFβ/BMP signaling and interrupted normal palatogenesis.
3.1 Results
3.1.1 Arrested palatal shelf elevation and elongation associated with the loss of TGFβR3
At E13.5, Tgfbr3-/- palatal shelves were shorter in width than those of the Tgfbr3+/+ mice (Fig. 1 A, B). At E14.5, the palatal shelves of Tgfbr3+/+ mice had elevated and were in apposition; however, the Tgfbr3-/- palatal shelves were not elevated and remained adjacent to the tongue (Fig. 1 C, D). Measurements of the palatal shelves at E14.5 demonstrated that the Tgfbr3-/- anterior and posterior palatal shelf heights were significantly reduced (Fig. 1 E). Tgfbr3-/- palatal shelf width was decreased anteriorly, but unaffected posteriorly (Fig. 1 F). Since abnormal tongue size may obstruct palatal shelf elevation, tongue width and height were also measured (Fig. 1 G, H). Tgfbr3-/- tongues showed no differences in height compared to Tgfbr3+/+ (Fig. 1 G); however, their widths were significantly decreased (Fig. 1 H). Roller bottle cultures served as an in situ model of palatal shelf elevation and fusion in order to overcome the problem of embryonic lethality by E15.5 (Fig. 1 I, J). Tgfbr3-/- cultures did not elevate their palatal shelves (Fig. 1 J) while the Tgfbr3+/+ cultures elevated and fused (Fig. 1 I). In vitro palatal shelf cultures demonstrated that apposed Tgfbr3+/+ palatal shelves were completely fused after 72 h in culture (Fig. 1 K); however, apposed Tgfbr3-/- palatal shelves could not complete fusion by dissolution of the MEE (Fig. 1 L). Additionally, loss of palatal elevation was not accompanied by altered hyaluronic acid (HA) production (data not shown). These data demonstrated that TGFβR3 was essential for normal palate development and global loss of this receptor resulted in failed palatal shelf elongation, elevation, and fusion.
Figure 1. Global deletion of TGFβR3 disrupted palate shelf elongation, elevation, and fusion.
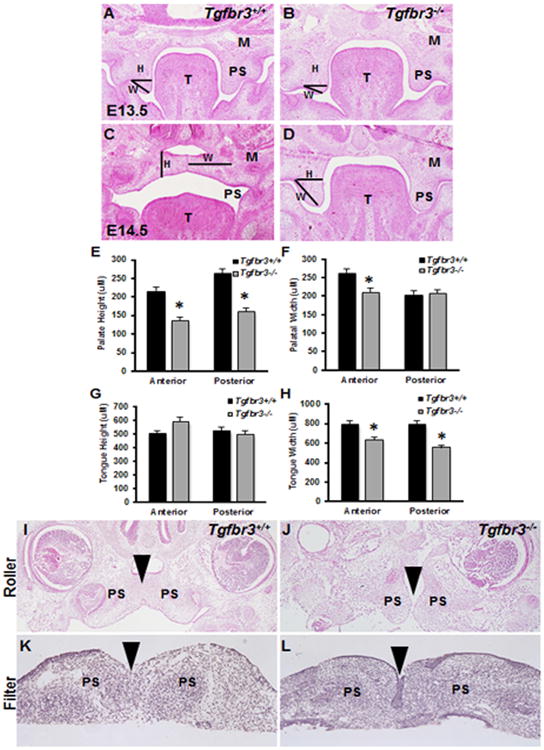
H&E staining of transverse sections through the developing palate of Tgfbr3+/+ (A, C) and Tgfbr3-/- (B, D) mice at E13.5 (A, B) and E14.5 (C, D) revealed disrupted palate shelf elongation and elevation coincident with the loss of TGFβR3. Palate height was reduced in Tgfbr3-/- mice, as determined by measuring line H in both anterior and posterior regions of the palate (E). Palate width was calculated from line W and was decreased anteriorly, but not posteriorly (F). Tongue height (G) was unaffected, but width (H) was concluded to be decreased in Tgfbr3-/- mice. Roller bottle cultures were established as an in situ model of palate shelf elevation, and the results confirmed that Tgfbr3-/- palate shelves cannot elevate like Tgfbr3+/+ controls (I, J). In vitro palatal shelf cultures were used to determine if fusion of palatal shelves would occur when in apposition. Tgfbr3+/+ palatal shelves were completely fused after 72h, whereas Tgfbr3-/- shelves were not (K, L). Analyses were performed on 3 individual littermate pairs n=3; Columns, mean fold change obtained from 3 separate experiments; bars, SEM; *=p<0.05. M=maxilla, PS=palate shelf, P=palate, T=tongue, H=height, W=width.
3.1.2 Altered proliferation and apoptosis within Tgfbr3-/- palatal shelves
At E13.5, there were no significant differences in the percent of proliferating cells within the anterior or posterior palatal shelves of Tgfbr3+/+ and Tgfbr3-/- embryos (Fig. 2 A-D, I). However, at this same developmental time point, Tgfbr3-/- embryos demonstrated increased cell death within their palatal shelves both anteriorly and posteriorly when compared to Tgfbr3+/+ embryos (Fig. 3 A-D, I). At E14.5, there were notably less proliferating cells within the palatal shelves of Tgfbr3-/- embryos when compared to Tgfbr3+/+ embryos both anteriorly and posteriorly (Fig. 2 E-H, J), but the percentage of cells undergoing programmed cell death was not statistically different between the palatal shelves of Tgfbr3+/+ and Tgfbr3-/- embryos (Fig. 3 E-H, J). These results suggested that changes in proliferation and apoptosis are associated with impeded palatogenesis in Tgfbr3-/- embryos.
Figure 2. Altered cell proliferation at E14.5 within Tgfbr3-/- palatal shelves.
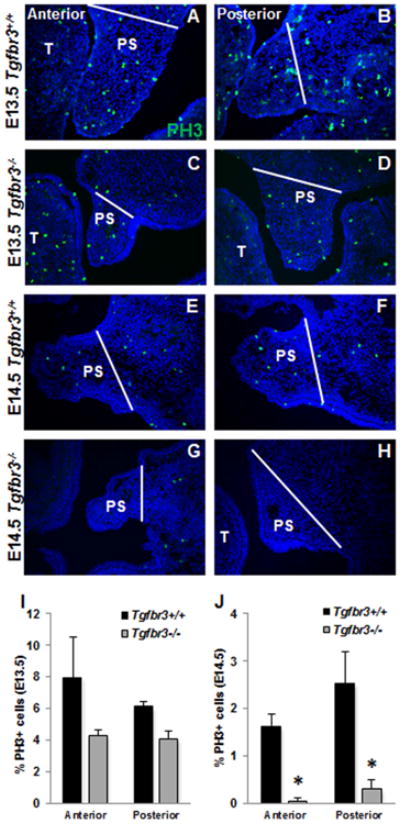
Immunohistochemistry to reveal PH3+ proliferative cells (green) within the palatal shelves did not show any notable differences between Tgfbr3+/+ and Tgfbr3-/- littermates at E13.5 (A-D, I), but demonstrated statistically significant decreases in the percentage of proliferating cells in Tgfbr3-/- embryos compared to Tgfbr3+/+ littermates at E14.5 (E-H, J). Analyses were performed on 3 individual littermate pairs n=3; Columns, mean fold change obtained from 3 separate experiments; bars, SEM; *=p<0.05. PS=palate shelf, T=tongue. White bars indicate lateral extent of palate shelf.
Figure 3. Tgfbr3-/- palatal shelves have increased cell apoptosis at E13.5.
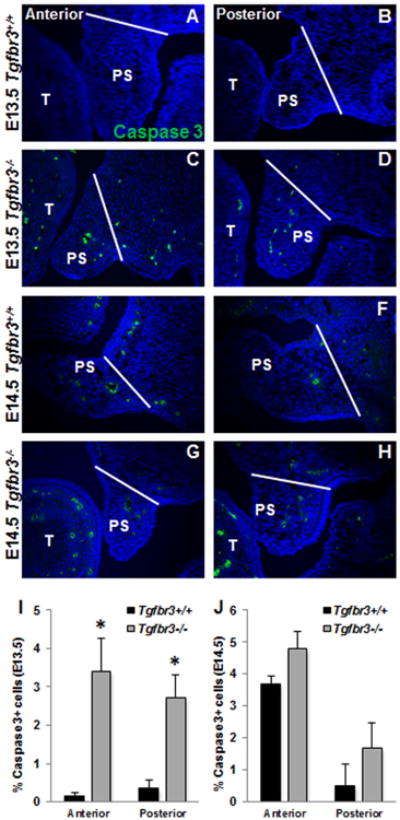
Immunohistochemistry to reveal apoptotic cells by cleaved caspase 3 localization (green) within the palatal shelves showed increased apoptosis in Tgfbr3-/- embryos compared to Tgfbr3+/+ littermates at E13.5 (A-D, I), but demonstrated similar levels of apoptotic cells in between Tgfbr3+/+ and Tgfbr3-/- littermates at E14.5 (E-H, J). ). Analyses were performed on 3 individual littermate pairs n=3; Columns, mean fold change obtained from 3 separate experiments; bars, SEM; *=p<0.05. PS=palate shelf, T=tongue. White bars indicate lateral extent of palate shelf.
3.1.3 Aberrant vascular development in Tgfbr3-/- palatal shelves
There is an obligate requirement of TGFβR3 for coronary vessel development (Compton et al., 2007); therefore, we investigated the necessity of TGFβR3 during palate vascularization. At E13.5, vasculogenesis had begun in Tgfbr3+/+ palatal shelves as shown by the organization of PECAM+ primary vascular channels (Fig. 4 A, B, white arrowheads), but Tgfbr3-/- palatal shelves had less PECAM localization and only rudimentary vascular formation (Fig. 4 C, D, white arrowheads). At E14.5, Tgfbr3+/+ palatal shelves exhibited robust PECAM localization within a well-formed vascular plexus (Fig. 4 E, F, white arrowheads). Yet, Tgfbr3-/- palatal shelves had notably less PECAM+ vessels demonstrating abnormal vascular development (Fig. 4 G, H, white arrowheads). Reduced Pecam expression was confirmed by qPCR analyses of mRNA collected from Tgfbr3-/- palates when compared to Tgfbr3+/+ mice at E13.5 and E14.5 (Fig. 4 I, J). No significant differences in SmαA expression were determined between Tgfbr3+/+ and Tgfbr3-/-(Fig. 4 I, J). Impaired vascular development implicated a role for TGFβR3 during palate vasculogenesis.
Figure 4. Irregular vascularization of Tgfbr3-/- palatal shelves.
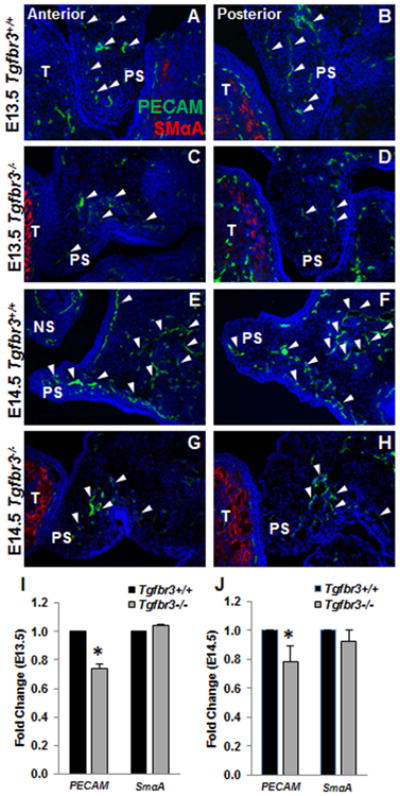
Immunohistochemistry to demonstrate the localization of PECAM (endothelial, white arrowheads) and SmαA (smooth muscle) protein in the vessels revealed notable vascular deficiencies in the Tgfbr3-/- palates (C, D, G, H) compared to Tgfbr3+/+ littermates (A, B, E, F) at both E13.5 (A-D) and E14.5 (E-H). n=3 for each time point. Analysis of gene expression by qPCR confirmed down-regulated expression of PECAM, but not SmαA, in Tgfbr3-/- mRNA relative to Tgfbr3+/+ littermates at both E13.5 (I) and E14.5 (J). Columns, mean fold change obtained from 3 separate experiments; bars, SEM; *=p<0.05 NS=nasal septum, PS=palate shelf, T=tongue.
3.1.4 Abnormal arterial and venous specification in Tgfbr3-/- palatal shelf vasculature
Based on the vascular alterations revealed in the developing palatal shelves, we examined vascular remodeling and specification in Tgfbr3+/+ and Tgfbr3-/- embryos. Evaluation of E13.5 palatal shelves showed minimal differences in LYVE1 (lymphatic, white arrowheads) and Notch1 (arterial, yellow arrowheads) localization during this developmental time point concurrent with the onset of vasculogenesis in the palatal shelves (Fig. 5 A-D); however, Lyve1 expression was significantly decreased in Tgfbr3-/- palatal mRNA (Fig. 5 I). Analyses at E14.5 demonstrated increased protein localization (Fig. 5 E-H) and mRNA expression (Fig. 5 J) for both LYVE1 and Notch1 in Tgfbr3-/- palatal shelves compared to Tgfbr3+/+ littermates. Furthermore, expression of EphB4 (venous) was significantly reduced while Alk1 (arterial) was upregulated in Tgfbr3-/- palatal mRNA (Fig. 5 J). The mis-expression of these critical regulators of vascular specification indicated vessel remodeling was at least in part, affected by the loss of TGFβR3.
Figure 5. Tgfbr3-/- palatal vasculature showed abnormal vascular specification of arteries, veins, and lymphatics.
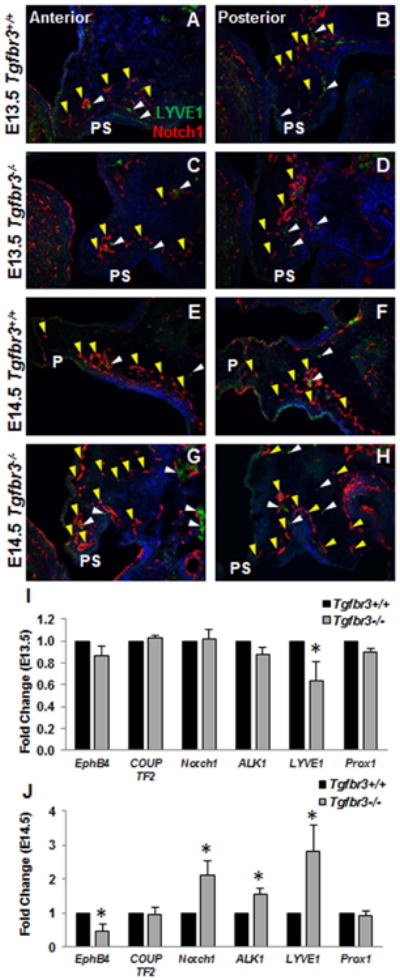
Immunohistochemistry for LYVE1 (lymphatic specific, white arrowheads) and Notch1 (arterial specific, yellow arrowheads) exposed differences in the remodeling phase of vascular development. At E13.5, no substantial changes in the localization of LYVE1 or Notch1 were seen between Tgfbr3+/+ or Tgfbr3-/- palate vessels (A-D). Notable increases in Notch1 and LYVE1 proteins were apparent in Tgfbr3-/- palatal vasculature compared to Tgfbr3+/+ littermates at E14.5 (E-H). n=3 for each time point. Gene expression changes were calculated by qPCR, and confirmed aberrant expression of LYVE1 at E13.5 (I) as well as altered EphB4 (venous), Notch1, Alk1 (arterial), and LYVE1 at E14.5 (J). Columns, mean fold change obtained from 3 separate experiments; bars, SEM; *=p<0.05 P=palate, PS=palate shelf.
3.1.5 Reduced ossification and expression of osteoblast gene determinants in Tgfbr3-/- palatal and maxillary mesenchyme
RUNX2, the critical protein supporting pre-osteoblast differentiation, localization is minimal at E13.5; yet, appeared reduced in the maxillas of Tgfbr3-/- embryos when compared to Tgfbr3+/+ littermates (Fig. 6 A-D). At E14.5 in Tgfbr3+/+ mice, RUNX2 is localized to the mesenchymal condensations that begin to form within the maxilla as the palatal shelves are fusing (Fig. 6 E, F). Tgfbr3-/- embryos have less RUNX2 staining in the maxilla when compared to Tgfbr3+/+ mice at E14.5 (Fig. 6 G, H). Additionally, decreased expression of the fundamental genes required for osteoblast development (Runx2, alk phos, osteocalcin, col1A1, col1A2) was seen in Tgfbr3-/- palatal and maxillary mRNA when compared to Tgfbr3+/+ mice at both E13.5 and E14.5 (Fig. 6 I, J). These data indicated that Tgfbr3-/- palatal and maxillary mesenchymal cells, which undergo intramembranous ossification to form the hard palate, have impaired osteoblast differentiation during palatogenesis. It remains unknown to what degree the initiation of maxillary ossification plays a role in palate formation.
Figure 6. Poor initiation of osteoblast fate in the palatal mesenchyme of Tgfbr3-/- mice.
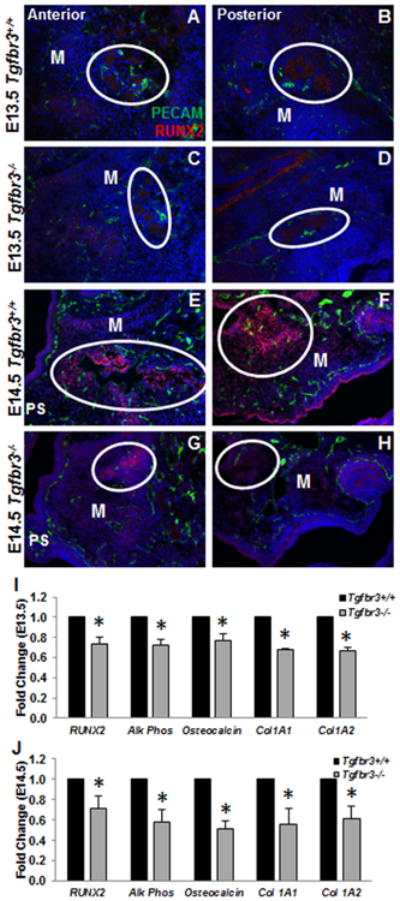
Immunohistochemistry performed on Tgfbr3+/+ or Tgfbr3-/- palatal sections showed localization of RUNX2 (pre-osteoblast) protein within the mesenchymal condensations that will initiate ossification of the palate and maxilla. The density of RUNX2 staining (circles) was reduced in Tgfbr3-/- mice maxillary prominences at both E13.5 (C, D) and E14.5 (G, H) relative to Tgfbr3+/+ littermates (A, B, E, F). n=3 for each time point. Analyses of critical osteoblast differentiation genes by qPCR at E13.5 and E14.5 demonstrated reduced expression of genes expressed from pre-osteoblast commitment throughout the establishment of bone (I, J). Columns, median fold change obtained from 3 separate experiments; bars, SEM; *=p<0.05. M=maxilla
Tgfbr3-/- mouse embryonic palatal mesenchymal (MEPM) cells cultured in OM+BMP2 showed significant decreases in mineralization (Fig. 7 A-E) and expression of essential osteogenic genes (Runx2, alk phos, and osteocalcin) (Fig. 7 F-H) when compared to Tgfbr3+/+ MEPM cells, similar to what was revealed in vivo (Fig 6 I, J). Overexpression of TGFβR3-FL (full length) prior to differentiation in Tgfbr3-/- MEPM cells rescued mineralization deficits (Fig. 8 A-C) and induced expression of Runx2, alk phos, and osteocalcin (Fig. 8 D-F) substantially greater than cells overexpressing GFP only. These data supported a critical role of TGFβR3 for osteoblast development during palate and maxillary ossification, and suggested that TGFβR3 supports MEPM cells ability to respond to OM+BMP2-induced differentiation in vitro.
Figure 7. Tgfbr3-/- MEPM cells failed to mineralize or induce osteoblast differentiation genes in vitro.
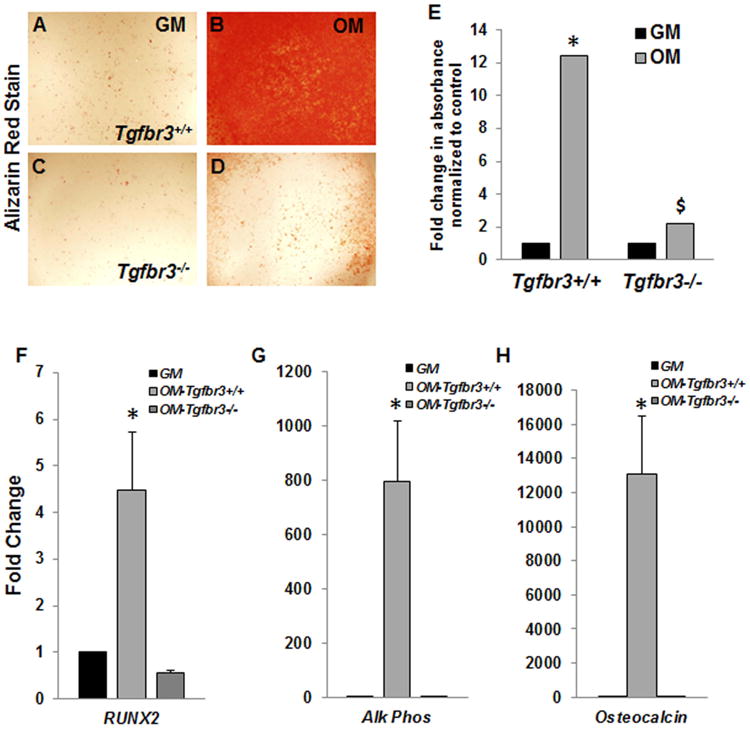
Tgfbr3+/+ and Tgfbr3-/- MEPM cells cultured in growth media (GM) or osteogenic media (OM+BMP2) were assayed for mineralization by incorporation of alizarin red (A-D). The alizarin red stain was solubilized and quantified (E), n=3; Columns, mean fold change obtained from 3 separate experiments; bars, SEM; *=p<0.05. qPCR revealed the induction of osteoblast differentiation markers in mRNA extracted from the cells following the differentiation time course (F-H). Differentiated Tgfbr3++/ cells (OM- Tgfbr3+/+) were compared to Tgfbr3+/+ cells incubated in GM, and differentiated Tgfbr3-/- (OM-Tgfbr3-/-) cells were analyzed relative to Tgfbr3-/- cells in GM. Tgfbr3+/+ cells induced robust expression of RUNX2, alk phos, and osteocalcin, which was in contrast to the Tgfbr3-/- cells (F-H). Columns, median fold change obtained from 3 separate experiments; bars, SEM; *=p<0.05 (relative to GM), $=p<0.05 (relative to Tgfbr3+/+ OM).
Figure 8. TGFβR3-FL overexpression rescues MEPM cell mineralization and differentiation in vitro.
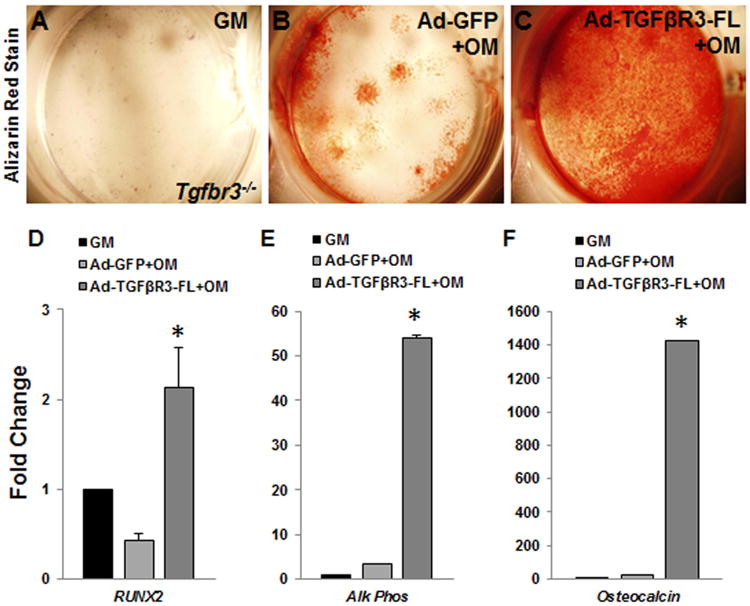
TGFβR3-FL adenovirus allowed the reintroduction of TGFβR3 signaling in Tgfbr3-/- MEPM cells. Cells were cultured in either growth media (GM) with no infection, OM+BMP2 following Ad-GFP infection, or OM+BMP2 subsequent to Ad-TGFβR3-FL infection. The alizarin red incorporation revealed that the cells overexpressing TGFβR3 were able to mineralize (A-C). Substantial induction of RUNX2, alk phos, and osteocalcin was determined from qPCR analysis in mRNA extracted from the TGFβR3 expressing cells. Both Ad-GFP-OM and Ad- TGFβR3-FL-OM cells were compared to Tgfbr3-/- incubated in GM. Columns, mean fold change obtained from 3 separate experiments; bars, SEM; *=p<0.05.
3.1.6 Mis-expression of TGFβ pathway members in Tgfbr3-/- palates
Reduced mRNA levels of TGFβ and BMP ligands and their receptors were seen in Tgfbr3-/- palatal shelves at both E13.5 and E14.5 (Fig. 9 A, B). Tgfb2 and Tgfb3 and the components of the canonical receptor complex (Alk5 and Tgfbr2) were appreciably down-regulated in Tgfbr3-/- palatal mRNA compared to Tgfbr3+/+ (Fig. 9 A). Bmp2 and Bmp4 and the BMP type 1 receptors (Alk2 and Alk3) were also notably decreased (Fig. 9 B). These results demonstrated that deletion of TGFβR3 down-regulated multiple genes required for TGFβ/BMP signaling and responsiveness and implied that TGFβR3 mediates the balance of these critical pathways.
Figure 9. Inadequate expression of TGFβ/BMP ligands and receptors in Tgfbr3-/- palates.
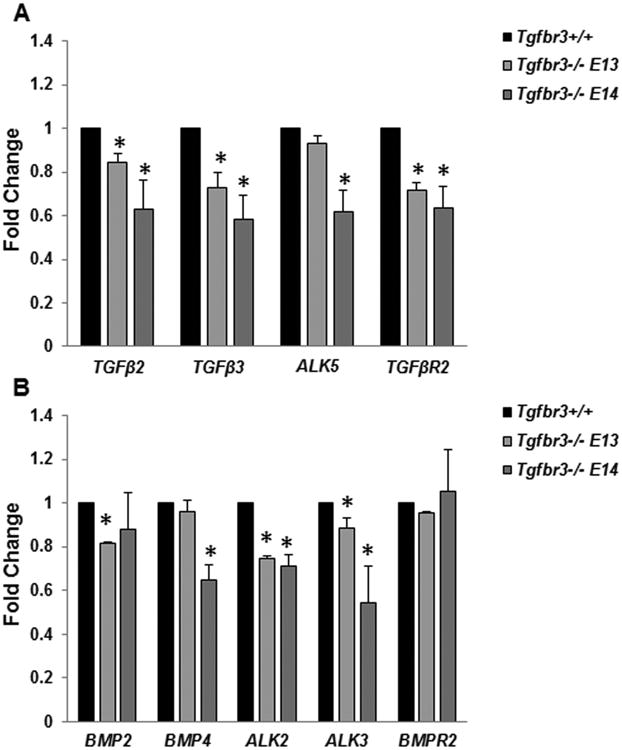
Gene expression differences in TGFβ/BMP pathway members between Tgfbr3+/+ or Tgfbr3-/- mRNA collected from the whole palate at E13.5 and E14.5 were calculated by qPCR. Significant reductions in ligands and receptors required for signaling and reception of both pathways were determined at both developmental time points. Columns, mean fold change obtained from 3 separate experiments; bars, SEM; *=p<0.05.
4.1 Discussion
The loss of TGFβR3 impedes palatal shelf elongation and elevation leading to cleft palate in Tgfbr3-/- mice at E14.5. Additionally, in vitro cultures revealed that apposed palatal shelves could not complete fusion without TGFβR3, similar to what was demonstrated with siRNA constructs targeting Tgfbr3 in palatal shelf cultures (Nakajima et al., 2007). Abnormal palate development was characterized by altered proliferation and apoptosis within the palatal shelves, histologic and gene expression findings of aberrant vascular formation, deficient osteoblast commitment both in vivo and in vitro, and reduced expression of Tgfb/Bmp genes. These results suggested that impaired signaling, which is absolutely critical for palate development, halted palate growth. Additionally, these findings revealed an essential role for TGFβR3 signaling during palatal vascular and bone development which may, in part, support palatal shelf elongation and elevation.
4.1.1 TGFβ/BMP signaling is indispensable for normal palatogenesis
Members of the TGFβ superfamily of signaling molecules regulate multiple cellular processes during craniofacial development. Interruption of the TGFβ/BMP signaling pathway lead to a wide variety of craniofacial malformations that range from cleft palate to severe facial deformities (Iwata et al., 2011). The loss of TGFβ3 resulted in cleft palate formation due to failure of medial edge epithelial (MEE) seam adhesion and subsequent degradation due to the loss of programmed cell death (Cui et al., 2003; Taya et al., 1999) and epithelial-mesenchymal transformation (Kaartinen et al., 1997). TGFβR1 (ALK5) and TGFβR2 are also required for normal palate development and conditional deletion in neural crest cells (Wnt1-cre) and the epithelium (K14- cre) led to cleft palate (Dudas et al., 2006; Ito et al., 2003; Xu et al., 2006). However, in each of these models, palatal shelf elongation and elevation still occurred. Altered BMPR1a (ALK3) signaling during palatogenesis by either conditional deletion in craniofacial primordia (Nestin-cre) (Liu et al., 2005) , over-expression in CNC cells (Li et al., 2013), or dominant-negative expression in CNC-derived mesenchyme (Mpz-cre) (Saito et al., 2012) resulted in cleft palate formation associated with altered mesenchymal cell proliferation, elevation and elongation deficits that did not support fusion of the palate shelves, in addition to reduced osteoblast differentiation. Cleft palate also occurred in mice with conditional deletion of Alk2 in neural crest cells (Wnt1-cre) due to delayed palate shelf elevation, although secondary to mandibular hypoplasia (Dudas et al., 2004b). While TGFβ/BMP signals are well-described as being required for appropriate palate formation, Tgfbr3-/- mice fail to complete palatogenesis before death by E15.5 with phenotypic features distinct from other models. Although cleft palate is common due to altered TGFβ/BMP signaling, this study identified a unique, previously unknown role of TGFβR3 in multiple aspects of palatal growth and development.
4.1.2 TGFβR3 is required for palatal vascular development and remodeling
There are multiple mouse models where cardiac and palate defects occur concomitantly due to mutations in genes regulating vascular formation, and up to 30% of children born with cleft palate also have cardiac anomalies (Shafi et al., 2003). During development, mesodermally derived hemangioblasts differentiate and organize into endothelial vessels forming the primary vascular plexus through vasculogenesis. Subsequently, sprouting angiogenesis allows expansion of the primary plexus and establishes the complexity needed to support the growing embryo. Finally, vessel stabilization occurs by the invasion of pericytes and smooth muscle cells as well as deposition of extracellular matrix. Previous studies have shown that TGFβ/BMP signaling is involved in both angiogenic sprouting and vessel stabilization, and it is appreciated that these pathways are indispensable during these developmental processes (Pardali et al., 2010). Tgfbr3-/- mice do not survive past E15.5 due to failed remodeling of the coronary primary vascular plexus (Compton et al., 2007); therefore, the vascular defects observed in the palate were suspected. The association between poor palate vascular development and reduced palatal shelf length in Tgfbr3-/- mice suggested that this is an intrinsic palatal defect associated with cleft palate formation. Interestingly, TGFβ3-cre;Alk5ko/flox mice have a cleft palate phenotype identical to TGFβ3-/- mice as well as cranial vascular defects, highlighting the importance of TGFβ signaling during cranial vascularization (Yang et al., 2008). Conversely, other vascular beds in Tgfbr3-/- mice appeared normal (Compton et al., 2007) which implied a distinct function for TGFβR3 signaling within the palate and heart.
As blood flow begins and embryonic tissues differentiate, the vascular plexus is remodeled, and the vessels are specified as arteries, veins, or lymphatics. Altered expression of genes associated with vessel identity revealed a shift in the balance favoring arterial and lymphatic development over venous specification in Tgfbr3-/- embryos (Fig. 5). These data demonstrated that the loss of TGFβR3 altered vessel remodeling and maturation and implicated this pathway in the transcriptional regulation of specification genes. Hereditary hemorrhagic telangiectasia (HHT), caused by mutations in endoglin (a TGFβ co-receptor) and ALK1 (a type I TGFβ receptor), is a vascular disorder in humans characterized by the loss of arterio-venous identity and weak vascular walls aberrantly invaded by vascular smooth muscle (Mancini et al., 2009; McAllister et al., 1994; Park et al., 2008). In addition, previous studies have shown that TGFβ signaling is required for lymphangiogenesis in the skin and the loss of TGFβ receptor expression led to decreased branching and complexity of the lymphatic network (James et al., 2013). Taken together, these data support the necessity of TGFβR3 for proper expansion, remodeling, and specification during palatal vascularization, but future studies will be aimed at the contribution of TGFβR3 signaling to drive these separate, but coupled processes.
4.1.3 TGFβR3 is required for osteoblast differentiation
The majority of palatal mesenchymal cells are derived from CNC cells migrating from the dorsal neural tube to the craniofacial prominences by E9.5 (Yoshida et al., 2008). Following fusion of the palatal shelves, mesenchymal condensations develop within multiple ossification centers and undergo intramembranous ossification forming the maxillary and palatine bones without the presence of a cartilaginous phase (Franz-Odendaal, 2011). The ossified areas continue to differentiate and expand until they become one congruent bone forming the upper jaw and establishing separation between the oral and nasal cavities. Conditional deletion of BMPR1a (ALK3) within the palatal mesenchyme (Osr2-cre) resulted in cleft palate associated with defects in palatal bone formation associated with reduced mesenchymal proliferation and condensation formation (Baek et al., 2011). Decreased mRNA expression and protein localization of osteoblast determinants in Tgfbr3-/- embryos suggested deficits within the entire osteoblast developmental program (Fig. 6). In vitro cultures of MEPM cells confirmed that TGFβR3 was necessary and sufficient for osteoblast differentiation, as measured by mineralization and gene expression changes in response to OM+BMP2 (Figs. 7, 8). These data established that TGFβR3 signaling supports pre-osteoblast cell commitment and early osteoblast development during elongation and elevation of the palatal shelves. Impending questions include whether TGFβR3 signaling directly or indirectly influences osteogenesis, and if the TGFβR3 pathway is upstream of the critical genes involved.
4.1.4 TGFβR3 modulates TGFβ and BMP signaling in palates
TGFβ/BMP signaling is required for normal palate development. Loss of TGFβ/BMP signaling, either by ligand deletion, or receptor loss, was associated with cleft palate formation due to aberrant cell cycle progression and altered gene expression (Dudas et al., 2004a; Dudas et al., 2004b; Ito et al., 2003; Kaartinen et al., 1995; Proetzel et al., 1995; Sanford et al., 1997; Taya et al., 1999). TGFβR3 has been previously implicated in the regulation TGFβ receptor recycling (Shi and Massague, 2003), and here our data supported a role for TGFβR3 in maintaining the expression of ligands and receptors within the TGFβ/BMP pathway in the developing palate. We showed that the loss of TGFβR3 resulted in perturbations of the TGFβ/BMP signaling program and suggested that TGFβR3 acts upstream of these pathways to sustain adequate levels for normal palatogenesis (Fig. 9, 10). TGFβR3 is uniquely positioned to regulate both TGFβ and BMP pathways by its ability to bind both ligands; furthermore, our findings and others suggest that precise coordination of both pathways is crucial to support vascular and osteoblast development within the palate (Fig. 10). Similarly, epicardial cell invasion required TGFβR3-mediated activation of both TGFβ and BMP canonical signaling pathways as well as alternate pathways via GIPC and Par6/Smurf/RhoA (Hill et al., 2012; Sanchez and Barnett, 2012). Taken together, these results revealed that the loss of TGFβR3 lessened the expression of key TGFβ/BMP molecules. Future goals include understanding the mechanisms by which TGFβR3 supports sufficient TGFβ/BMP signaling during palate formation.
Figure 10. TGFβR3 maintains the balance of GFβ/BMP signals during vascular and osteoblast development that supports palatogenesis.
5.1 Conclusion
Cleft palate formation remains a common craniofacial anomaly that is associated with a high burden of care, the features of which have not changed significantly in the last 50 years. Increased understanding of the developmental programs active during palate formation are key to formulate potential avenues of more effective clinical care. The TGFβ pathway has been used as a model to study cleft palate, yet little was known about the role of TGFβR3 during palate formation. These data demonstrated that TGFβR3 is required for normal palatogenesis while also mediating vascular and bone development (Fig. 10). The importance of both TGFβ and BMP signals during palatogenesis is well understood. Our studies revealed that the loss of TGFβR3 reduced the expression of several ligands and receptors in the TGFβ/BMP family. This revealed that even slight alterations in TGFβ/BMP signaling will interrupt vascular and osteogenic differentiation during palate formation and that TGFβR3 is essential for maintaining the expression of these molecules (Fig. 9, 10). Revealing the mechanisms of TGFβR3's signaling pathway during the parallel processes involved in palate formation will expand our understanding of palate development and identify new targets aimed at improving future therapy for patients with cleft palate deformities.
6.1 Experimental procedures
6.1.1 Murine Model
Tgfbr3+/+ and Tgfbr3-/- mice were analyzed at E13.5 and E14.5 via timed pregnancies. At least three littermate pairs were analyzed in all experiments. All procedures and protocols were done in accordance with a Vanderbilt IACUC approved protocol.
6.1.2 Tissue preparation
Embryos were harvested at E13.5 and E14.5 genotyped, and processed as previously described (Humphreys et al., 2012). Briefly, heads were fixed in 4% paraformaldehyde (PFA) for 30 minutes to 1 hour and frozen in optimal cutting temperature (OCT) media after sucrose dehydration. All staining was performed on 8 μm thick coronal sections that were thawed and rehydrated in phosphate buffered saline (PBS).
6.1.3 H&E staining
8 μm sections of paraffin embedded tissue were processed as previously described (Humphreys et al., 2012). Following rehydration, sections were stained in Meyer's hematoxylin for 5 min, re-dehydrated, and counter-stained with eosin according to standard protocols. Palatal measurements were analyzed as previously described (Goudy et al., 2010). Briefly, all images were taken with a digital caliper placed on the image. The measurements were then determined in photoshop using the digital caliper as the standard. Palate length is defined as the length from the hinge of the palate shelf to the tip at the MEE (Rice et al., 2004).
6.1.4 Roller bottle cultures
Roller culture of the palatal shelves was performed as according to the previously published protocol (Goudy et al., 2010). Tgfbr3+/+ and Tgfbr3-/- littermate embryos were dissected at E13.5 in cold PBS under sterile conditions. The mandibles and brain tissue were removed from the midface of each embryo. The remaining midface including the palate shelves was placed in a roller bottle with 3 ml of serum-free BGJb. The cultures were incubated for 72 hours at 37 ° with 95% O2 and 5% CO2 with media changes every 24 hours. The palatal cultures were fixed, sectioned, and stained as described above.
6.1.5 Palatal shelf culture
Filter cultures of the palatal shelves were performed as previously published (Goudy et al., 2010). Tgfbr3+/+ and Tgfbr3-/- littermate embryos were dissected at E13.5 in cold PBS under sterile conditions. The palatal shelves were removed and placed in apposition on a 0.4 μm filter transwell with 1 ml of serum-free BGJb in the lower chamber. The cultures were incubated for 72 hours at 37 ° with 95% O2 and 5% CO2 with media changes every 24 hours. The palatal cultures were fixed, sectioned, and stained as described above.
6.1.6 Immunofluorescence
After allowing slides to dry at room temperature for 15 minutes, slides were fixed in acetone (-80°C) for 5 minutes and allowed to dry for 8 minutes. Slides were washed 3 times in PBS for 3-5 minutes each wash and permeabilized in 3 successive changes of 0.1% Tween-20 for 3-5 minutes. Sections were blocked with 10% donkey serum for 1 hour at room temperature and incubated with primary antibodies: PH3 (Cell signaling), Cleaved Caspase 3 (Cell signaling), CD-31 (BD Pharmingen), SmαA (Sigma), Lyve1 (Upstate), Notch1 (Abcam), RUNX2 (Abcam) diluted in 1% donkey serum overnight at 4°C. The following day the slides are washed with PBS followed by 0.1% Tween-20 as previously stated and incubated with secondary antibodies (Invitrogen) for 1 hour at room temperature. Finally they are washed in PBS followed by dH2O and counterstained with hard mount DAPI (Vectastain).
All imaging was performed on a Nikon E800 microscope, and images were obtained with SPOT imaging software (Diagnostic Instruments, Inc.).
6.1.7 qPCR
To determine changes in gene expression, we used qPCR as previously described (Hill et al., 2012). Total RNA was isolated using the TRIzol reagent (Invitrogen) according to the manufacturer's protocol. cDNA was generated from 1 μg total RNA using oligo-dT primers and Superscript III polymerase (Invitrogen). Primer pairs are shown in Table 1. Real-time PCR analysis was done with iQ SYBR green supermix (Bio-Rad) in the Bio-Rad iCycler for 40 cycles. The expression levels are calculated using the ΔΔCT method. The threshold cycle (CT) represents the PCR cycle at which an increase of the reporter fluorescence above the baseline is first detected. The fold change in expression levels, R, is calculated as follows: R=2-ΔΔCT (where R = 2 (ΔCT treated-ΔCT control)) to normalize the abundance of all transcripts to the level of GAPDH RNA expression.
Table 1. qPCR primer sequences.
| Gene | SENSE PRIMER (5′→3′) | ANTI-SENSE PRIMER (5′→3′) |
|---|---|---|
| GAPDH | ATGACAATGAATACGGCTACAG | TCTCTTGCTCAGTGTCCTTG |
| Pecam | TGGTTGTCATTGGAGTGGTC | TTCTCGCTGTTGGAGTTCAG |
| SmαA | AGCCAGTGAAGGTGCCTGAGAAC | TGCCCAAAGCCATTAGAGTCCTC |
| EphB4 | AATGTCACCACTGACCGTGA | TCAGGAAACGAACACTGCTG |
| Coup Tf-II | GCAAGTGGAGAAGCTCAA | CACACTGGGACTTTTCCT |
| Notch1 | ATGTCAATGTTCGAGGACCAG | TCTGAGTCTTCCCCTTCTGG |
| Alk1 | ACCCAATGACCCCAGTTT | GTACCAGCACTCTCTCATCA |
| Lyve1 | CAGCATTCAAGAACGAAGCAG | GCCTTCACATACCTTTTCACG |
| Prox1 | TTCTTTTACACCCGCTACCC | TTGACGCGCATACTTCTCC |
| RUNX2 | CCCAGCCACCTTTACCTACA | TATGGAGTGCTGCTGGTCTG |
| Alk Phos | GCTGATCATTCCCACGTTTT | CTGGGCCTGGTAGTTGTTGT |
| Osteocalcin | TGCTTGTGACGAGCTATCAG | GAGGACAGGGAGGATCAAGT |
| Col1A1 | AAGGATACAGTGGATTGCAGG | TCTACCATCTTTGCCAACGG |
| Col1A2 | CATAAAGGGTCATCGTGGCT | TTGAGTCCGTCTTTGCCAG |
| TGFβ2 | TGCTAACTTCTGTGCTGGG | GCTTCGGGATTTATGGTGTTG |
| TGFβ3 | CAGGATCTAGGCTGGAAATGG | GGGTTCAGGGTGTTGTATAGTC |
| ALK5 | CCTTCTGATCCATCGGTTGA | CCATTGGCATACCAGCAT |
| TGFβR2 | GGAGAAGTGAAGGATTACGAGC | CACACGATCTGGATGCCC |
| BMP2 | TTATCAGGACATGGTTGTGGAG | GGGAAATATTAAAGTGTCAGCTGG |
| BMP4 | GTAGTGCCATTCGGAGCG | ATCAGCATTCGGTTACCAGG |
| ALK2 | AGAGGGTCGATATTTGGGC | AACTTGGGTCATTGGGAAC |
| ALK3 | ACCATTTCCAGCCCTACA | TCACTGGGCACCATGTT |
| BMPR2 | TTCTCTGGATCTTTCAGCCAC | CCTGATTTGCCATCTTGTGTTG |
6.1.8 Mouse embryonic palatal mesenchymal cell culture
Primary mouse embryonic palatal mesenchymal (MEPM) cells were generated from E13.5 embryos. Embryos were harvested and the maxilla was dissected and incubated in trypsin at 37°C with 5% CO2 for 30 minutes. The epithelium was removed from the mesenchyme, and the tissue was pipetted up and down vigorously until the cells were dispersed. The cells were filtered through a 100 μm mesh and cultured in DMEM/F12 supplemented with 10% FBS and 100 μg/mL penicillin/streptomycin.
6.1.9 Osteoblast mineralization
The capacity of MMEM cells to mineralize surrounding matrix was tested by providing confluent monolayers osteogenic media (OM): α-MEM containing 2.5% FBS, 100 μg/mL penicillin/streptomycin, 100 μg/mL ascorbic acid, 5 mM β-glycerophosphate, and 100 ng/mL BMP2 (R&D Systems). To assure that osteogenic media was essential for matrix mineralization, control wells were incubated in growth media (GM): α-MEM containing 2.5% FBS, 100 μg/ml penicillin/streptomycin, and vehicle. Cultures were incubated for 16 days at 37°C with 5% CO2 with changes of media every 2 days. Cell cultures were washed with PBS twice and fixed in 4% PFA for 30 minutes at room temperature. The monolayers grown in each well of a 12 well tissue culture plate were washed twice with dH2O prior to the addition of 500 μL of 40 mM alizarin red staining (ARS) solution and incubated for 20 minutes at room temperature with gentle rocking. The monolayers were then washed 4 times with excess dH2O for 4 minutes at room temperature while shaking. The plates were then tilted and left at an angle to remove all excess water from the well. Plates were photographed and stored at -20°C until solubilization of the dye. To quantify ARS, 400 μL of 10% acetic acid was added to each well and incubated for 30 minutes at room temperature with shaking. The monolayers were transferred to a 1.5 mL microfuge tube by scraping each well with a cell scraper. The tubes were vortexed for 30 minutes and 250 μL of mineral oil was added to each tube. Each tube was heated to 85°C for 10 minutes, cooled on ice for 5 minutes, and centrifuged at 20,000 g for 15 minutes. 250 μL of the acetic acid phase was transferred to a new tube and 100 μL of 10% ammonium hydroxide was added to neutralize the acid. Aliquots of each sample were read on SpectraMax M5 (Molecular Devices) plate reader in triplicate at 405 nm in 96-well format using opaque-walled, transparent-bottomed plates Data was collected with Soft Max Pro (Molecular Devices) software.
6.1.10 Adenovirus
Adenoviruses were generated using the pAdEasy system (He et al., 1998). Viruses were titered by performing serial dilutions of the concentrated virus and counting the number of GFP-expressing HEK293 cells after 18-24 h. The following adenoviruses co-expressing GFP were used: pAdTrack-GFP (control) and full length TGFβR3 (FL). Cells were plated at a density of 100,000 cells per well of a 24-well plate in MEPM cells media and allowed to adhere overnight at 37°C with 5% CO2. The following day, virus was added directly to the cells at a final concentration of 108 PFU/ml. 24 h later, the cells were given either GM or OM to induce osteoblast differentiation as described above.
6.1.11 Statistical Analysis
Data are presented as the mean of three independent experiments ± SEM for three Tgfbr3+/+ and Tgfbr3-/- littermate pairs, unless otherwise specified. Paired students t-tests were performed to establish significance, which was determined by a p-value of <0.05.
Acknowledgments
We would like to thank the expert technical assistance from Yan Zhao and Xiaomin Fu.
Grant Sponsor and Grant Number: NIH 5K08DE17953-5
Abbreviations
- ALK
Activin Receptor-Like Kinases
- ARS
Alizarin red staining
- BMP
Bone Morphogenetic Protein
- BMPR
Bone Morphogenetic Protein Receptor
- CNC
cranial neural crest
- GIPC
Gaip Interacting Protein C
- GM
growth media
- M
maxilla
- MEE
medial edge epithelium
- MEPM
mouse embryonic palate mesenchymal
- NS
nasal septum
- OM
osteogenic media
- P
palate
- PS
palatal shelf
- SEM
standard error of the mean
- TGFβ
transforming growth factor beta
- TGFβR1
Type I TGFβ receptor
- TGFβR2
Type II TGFβ receptor
- TGFβR3
Type III TGFβ receptor
- TGFβR3-FL
Type III TGFβ receptor-full length
Contributor Information
Cynthia R. Hill, Email: cynthia.r.allison@vanderbilt.edu.
Britni H. Jacobs, Email: britni.jacobs@vanderbilt.edu.
Christopher B. Brown, Email: chris.brown@vanderbilt.edu.
Joey V. Barnett, Email: joey.barnett@vanderbilt.edu.
Steven L. Goudy, Email: steven.goudy@vanderbilt.edu.
References Cited
- Baek JA, Lan Y, Liu H, Maltby KM, Mishina Y, Jiang R. Bmpr1a signaling plays critical roles in palatal shelf growth and palatal bone formation. Dev Biol. 2011;350:520–531. doi: 10.1016/j.ydbio.2010.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Maxson RE., Jr Recent advances in craniofacial morphogenesis. Dev Dyn. 2006;235:2353–2375. doi: 10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- Compton LA, Potash DA, Brown CB, Barnett JV. Coronary vessel development is dependent on the type III transforming growth factor beta receptor. Circ Res. 2007;101:784–791. doi: 10.1161/CIRCRESAHA.107.152082. [DOI] [PubMed] [Google Scholar]
- Cui XM, Chai Y, Chen J, Yamamoto T, Ito Y, Bringas P, Shuler CF. TGF-beta3-dependent SMAD2 phosphorylation and inhibition of MEE proliferation during palatal fusion. Dev Dyn. 2003;227:387–394. doi: 10.1002/dvdy.10326. [DOI] [PubMed] [Google Scholar]
- Cui XM, Shuler CF. The TGF-beta type III receptor is localized to the medial edge epithelium during palatal fusion. Int J Dev Biol. 2000;44:397–402. [PubMed] [Google Scholar]
- Doetschman T, Georgieva T, Li H, Reed TD, Grisham C, Friel J, Estabrook MA, Gard C, Sanford LP, Azhar M. Generation of mice with a conditional allele for the transforming growth factor beta3 gene. Genesis. 2012;50:59–66. doi: 10.1002/dvg.20789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas M, Kim J, Li WY, Nagy A, Larsson J, Karlsson S, Chai Y, Kaartinen V. Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev Biol. 2006;296:298–314. doi: 10.1016/j.ydbio.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas M, Nagy A, Laping NJ, Moustakas A, Kaartinen V. Tgf-beta3-induced palatal fusion is mediated by Alk-5/Smad pathway. Dev Biol. 2004a;266:96–108. doi: 10.1016/j.ydbio.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Dudas M, Sridurongrit S, Nagy A, Okazaki K, Kaartinen V. Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech Dev. 2004b;121:173–182. doi: 10.1016/j.mod.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Ferguson MW. Palate development. Development. 1988;103 Suppl:41–60. doi: 10.1242/dev.103.Supplement.41. [DOI] [PubMed] [Google Scholar]
- Franz-Odendaal TA. Induction and patterning of intramembranous bone. Front Biosci (Landmark Ed) 2011;16:2734–2746. doi: 10.2741/3882. [DOI] [PubMed] [Google Scholar]
- Goudy S, Law A, Sanchez G, Baldwin HS, Brown C. Tbx1 is necessary for palatal elongation and elevation. Mech Dev. 2010;127:292–300. doi: 10.1016/j.mod.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CR, Sanchez NS, Love JD, Arrieta JA, Hong CC, Brown CB, Austin AF, Barnett JV. BMP2 signals loss of epithelial character in epicardial cells but requires the Type III TGFbeta receptor to promote invasion. Cellular signalling. 2012;24:1012–1022. doi: 10.1016/j.cellsig.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys R, Zheng W, Prince LS, Qu X, Brown C, Loomes K, Huppert SS, Baldwin S, Goudy S. Cranial neural crest ablation of Jagged1 recapitulates the craniofacial phenotype of Alagille syndrome patients. Hum Mol Genet. 2012;21:1374–1383. doi: 10.1093/hmg/ddr575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Yeo JY, Chytil A, Han J, Bringas P, Jr, Nakajima A, Shuler CF, Moses HL, Chai Y. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development. 2003;130:5269–5280. doi: 10.1242/dev.00708. [DOI] [PubMed] [Google Scholar]
- Iwata J, Hacia JG, Suzuki A, Sanchez-Lara PA, Urata M, Chai Y. Modulation of noncanonical TGF-beta signaling prevents cleft palate in Tgfbr2 mutant mice. J Clin Invest. 2012;122:873–885. doi: 10.1172/JCI61498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata J, Parada C, Chai Y. The mechanism of TGF-beta signaling during palate development. Oral Dis. 2011;17:733–744. doi: 10.1111/j.1601-0825.2011.01806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James JM, Nalbandian A, Mukouyama YS. TGFbeta signaling is required for sprouting lymphangiogenesis during lymphatic network development in the skin. Development. 2013;140:3903–3914. doi: 10.1242/dev.095026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen V, Cui XM, Heisterkamp N, Groffen J, Shuler CF. Transforming growth factor-beta3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev Dyn. 1997;209:255–260. doi: 10.1002/(SICI)1097-0177(199707)209:3<255::AID-AJA1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Levi G, Mantero S, Barbieri O, Cantatore D, Paleari L, Beverdam A, Genova F, Robert B, Merlo GR. Msx1 and Dlx5 act independently in development of craniofacial skeleton, but converge on the regulation of Bmp signaling in palate formation. Mech Dev. 2006;123:3–16. doi: 10.1016/j.mod.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Li L, Wang Y, Lin M, Yuan G, Yang G, Zheng Y, Chen Y. Augmented BMPRIA-mediated BMP signaling in cranial neural crest lineage leads to cleft palate formation and delayed tooth differentiation. PLoS One. 2013;8:e66107. doi: 10.1371/journal.pone.0066107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Sun X, Braut A, Mishina Y, Behringer RR, Mina M, Martin JF. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development. 2005;132:1453–1461. doi: 10.1242/dev.01676. [DOI] [PubMed] [Google Scholar]
- Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- Lopez-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massague J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-beta receptor system. Cell. 1991;67:785–795. doi: 10.1016/0092-8674(91)90073-8. [DOI] [PubMed] [Google Scholar]
- Mancini ML, Terzic A, Conley BA, Oxburgh LH, Nicola T, Vary CP. Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Dev Dyn. 2009;238:2479–2493. doi: 10.1002/dvdy.22066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- Murray JC, Schutte BC. Cleft palate: players, pathways, and pursuits. J Clin Invest. 2004;113:1676–1678. doi: 10.1172/JCI22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima A, Ito Y, Asano M, Maeno M, Iwata K, Mitsui N, Shimizu N, Cui XM, Shuler CF. Functional role of transforming growth factor-beta type III receptor during palatal fusion. Dev Dyn. 2007;236:791–801. doi: 10.1002/dvdy.21090. [DOI] [PubMed] [Google Scholar]
- Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20:556–567. doi: 10.1016/j.tcb.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, Park A, Wu X, Kaartinen V, Roman BL, Oh SP. ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood. 2008;111:633–642. doi: 10.1182/blood-2007-08-107359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 2004;113:1692–1700. doi: 10.1172/JCI20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H, Yamamura K, Suzuki N. Reduced bone morphogenetic protein receptor type 1A signaling in neural-crest-derived cells causes facial dysmorphism. Dis Model Mech. 2012;5:948–955. doi: 10.1242/dmm.009274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez NS, Barnett JV. TGFbeta and BMP-2 regulate epicardial cell invasion via TGFbetaR3 activation of the Par6/Smurf1/RhoA pathway. Cell Signal. 2012;24:539–548. doi: 10.1016/j.cellsig.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez NS, Hill CR, Love JD, Soslow JH, Craig E, Austin AF, Brown CB, Czirok A, Camenisch TD, Barnett JV. The cytoplasmic domain of TGFbetaR3 through its interaction with the scaffolding protein, GIPC, directs epicardial cell behavior. Dev Biol. 2011;358:331–343. doi: 10.1016/j.ydbio.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafi T, Khan MR, Atiq M. Congenital heart disease and associated malformations in children with cleft lip and palate in Pakistan. Br J Plast Surg. 2003;56:106–109. doi: 10.1016/s0007-1226(03)00044-4. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shiomi N, Cui XM, Yamamoto T, Saito T, Shuler CF. Inhibition of SMAD2 expression prevents murine palatal fusion. Dev Dyn. 2006;235:1785–1793. doi: 10.1002/dvdy.20819. [DOI] [PubMed] [Google Scholar]
- Taya Y, O’Kane S, Ferguson MW. Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development. 1999;126:3869–3879. doi: 10.1242/dev.126.17.3869. [DOI] [PubMed] [Google Scholar]
- Van Laer L, Dietz H, Loeys B. Loeys-dietz syndrome. Adv Exp Med Biol. 2014;802:95–105. doi: 10.1007/978-94-007-7893-1_7. [DOI] [PubMed] [Google Scholar]
- Xu X, Han J, Ito Y, Bringas P, Jr, Urata MM, Chai Y. Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Dev Biol. 2006;297:238–248. doi: 10.1016/j.ydbio.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Yang LT, Li WY, Kaartinen V. Tissue-specific expression of Cre recombinase from the Tgfb3 locus. Genesis. 2008;46:112–118. doi: 10.1002/dvg.20372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Vivatbutsiri P, Morriss-Kay G, Saga Y, Iseki S. Cell lineage in mammalian craniofacial mesenchyme. Mech Dev. 2008;125:797–808. doi: 10.1016/j.mod.2008.06.007. [DOI] [PubMed] [Google Scholar]
- You HJ, How T, Blobe GC. The type III transforming growth factor-beta receptor negatively regulates nuclear factor kappa B signaling through its interaction with beta-arrestin2. Carcinogenesis. 2009;30:1281–1287. doi: 10.1093/carcin/bgp071. [DOI] [PMC free article] [PubMed] [Google Scholar]