

Abstract
Sustained high levels of circulating catecholamines are reported to induce cardiotoxicity. Isoproterenol (ISP), a synthetic catecholamine has been widely employed to induce myocardial injury, though the role of inflammation and apoptosis is not well established. This study was designed to investigate the underlying mechanism of oxidative damage, inflammatory signaling, cell death in ISP induced myocardial infarction in rats. Wistar albino rats were divided in two groups: group I (sham control) and group II (ischemic control). ISP (85 mg/kg, s.c.) was administered at an interval of 24 h to group II for two consecutive days. On day third, after 48 h of the first injection of ISP, blood was collected from retro orbital plexus of rat eyes to estimate the biochemical parameters. Glutathione (GSH) and superoxide dismutase (SOD) were measured for antioxidant status. Similarly, malondialdehyde (MDA) was measured as an index of lipid peroxidation. Cardiac markers (SGOT, CK-MB, TropI and LDH) and pro-inflammatory cytokines (IL-6, CRP and TNF-α) were also estimated in ISP-induced rats. At the end of experiments animals were sacrificed for histopathological studies. GSH and SOD showed significant decrease after ISP challenge as compared to sham (control) group (p < 0.01) while MDA level, increased significantly (p < 0.01). ISP, also increased the level of cardiac markers and markers of inflammation significantly (p < 0.01), which was further verified by histopathological studies of the heart tissues. The study confirmed that ISP causes detrimental changes in the myocardium by altering cardiac and inflammatory markers, which leads to severe necrosis. The deleterious effects produced by ISP substantiate its suitability as a novel animal model for evaluation of cardioprotective agents/drugs.
Keywords: Oxidative stress, Myocardial ischemia, Cardiac markers, Cytokines, Isoproterenol
Introduction
Acute myocardial infarction (AMI) and heart failure represent leading cause of mortality worldwide [1]. AMI is linked with oxidative stress and associated inflammatory response promoting metabolic derangement, cardiac dysfunction and heart failure [2]. The rising burden of AMI has naturally prompted screening and evaluation of the potential therapeutic/preventive agents. Several animal models of cardiovascular pathology, contributing towards understanding and treatment of a broad range of conditions have been developed [2]. However, recent years have witnessed the important changes in our clinical approaches to AMI, which is increasing recognition that the pathophysiology of human AMI is a process occurring at many levels, not just within the epicardial coronary artery, but also within the microvasculature and the myocardium. These changes in our understanding of AMI have implications for the relevance of exploring the mechanisms responsible for AMI in animal models. The model of Isoproterenol (ISP) overdose in rodents, especially in rats, is one of the most popular experimental models used for AMI [3–6]. The advantage of this model is that ISP, a cardiotoxicant, produces AMI which mimics the resemblance to human MI. The experiments are technically easy to perform and, most importantly, it is a clinically relevant model. ISP chemically known as 4-[1-hydroxy-2-(propan-2-ylamino) ethyl] benzene-1, 2-diol is a synthetic catecholamine and a powerful nonselective β-agonist (Fig. 1). An insight into the pathogenesis of ISP-induced myocardial injury reveals the occurrence of oxidative damage, lipid peroxidation, altered hemodynamics and left ventricular function following hypertrophy [7], inflammatory cell infiltration, fibrosis and necrosis [6, 8–10]. MI induced by ISP shows notable metabolic and morphologic aberrations in the myocardium of experimental animals [6] and also inflicted complex biochemical and structural changes leading to cell damage and necrosis [4, 11, 12].
Fig. 1.

Chemical structure of Isoproterenol (ISP)
Although, the exact mechanism of ISP-induced myocardial damage is still controversial. However, a mismatch of oxygen supply versus demand following coronary hypotension and myocardial hyperactivity may explain it in better manner for its complex morphological alterations observed in the coronary vasculature [13]. Therefore, even after several years of researches, there is paucity of substantial information on mechanisms of ISP induced cardiotoxicity particularly on biochemical defense, inflammatory signaling and apoptotic mode of cell death and thus the interpretation of in vivo data is often difficult. The present study was undertaken to evaluate the effect of ISP on myocardial antioxidant status, cardiac markers, pro-inflammatory cytokines and histopathological alterations in rats.
Materials and Methods
Animals
Wistar albino rats, weighing 150–200 g and 10–12 weeks old, were used in the study. The study protocol was reviewed and approved by the Institutional Animal Ethical Committee of University College of Medical Sciences, Delhi and conforms to the Indian National Science Academy guidelines for the use and care of experimental animals in research. Animals were obtained from the central animal house facility of University College of Medical Sciences, Delhi. The rats were housed in polyacrylic cages (38 × 23 × 10 cm3) with not more than four animals per cage. They were housed under standard laboratory conditions with natural light and dark cycles (approximately 12 h light/12 h dark) and maintained at humidity of 55 ± 5 % and an ambient temperature of 22 ± 2 °C. The animals were allowed free excess to standard pellet diet (Durga Brothers Pvt. Ltd.) and tap water ad libitum.
Chemicals and Drugs
ISP was purchased from Sigma Chemical Company, St. Louis, USA and all other chemicals used were of analytical grade. Creatine kinase-MB (CK-MB), lactate dehydrogenase (LDH) and serum glutathione oxaloacitate transaminase (SGOT) assay kits were procured from Spinreact SA, Spain. Troponin I (TropI), interleukin-6 (IL-6), C-reactive protein (CRP) and tumor necrosis factor-alpha (TNF-α) ELISA kit were purchased from Calbiotech, USA. Immuno-histo staining kit based on horseradish peroxidase (HRP) polymer detection system was purchased from Thermo Fisher Scientific, USA and mouse monoclonal primary antibodies for Bax and Bcl-2 were obtained from Santa Cruz Biotechnology, USA, TUNEL assay kit was purchased from Roche Diagnostic USA.
Induction of Myocardial Ischemia
ISP was freshly prepared in normal saline and injected subcutaneously at a dose of 85 mg/kg b.w. to the rats after a week of acclimatization on day 1 and 2 at an interval of 24 h [6, 11, 12].
Experimental Protocol
Animals were randomly divided into two groups, each containing eight rats (n = 8). Rats of Group I were designated normal control and were orally administered normal saline (1 ml/kg). In addition, they were injected normal saline subcutaneously. Rats of Group II, designated as ischemic control, were administered normal saline + ISP injection (85 mg/kg) once daily for two consecutive days at an interval of 24 h. Blood samples were collected from retro orbital plexus of the animals on day first and third to perform biochemical studies. After blood sampling on third day, the rats were anesthetized with sodium pentobarbital (30 mg/kg, i.p.). To carry out immunohistochemical and histological studies, the heart was immediately dissected out, washed in ice-cold saline and stored in 10 % buffered formalin.
Biochemical Studies
Serum malondialdehyde (MDA) levels were estimated using the method previously described by Satoh [14]. Activity of superoxide dismutase (SOD) in erythrocytes was assayed as described by Marklund and Marklund [15] and modified by Nandi and Chatterjee [16]. Erythrocyte’s reduced glutathione (GSH) content was estimated by the method of Beutler et al. [17].
Estimation of Cardiac Markers
The cardiac function or myocardial injury markers such as CK-MB, SGOT and LDH in serum were estimated spectrophotometrically by using commercially available kits. Cardiac Troponin-I was measured in serum by standard ELISA method.
Estimation of Markers of Inflammation
Markers of inflammation such as IL-6, CRP and TNF-α in serum were measured by standard ELISA method.
Immuno-Staining for the Localization of Bax and Bcl-2 Proteins
Mouse monoclonal anti-Bcl-2 and Bax antibodies were used to recognise Bcl-2/Bax in tissue by immunohistochemical staining. The Ultravision ONE HRP polymer detection system locates primary antibody by a universal secondary antibody polymer formulation. The amino acid polymer is conjugated to HRP and the Fab fragments of secondary antibody. The polymer complex is visualized with an appropriate chromogen/substrate. Briefly, formalin-fixed paraffin-embedded myocardial sections were subjected to the immunohistochemical procedure for the localization of Bax and Bcl-2 proteins using specific mouse monoclonal primary antibodies. Sections are first blocked and then incubated in primary antibody followed by UltraVision One HRP polymer. The target protein (Bax/Bcl-2) was visualized by incubation in peroxidase substrate (H2O2) using 3,3′ diaminobenzidine (DAB) as the chromogen.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labelling (TUNEL assay)
Myocardial apoptosis was quantified by detection of DNA fragmentation using the TUNEL technique [18]. Briefly, the enzyme terminal deoxynucleotidyl transferase was used to incorporate residues of digoxigenin nucleotide into 3′ OH ends of DNA fragments. The free end of cellular DNA was labelled by incubating the specimens in streptavidin conjugated to horse radish peroxidase enzyme and peroxidase substrate. The signal of terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) assay was used to identify apoptotic cells using secondary reaction with antibodies and DAB chromogen. The slides were counterstained in methyl green and total cell counts and TUNEL positive cells in the specimens were determined by means of a light microscope. The cells with clear nuclear labelling were defined as TUNEL positive cells.
Histopathological Studies
Myocardial tissue fixed in buffered formalin was processed for paraffin embedding, sectioned at 5 μm and mounted onto microscope slides. These sections were stained with hematoxylin and eosin (H&E), and visualized under light microscope to study the histo-architectural changes of the myocardium.
Statistical Analysis
The results were expressed as mean ± SD. and statistical differences between mean values were determined by student t test. A value of p < 0.05 was considered statistically significant.
Results
Effect of ISP on Oxidative Stress Parameters and Antioxidant Status
ISP administration increases the level of lipid peroxidation marker MDA (p < 0.01), while endogenous antioxidants GSH and SOD decreases significantly (p < 0.01), which is presented in Fig. 2.
Fig. 2.

Effect of ISP on oxidative stress markers, a MDA, b GSH, c SOD. Values are expressed as mean ± SD; n = 8, *p < 0.05
Effect of ISP on Cardiac Markers
The level of cardiac markers in the serum increases significantly (p < 0.01) after ISP administration. SGOT, CK-MB, LDH and TropI increases in ischemic control group after ISP administration as compared to healthy control group (Fig. 3).
Fig. 3.

Effect of ISP on cardiac marker enzymes, a SGOT, b CK-MB, c TropI, d LDH. Values are expressed as mean ± SD; n = 8, *p < 0.05
Effect of ISP on Markers of Inflammation
Markers of inflammation were increased significantly (p < 0.01) after ISP administration. IL-6, CRP and TNF-α levels in the serum were increased after ISP challenge as compared to healthy control (Fig. 4).
Fig. 4.

Effect of ISP on pro-inflammatory cytokines, a IL-6, b CRP, c TNF-α. Values are expressed as mean ± SD; n = 8, *p < 0.05
Effect of ISP on Expression of Bax and Bcl-2 Protein
Bax is pro-apoptotic protein which is absent/or very less in normal rat myocardium. The localization of Bax protein is indicated by purple positive immune-reactivity. ISP administration increases the Bax expression (Fig. 5). Anti-apoptotic protein Bcl-2 was expressed in healthy control rats, the localization of Bcl-2 protein is indicated by dark brown positive immunoreactivity. ISP control rats shows reduced expression of Bcl-2, while normal rats have greater expression of Bcl-2 (Fig. 6).
Fig. 5.
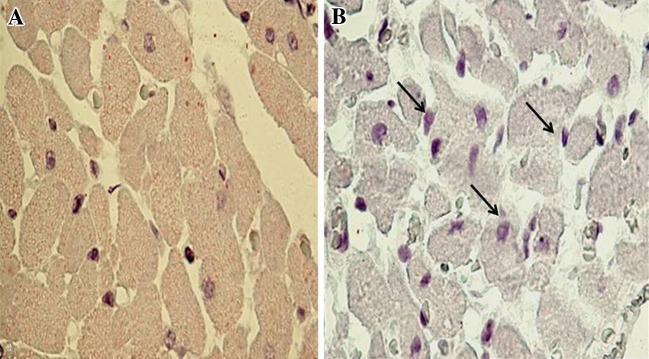
Representative photomicrographs of ventricular tissue stained for Bax protein (×400) from normal and ISP control groups. The localization of Bax protein is indicated by purple positive immunoreactivity, a healthy control rats, showing slight Bax immunoreactivity (magnification ×400) b ISP control rats, showing increased expression for Bax protein (magnification ×400)
Fig. 6.
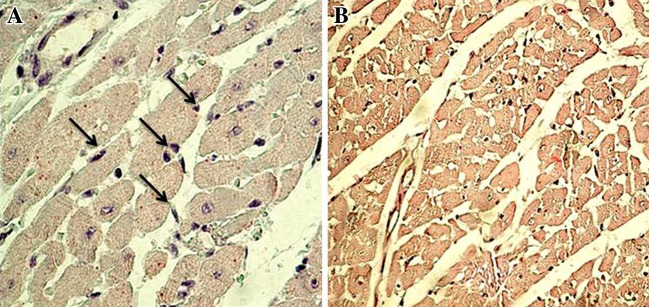
Immunohistochemical findings of Bcl-2 protein in the rat myocardium from normal and ISP control group. The localization of Bcl-2 protein is indicated by dark brown positive immunoreactivity, a Healthy control rats, showing positive Bcl-2 immunoreactivity in the myocytes (×400) b ISP control rats, shows lesser Bcl-2 immunoreactivity (400×)
Effect of ISP on Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labelling (TUNEL Assay)
TUNEL assay was done for the determination of apoptotic nuclei. TUNEL positive nuclei are stained as brown to black nuclei. ISP administration increases the apoptotic nuclei which was evident by TUNEL assay (Fig. 7).
Fig. 7.
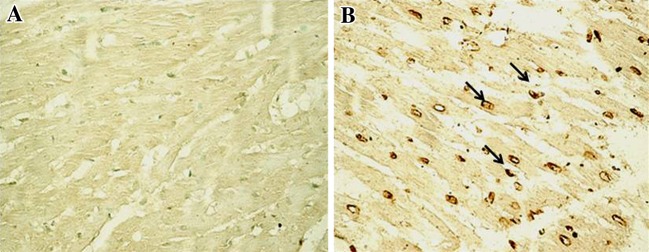
Representative photomicrographs of ventricular tissue stained for nick-end labeling (TUNEL) for DNA breaks (200×) from normal and ISP control groups. TUNEL positive nuclei are stained as brown to black nuclei. a Healthy control rats, showing no TUNEL positive cells (200×) b ISP control rats, showing TUNEL positive cells, indicated by brown to black staining (200×)
Effect of ISP on Histology of Myocardium
Microscopic sections of the heart tissue revealed that the histology of myocardium is deteriorated after ISP administration as compared to normal rat myocardium. ISP treated rat heart showing extensive myocardial necrosis and inflammatory cell infiltration, which was evident after H&E staining (Fig. 8).
Fig. 8.
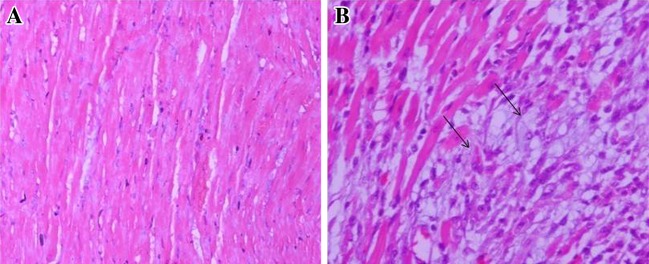
Photomicrographs of the heart tissue, a Microscopic section of normal control group rat heart showing normal architecture of cardiac muscle (H&E 100×); b ISP treated group rat heart showing extensive myocardial necrosis and inflammatory cell infiltration (H&E 100×)
Discussion
In the present study we have observed that ISP administration leads to deleterious change in the myocardium. ISP has been shown to impair the in vivo cardiac performance and induce cardiac necrosis. In myocardium, the tissue defense system consists of antioxidant enzymes including SOD, CAT, GPx and GSH [2]. SOD plays a major role in controlling mitochondrial reactive oxygen species (ROS) generated during normal oxidative phosphorylation and protects cells against oxidative stress by catalytic removal of superoxide radicals and conversion to hydrogen peroxide [2]. ISP challenge in rats caused a significant decrease in SOD, is suggestive of occurrence of oxidative stress and impaired mitochondrial energetic, required for normal cardiac function. GSH content was also decreased which play a critical role in regulating ROS in myocardium. GPx protects cellular and sub-cellular membranes from peroxidative damage by catalyzing removal of hydrogen peroxide via oxidation of GSH that is recycled from oxidized glutathione by the NADPH-dependent glutathione reductase. The reduced activity of GSH in the present study might be due to increased oxidative stress. GSH plays an important role in the regulation of cell functions and protect cells from ROS as well as scavenging ROS by reacting with superoxide radicals and neutralized them [11]. Myocardium showed enhanced susceptibility to lipid peroxidation, a type of oxidative degeneration of polyunsaturated fatty acids which is recognized as one of the basic deteriorative reactions during myocardial ischemia [4]. ISP induced rise in MDA level has been suggested that it might be due to enhanced formation of free radicals and severe oxidative stress. Cardiac markers are the useful tool for detection of myocyte damage and have great clinical importance [19]. ISP produced alteration in the endogenous antioxidants due to severe stress. Changes in cardiac markers (SGOT, CK-MB, TROP I and LDH) and markers of inflammation (IL-6, CRP and TNF-α) have been observed in this study. SGOT, CK-MB and TropI are the early marker for detection of myocardial ischemic injury, while LDH is a delayed marker of MI, begins to rise in 12–24 h following injury with peak levels in 2–3 days. Rise in the levels of the cardiac marker enzymes in the serum after 48 h of ISP administration clearly demonstrate the severe ischemic injury which is supported by the previous reports [11, 20]. When ISP is administered to rats, the myocardial cells containing CPK-MB isoenzyme and LDH are damaged or destroyed due to deficient oxygen supply or glucose. The integrity of cell membrane gets disordered and it might become more porous and permeable or may rupture that result in the leakage of these enzymes (SGOT, CPK-MB, TropI and LDH), which were increased after ISP administration. Serum level of cardiac TropI is closely associated with the degree of myocardial injury [21, 22]. ISP increased in the level of markers of inflammation viz. IL-6, CRP and TNF-α, which mainly evolve in the serum during inflammation as evident during ISP challenge. The intracellular changes occurred in due course of ischemic injury such as accumulation of H+ and Ca2+ as well as the disruption of mitochondrial membrane potential, lead to the formation of oxygen free radical or ROS. ROS accumulation and the subsequent activation of pro-inflammatory pathways play an important role in ischemic injury. Therefore a crucial mediator of ischemic injury is excessively generated derived free radicals, leading to different forms of oxygen species [23]. Reactive oxygen intermediates cause direct damage to cellular DNA, protein, and lipids, in addition to activating pathways of stress response. This nonspecific injury initiates a cytokine-mediated cascade, which results in the production of tumor necrosis factor alpha (TNF-α) [24]. Subsequent to excessive TNF-α expression the stimulation of cardiomyocyte TNF receptor type-1 induces contractile dysfunction, hypertrophy, fibrosis and cell death. Additionally, the increase in the intracellular Ca2+ concentration with the generation of calcium pyrophosphate complexes and the formation of uric acid is a potent stimulator of inflammation. Calcium phosphate complexes and uric acid belong to a group of so called danger signals, and can bind to intracellular protein complexes called inflammasomes [25]. The inflammasomes include different adaptor molecules that mediate an increase of the production and secretion of IL-6 and the stimulated danger signals eventually stimulating the secretion of further pro-inflammatory cytokines and chemokines through the activation of NF-κB [26]. Ischemia leads to significant metabolic and hemodynamic changes followed by inflammatory processes. Inflammatory signaling in cardiomyocytes also may be initiated through canonical pathways involving extracellular cytokine stimulation of cell surface receptors. Cytokines may be released from neighboring cardiomyocytes as a result of inflammatory activation, thus amplifying the inflammatory signal to multiple cells. Cytokines also may be released from immune cells recruited to the myocardium. To date, this is the first study which shown the changes in markers of inflammation along with several other biochemical parameters.
In the present study, TUNEL positivity and the immunohistochemical localization of Bax (an inducer of apoptosis) and Bcl-2 proteins (inhibitors of apoptosis) were studied to delineate the involvement of apoptosis in ischemic injury induced by ISP. Over the past several years, a large number of studies have demonstrated apoptosis, an important mode of cell death in the development and progression of cardiovascular diseases [27]. In the myocardium, apoptosis has been detected in a number of cardiac pathologies including myocardial infarction [28]. The progressive loss of cardiomyocytes by apoptosis causes further deterioration of cardiac function, hence targeting apoptosis, a biochemically-regulated process appears to be a novel and effective therapeutic strategies to limit the amount of tissue damage in myocardial injury [12]. In this study, ISP significantly induced apoptosis as it down regulated the expression of the anti-apoptotic protein, Bcl-2 and up regulated the expression of the pro-apoptotic protein Bax in association with induction in the expression of caspase-3 enzyme and percentage of TUNEL-positive cells in the ischemic myocardium. The possible mechanism of cell death by ISP is by induction of calcium paradox and oxidative stress. Substantial evidence indicates that caspase-3 plays a critical regulatory role in the signal transduction pathway leading to apoptosis [29]. Caspase-3 is a central effector caspase in many cells and mediates the cleavage of itself, other downstream caspases and other caspase substrates [30]. Moreover, free radicals have been demonstrated to directly activate calcium and magnesium dependent endonuclease, thus resulting in DNA fragmentation and cell apoptosis.
ISP-induced myocardial ischemia model is simple, noninvasive, economical and rapid model of choice to induce MI, requiring minimal surgery and leaving the pericardium intact. As reported previously that ISP administration consistently produce cardiac dysfunction and left ventricular dilation similar to those found in the severely infarcted heart. Findings of present study clearly demonstrate that ISP-induced oxidative stress contributes to biochemical and hemodynamic impairment along with histopathological damage. ISP-induced myocardial injury model can be explicitly used for evaluating cardioprotective agents and may provide an integrated insight of mechanistic evaluation of cardio protectants in toxicological and pharmacological studies against ISP-induced myocardial necrosis. Unfortunately, the model is being used as a tool by investigators who are not necessarily experts in ISP toxicity leading to frequent misinterpretation of data. Hence, the results presented would deliberate some of the established and controversial mechanisms and the potential pitfalls, one should be aware of when using this model.
Findings of present study clearly demonstrate that ISP-induced oxidative stress contributes to impairment of cardiac performance and deleterious changes in myocardium. Previous reports supports that ISP-induced myocardial injury, especially in rats could be a clinically relevant model that is suitable to test the efficacy of cardioprotective natural products and other compounds in vivo; hence this study may provide a clear insight of the deleterious changes in myocardium caused by ISP. Given the extensive knowledge of the mechanisms of ISP-induced myocardial injury, the model can also be used to investigate mechanisms of therapeutic action. Importantly, when the full knowledge of ISP-induced patho-mechanisms is applied to the testing of new compounds, we expect to obtain a realistic picture of the therapeutic potential of the therapeutic or preventive agents may offer protection and their underlying mechanisms. This information will be certainly critical before considering a potential clinical application of new drug entities.
References
- 1.Weir RA, McMurray JJ. Epidemiology of heart failure and left ventricular dysfunction after acute myocardial infarction. Curr Heart Fail Rep. 2006;3:175–180. doi: 10.1007/s11897-006-0019-5. [DOI] [PubMed] [Google Scholar]
- 2.Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655–673. doi: 10.1097/00004872-200018060-00002. [DOI] [PubMed] [Google Scholar]
- 3.Ojha SK, Nandave M, Arora S, Narang R, Dinda AK, Arya DS. Chronic administration of Tribulus terrestris Linn. extract improves cardiac function and attenuates myocardial infarction in rats. Int. J Pharmacol. 2008;4:1–10. [Google Scholar]
- 4.Mainzen Prince PS, Kumar R, Selvakumari CJ. Effects of gallic acid on brain lipid peroxide and lipid metabolism in streptozotocin-induced diabetic Wistar rats. J Biochem Mol Toxicol. 2011;25:101–107. doi: 10.1002/jbt.20365. [DOI] [PubMed] [Google Scholar]
- 5.Punithavathi VR, Shanmugapriya K, Mainzen Prince PS. Protective effects of rutin on mitochondrial damage in isoproterenol-induced cardiotoxic rats: an in vivo and in vitro study. Cardiovas Toxicol. 2010;10:181–189. doi: 10.1007/s12012-010-9077-8. [DOI] [PubMed] [Google Scholar]
- 6.Rona G, Chappel CI, Balazs T, Gaudry R. An infarct-like myocardial lesion and other toxic manifestations produced by isoproterenol in the rat. AMA Arch Pathol. 1959;67:443–445. [PubMed] [Google Scholar]
- 7.Beznak M. Hemodynamics during the acute phase of myocardial damage caused by isoproterenol. Can J Biochem Physiol. 1962;40:25–30. doi: 10.1139/o62-004. [DOI] [PubMed] [Google Scholar]
- 8.Shizukuda Y, Buttrick PM, Geenen DL, Borczuk AC, Kitsis RN, Sonnenblick EH. beta-adrenergic stimulation causes cardiocyte apoptosis: influence of tachycardia and hypertrophy. Am J Physiol. 1998;275:961–968. doi: 10.1152/ajpheart.1998.275.3.H961. [DOI] [PubMed] [Google Scholar]
- 9.Nirdlinger EL, Bramante PO. Subcellular myocardial ionic shifts and mitochondrial alterations in the course of isoproterenol-induced cardiopathy of the rat. J Mol Cell Cardiol. 1974;6:49–60. doi: 10.1016/0022-2828(74)90006-6. [DOI] [PubMed] [Google Scholar]
- 10.Li L, Zhang LK, Pang YZ, Pan CS, Qi YF, Chen L, et al. Cardioprotective effects of ghrelin and des-octanoyl ghrelin on myocardial injury induced by isoproterenol in rats. Acta Pharmacol Sin. 2006;27:527–535. doi: 10.1111/j.1745-7254.2006.00319.x. [DOI] [PubMed] [Google Scholar]
- 11.Goyal SN, Arora S, Sharma AK, Joshi S, Ray R, Bhatia J, et al. Preventive effect of crocin of Crocus sativus on hemodynamic, biochemical, histopathological and ultrastuctural alterations in isoproterenol-induced cardiotoxicity in rats. Phytomedicine. 2010;17:227–232. doi: 10.1016/j.phymed.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Mohanty IR, Arya DS, Gupta SK. Wthania somnifera provides cardioprotection and attenuates ischemia-reperfusion induced apoptosis. Clin Nutr. 2008;27:635–642. doi: 10.1016/j.clnu.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Yeager JC, Lams SG. The hemodynamics of isoproterenol induced cardiac failure in the rat. Circ Shock. 1981;8:151–163. [PubMed] [Google Scholar]
- 14.Satoh K. Serum lipid peroxide in cerebrovascular disorder determined by a new colorimetric method. Clin Chem Acta. 1978;90:37–43. doi: 10.1016/0009-8981(78)90081-5. [DOI] [PubMed] [Google Scholar]
- 15.Marklund S, Marklund G. Involvement of the superoxide anion radical in the autooxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem. 1974;47:469–474. doi: 10.1111/j.1432-1033.1974.tb03714.x. [DOI] [PubMed] [Google Scholar]
- 16.Nandi A, Chatterjee B. Assay of superoxide dismutase activity in animal tissues. J Biosci. 1988;13:305–315. doi: 10.1007/BF02712155. [DOI] [Google Scholar]
- 17.Beutler E, Duron O, Kelly BM. Improved method for the determination of blood glutathione. J Lab Clin Med. 1963;61:882–888. [PubMed] [Google Scholar]
- 18.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:01–493. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaffe AS, Landt Y, Parvin CA, Abendschein DR, Geltman EM, Ladenson JH, et al. Comparative sensitivity of cardiac troponin I and lactate dehydrogenase isoenzymes for diagnosing acute myocardial infarction. Clin Chem. 1996;42:1770–1776. [PubMed] [Google Scholar]
- 20.Priscilla DH, Prince PS. Cardioprotective effect of gallic acid on cardiac troponin-T, cardiac marker enzymes, lipid peroxidation products and antioxidants in experimentally induced myocardial infarction in Wistar rats. Chem Biol Interact. 2009;179:118–124. doi: 10.1016/j.cbi.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 21.Bertinchant JP, Robert E, Polge A, Marty-Double C, Fabbro-Peray P, Poirey S, et al. Comparison of the diagnostic value of cardiac troponin I and T determinations for detecting early myocardial damage and the relationship with histological findings after isoprenaline-induced cardiac injury in rats. Clin Chim Acta. 2000;298:13–28. doi: 10.1016/S0009-8981(00)00223-0. [DOI] [PubMed] [Google Scholar]
- 22.Wallace KB, Hausner E, Herman EH, Holt GD, MacGregor JT, Metz AL, et al. Serum troponin as biomarkers of drug-induced cardiac toxicity. Toxicol Pathol. 2004;32:106–121. doi: 10.1080/01926230490261302. [DOI] [PubMed] [Google Scholar]
- 23.Farvin KHS, Anandan R, Kumar SHS, Shiny KS, Mathew S, Sankar TV, et al. Cardioprotective effect of squalene on lipid profile in isoprenaline-induced myocardial infarction in rats. J Med Food. 2006;9:531–536. doi: 10.1089/jmf.2006.9.531. [DOI] [PubMed] [Google Scholar]
- 24.Kumar S, Seth S, Jaiswal A, Enjamoori R, Dinda AK, Ray R, et al. Chronic β-adrenergic activation-induced left ventricular systolic dysfunction is associated with systemic release of TNF-α and IL-1-β in rats. Pharmacol Rep. 2009;61:870–876. doi: 10.1016/S1734-1140(09)70143-4. [DOI] [PubMed] [Google Scholar]
- 25.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:4–594. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 26.Arslan F, Smeets MB, O’Neill LA, Keogh B, McGuirk P, Timmers L, et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation. 2010;121:80–90. doi: 10.1161/CIRCULATIONAHA.109.880187. [DOI] [PubMed] [Google Scholar]
- 27.Haunstetter A, Izumo S. Apoptosis: basic mechanism and implication for cardiovascular disease. Circ Res. 1998;82:1111–1129. doi: 10.1161/01.RES.82.11.1111. [DOI] [PubMed] [Google Scholar]
- 28.Haunstetter A, Izumo S. Future perspectives and potential implications of cardiac myocyte apoptosis. Cardiovas Res. 2000;45:795–801. doi: 10.1016/S0008-6363(99)00264-3. [DOI] [PubMed] [Google Scholar]
- 29.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 30.Narula J, Pandey P, Arbustini E, Haider N, Narula N, Kolodgie FD, et al. Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc Natl Acad Sci USA. 1999;96:8144–8149. doi: 10.1073/pnas.96.14.8144. [DOI] [PMC free article] [PubMed] [Google Scholar]