

Abstract
The aim of this work is to investigate penetration enhancers in proniosomes as a transdermal delivery system for nisoldipine. This was performed with the goal of optimising the composition of proniosomes as transdermal drug delivery systems. Plain proniosomes comprising sorbitan monostearate, cholesterol, ethanol and a small quantity of water were initially prepared. Subsequently, proniosomes containing lecithin or skin penetration enhancers were prepared and evaluated for transdermal delivery of nisoldipine. The plain proniosomes significantly enhanced the transdermal flux of nisoldipine to reach 12.18 μg cm−2 h−1 compared with a saturated aqueous drug solution which delivered the drug at a rate of 0.46 μg cm−2 h−1. Incorporation of lecithin into such proniosomes increased the drug flux to reach a value of 28.51 μg cm−2 h−1. This increase can be attributed to the penetration enhancing effect of lecithin fatty acid components. Replacing lecithin oleic acid (OA) produced proniosomes of comparable efficacy to the lecithin containing system. The transdermal drug flux increased further after incorporation of propylene glycol into the OA based proniosomes. Similarly, incorporation of isopropyl myristate into plain proniosomes increased drug flux. The study introduced enhanced proniosomes as a promising transdermal delivery carrier and highlighted the role of penetration enhancing mechanisms in enhanced proniosomal skin delivery. The study opened the way for another line of optimisation of niosome proconcentrates.
Keywords: Nisoldipine, Niosome proconcentrates, Oleic acid transdermal, Isopropyl myristate, Transdermal vesicles, Penetration enhancer
1. Introduction
The strategy of using lipid vesicles to improve drug delivery to and across the skin has gained interest. These vesicles included traditional liposomes (Mezei and Gulasekharam, 1980), transfersomes (ultradeformable vesicles), and ethosomes (Cevc and Blume, 1992; Touitou et al., 2000; El Maghraby et al., 2001). However, most of liposomes were reported to have stability problem and high cost. The stability problems of liposomes are in the form of loss of entrapped drug, change in the size upon storage as well as chemical degradation of the lipid components (Sharma and Sharma, 1997). Accordingly, niosomes which are surfactant based vesicles that are more stable (chemically) and less expensive than liposomes were introduced (Schreier and Bouwstra, 1994; Manosroi et al., 2008). However, although niosomes exhibit more chemical stability during storage, there may be a physical stability problem upon storage of niosome dispersion. Proniosomes were prepared as dry powder for reconstitution before use as a means of preserving the chemical and physical integrity of vesicles (Hu and Rhodes, 2000). For the transdermal delivery purpose proniosomes were prepared as gel like concentrated niosomes suitable for topical application (El Maghraby and Williams, 2009). These gel like structures have the advantage of being suitable for scaling up while maintaining the skin penetration enhancing abilities and better physicochemical stability. However, the published data on proniosomes are rare and are not systematic with many factors requiring further investigation. These factors include the effect of composition and the possibility of incorporating traditional skin penetration enhancers into proniosomes.
Accordingly, the main aim of this study is to investigate the effect of incorporation of skin penetration enhancers in proniosomes on the transdermal delivering ability of this system. To achieve this objective, nisoldipine a second-generation dihydropyridine calcium antagonist was selected as the test drug. This selection was based on the fact that the drug suffers from low and variable bioavailability after oral administration. This low bioavailability was attributed to the extensive first pass metabolism (Zannad, 1995; El Maghraby and Elsergany, 2014). This together with its high potency made the drug an excellent candidate for transdermal delivery. The physicochemical properties of nisoldipine were also considered in the selection of the drug candidate for this study. Nisoldipine has a molecular weight of 388.4 Daltons and is highly lipophilic as indicated from its high partition coefficient (log P = 3.63) (Marinkovic et al., 2003; El Maghraby and Elsergany, 2014). The molecular weight (below 500 Daltons) and lipophilicity provided nisoldipine with high potential for transdermal delivery. The lipophilic nature of the drug is another advantage for the current study due to expected high entrapment efficiency in proniosomes and other vesicular systems (El Maghraby et al., 2001). The study will thus provide dual functions as it will add a new factor in optimised transdermal delivery from provesicular systems and develop a method for enhanced nisoldipine transdermal delivery.
2. Materials and methods
2.1. Materials
Sorbitan monostearate (Span 60), cholesterol and lecithin were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Nisoldipine was purchased from Jinan Jianfeng Chemical Co. Ltd., China. Methanol, acetonitrile (HPLC – grade) and propylene glycol were obtained from BDH, England. Ethanol (96%) and oleic acid were from El-Nasr Pharmaceutical Chemicals Company, Egypt. Isopropyl myristate (IPM) was a gift from Sigma for Pharmaceutical Industries, Egypt.
2.2. Preparation of proniosomes
Table 1 presents the composition of the tested proniosome formulations. The surfactant mixture (Span 60 – cholesterol), the drug, ethanol and penetration enhancer (if any) were mixed and heated to 65 + 1 °C for 5 min in a stoppered vessel. This provided a clear liquid system. The aqueous phase was added and the mixture was warmed upto clarity. The mixture was allowed to cool down by continuous mixing at room temperature till the formation of a proniosome gel (Vora et al., 1998).
Table 1.
The composition of the tested formulations (expressed in grams per formulation) and the entrapment efficiency of the drug into proniosomes.
| Formulation | Surfactant system⁎ | Ethanol | Aqueous phase⁎⁎ | IPM | OA | PG | Entrapment efficiency (%) |
|---|---|---|---|---|---|---|---|
| P | 5 | 5 | 4 | – | – | – | 84.1 ± 0.3 |
| P/Lec. | 5 | 5 | 4 | – | – | – | 80.3 ± 0.3 |
| P/IPM | 5 | 5 | 4 | 1 | – | – | 87.7 ± 0.2 |
| P/OA | 5 | 5 | 4 | – | 1 | – | 88.2 ± 0.2 |
| P/OA-PG0.5 | 5 | 5 | 4 | – | 1 | 0.5 | 83.5 ± 1.5 |
| P/OA-PG1 | 5 | 5 | 4 | – | 1 | 1.0 | 84.5 ± 0.01 |
| P/OA-PG1.5 | 5 | 5 | 4 | – | 1 | 1.5 | 84.8 ± 0.2 |
The composition of the surfactant system in all formulations was Span 60 and cholesterol (4.5:0.5) except the formulation (P/lec) which contained Span 60, cholesterol and lecithin (3.5:0.5:1) respectively.
The aqueous phase was 0.1% glycerol in water. The amount of the drug added to each formulation was 0.3 g.
2.3. Determination of entrapment efficiency
To determine the entrapment efficiency of the drug into proniosomes, the formulation was hydrated to develop the corresponding niosome. This was achieved by hydrating the proniosome gel (1 g) using 10 ml distilled water with the aid of mechanical stirring for 30 min. The resulting niosomes were subjected to 30 min of bath sonication. Immediately after hydration of proniosomes, the niosome dispersion was incubated in a dialysis sac (Cellulose tubing, cut off 12,000 Daltons, Sigma diagnostics, St. Louis, MO, USA) and dialysed against 100 ml of 40% v/v ethanol in water for 4 h. This dialysis fluid was selected to ensure sink conditions. The amount of the drug found in the dialysate was taken as a measure of the free drug. The entrapment efficiency was calculated by the following equation (Trotta et al., 2002; Foco et al., 2005; Maestrelli et al., 2005):
where Ct is the total concentration of the drug and Cf is the concentration of the free drug.
2.4. Viscosity measurements
The flow behaviour and viscosity of the tested formulations were determined using a DV III rotating Brookfield viscometer using spindle RV-3 (Brookfield Engineering Laboratories Inc., Stoughton, MA, USA).
2.5. Determination of drug release
Drug release from vesicles is temperature dependent, generally being greatest around the phase transition temperature of the lipid (Papahadjopoulos et al., 1973). According to our experimental conditions the skin surface was maintained at 32 °C. Accordingly, the drug release studies were conducted at 32 °C to give a chance for correlation between drug release and skin permeation data.
The release study employed the vertical glass Franz diffusion cells which have a diffusional surface area of 2.27 cm2 with the receptor compartment of 14.5 ml volume. The dialysis membrane (Cellulose tubing, Sigma diagnostics, St. Louis, MO, USA) was soaked in distilled water overnight before cutting into suitable pieces. This soaking was conducted to ensure complete swelling of the membrane to provide a constant pore diameter throughout the experiment. The membrane was then mounted between the donor and receptor compartments before filling the receptor compartment with 40% v/v ethanol in water. This receptor fluid was selected to maintain sink conditions which was confirmed by recording linear release profiles which did not tail off (see the results). The diffusion cells were incubated into a thermostatically controlled water bath with its temperature being adjusted to maintain the temperature of the membrane surface at 32 + 1 °C to mimic in vivo conditions. Proniosomes (2.5 g) were loaded into the donor compartments. Receptor samples were taken at different time intervals and analysed for the drug by HPLC. These samples were replaced with fresh receptor. The cumulative amount of drug released was calculated as a function of time (El Maghraby, 2010).
2.6. Preparation of skin samples
Ideally skin permeation experiments should employ human skin. Unfortunately, it is difficult to obtain good human skin samples. Accordingly, the rabbit ear model which has been extensively used for the investigation of transdermal delivery of a variety of lipophilic drugs like our drug was employed in this study (Corbo et al., 1990; Touitou et al., 2000; El Maghraby, 2012). Full thickness skin obtained from the inner side of freshly excised ears of 8 male rabbits (weighing 2–3 kg) was used. The skin was peeled from the underlying cartilage after cutting along the tips of ears. The skin samples were mounted immediately on the diffusion cells (El Maghraby, 2008).
2.7. Skin permeation studies
As for the release studies, the vertical glass Franz diffusion cells were employed in the skin permeation experiments. The skin was mounted with the stratum corneal side uppermost on the diffusion cells. To ensure sink conditions, 40% v/v ethanol in water was used as receptor. The diffusion cells were incubated into a thermostatically controlled water bath with its temperature being adjusted to maintain the temperature of the skin surface at 32 + 1 °C to mimic vivo conditions. After overnight equilibration, the tested formulation of proniosomes (2.5 g) was loaded into the donor compartments which were occluded with aluminium foil. Receptor samples were taken at different time intervals and analysed for the drug by HPLC. These samples were replaced with fresh receptor. The study ensured that skin obtained from the same rabbit was used for the test and control (El Maghraby, 2010).
2.8. Chromatography
The drug concentrations in all samples were determined using an HPLC analysis. This employed a high pressure liquid chromatograph (Waters™ 600 controller, USA) equipped with a variable wavelength detector (Waters™ 486, Tunable Absorbance Detector, USA) and an automatic sampling system (Waters™ 717 Plus Autosampler, USA). This was under computer control. Separation was accomplished on a reversed phase column 15 cm × 4.6 mm (i.d.) C18, μ Bondapak™, Waters, with an average particle size of 10 μm.
The mobile phase was a mixture of methanol, acetonitrile and water (35:30:35) flowing at 1.3 ml/min. The column effluent was monitored at 238 nm (El Maghraby and Elsergany, 2014). The chromatographic data analysis was performed with the Millinium™ Programme (Waters, USA). The receptor samples (30 μl) were injected directly into the HPLC system.
2.9. Data analysis
The cumulative amounts of the drug permeated were plotted as a function of time to produce the permeation profiles. The plots were typical steady state profiles which are expected after occlusive application of formulations containing excess amounts of the drug (Fig. 1). These profiles were used to calculate the transdermal drug flux (J), which was obtained from the slope of the regression line fitted to the linear portion of the profile. Extrapolation of this line will intercept with the x axis at a time equal to the lag time.
Figure 1.
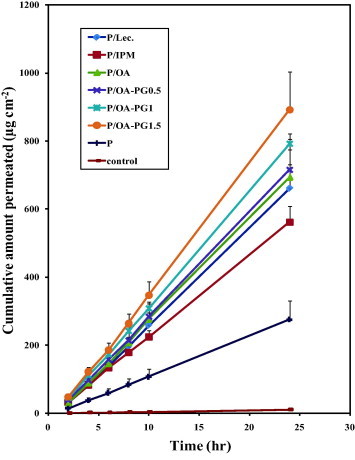
The transdermal permeation profiles of nisoldipine after the application of different proniosome formulations to rabbit skin in vitro. The control was an saturated aqueous solution of the drug. Formulation details are shown in Table 1.
2.10. Statistical analysis
Students’ t-test was used to test for significance.
3. Results
3.1. Entrapment efficiency
The recorded entrapment efficiency values are presented in Table 1. The entrapment efficiency ranged from 80.3 + 0.3% to 88.2 + 0.2%. With regard to the entrapment efficiency of individual formulations, the plain proniosome formulation (P) entrapped 84.1% of the added drug. This value was increased further after incorporation of oleic acid or IPM into the formulations. In contrast, incorporation of lecithin resulted in a reduction of the entrapment efficiency of the drug into proniosomes compared to that of the plain proniosomes. Addition of propylene glycol with oleic acid reduced the entrapment efficiency to become comparable to that of the standard proniosomes (P).
3.2. Viscosity determinations
The flow behaviour of the tested formulations followed a non-Newtonian system with a shear thinning behaviour. This shear thinning behaviour is shown in Fig. 2 which reveals a reduction in the viscosity by increasing the shear rate. Having a non-Newtonian flow, the viscosity was calculated at a fixed rpm (50 rpm). The recorded viscosity values are presented in Table 2. The plain proniosomes were the most viscous with incorporation of penetration enhancers into proniosomes reducing the viscosity. Surprisingly, addition of propylene glycol to oleic acid containing the formulation decreased the viscosity reducing effect of oleic acid but the viscosity of the resulting formulation remained lower than the plain formulation.
Figure 2.
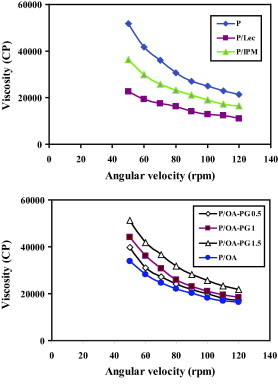
The viscosity of the tested proniosome formulations as a function of the angular velocity. Formulation details are shown in Table 1.
Table 2.
Characteristics, transdermal permeation parameters and release rate of nisoldipine obtained after the application of nisoldipine in the form of proniosome formulations and saturated aqueous solution (control).
| Drug formulation | Viscosity (cp) | Release rate (μg cm−2 h−1) | Flux (μg cm−2 h−1) | Lag time (h) |
|---|---|---|---|---|
| P | 51,800 (1633) | 15.3 (1.12) | 12.18 (2.32) | 0.99 (0.11) |
| P/lec | 22,533 (467) | 45.01 (3.28) | 28.51 (2.83) | 0.94 (0.36) |
| P/IPM | 36,400 (1562) | 29.1 (1.94) | 23.97 (1.92) | 0.59 (0.07) |
| P/OA | 33,733 (929) | 34.3 (3.82) | 30.21 (3.46) | 0.93 (0.37) |
| P/OA-PG0.5 | 39,650 (4854) | 43.2 (1.16) | 30.86 (2.94) | 0.97 (0.43) |
| P/OA-PG1 | 44,067 (1023) | 42.02 (1.98) | 34.08 (1.35) | 0.84 (0.17) |
| P/OA-PG1.5 | 51,733 (2034) | 41.19 (2.97) | 38.67 (5.07) | 0.92 (0.21) |
| Control | ND | 9.7 (0.8) | 0.46 (0.03) | 1.7 (0.1) |
Values between brackets are S.D. (n = 3). Formulation details are shown in Table 1.
3.3. Drug release
The release profiles of nisoldipine obtained from different proniosomes are shown in Fig. 3. The recorded release profiles were linear with lag time in all cases. The calculated release rates are presented in Table 2. All proniosomes revealed significantly higher release rates compared with the saturated aqueous solution of the drug. With respect to the effect of composition of proniosomes on the drug release, incorporation of various penetration enhancers resulted in a significant increase in the rate of nisoldipine release (P < 0.05) compared with the plain formulation. Lecithin containing formulation showed the highest release rate.
Figure 3.
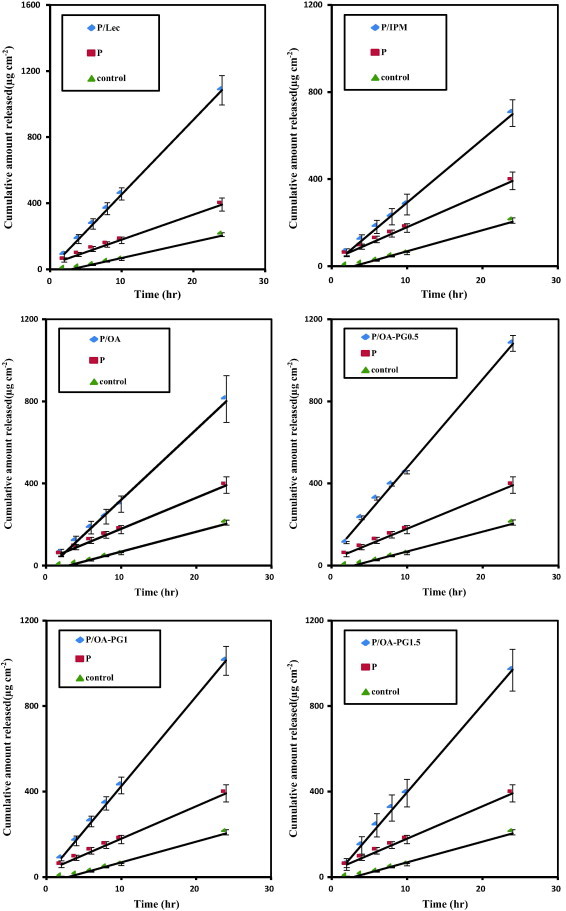
The in vitro release profiles of nisoldipine from different proniosome formulations. The control was an saturated aqueous solution of the drug. Formulation details are shown in Table 1.
3.4. Proniosomal skin delivery of nisoldipine
The transdermal permeation profiles of nisoldipine from different proniosomes formulations are shown in Fig. 1. These profiles were typical steady-state profiles with a lag time which is expected after occlusive application of formulations containing the drug at saturation with excess drug to maintain saturation. The calculated permeation parameters are presented in Table 2. All proniosomal formulations delivered the drug through the skin at a significantly higher rate compared to the saturated aqueous control. This is clear from the recorded transdermal flux values which were significantly higher in the case of proniosomes. The increase in transdermal flux was associated with a trend of reduction in the lag time (Table 2). The trend of lag time reduction after proniosomal application in the current study indicates increased diffusivity and supports skin penetration enhancement of components. Incorporation of lecithin into the proniosomes resulted in a significant increase in transdermal drug flux compared to the plain formulation (P < 0.05).
With respect to the effect of penetration enhancers, incorporation of oleic acid into proniosomes resulted in significant increase in the transdermal flux of nisoldipine compared with the plain proniosomes (P < 0.05). The efficacy of the oleic acid containing system was comparable to that of the lecithin based formulation (Table 2). The efficacy of oleic acid based proniosomes became even greater after incorporation of propylene glycol into proniosomes. This is clear after comparing the transdermal drug flux of oleic acid based proniosomes in the presence and absence of increasing concentrations of propylene glycol (Table 2). The efficacy of oleic acid/propylene glycol combination was even better than that of lecithin after incorporation into proniosomes.
As for oleic acid, incorporation of IPM into proniosomes resulted in a significant increase in the transdermal delivery efficiency compared with the corresponding plain proniosomes. However, the efficacy of the IPM based system was lower than that of lecithin or oleic acid containing proniosomes.
4. Discussion
The recorded entrapment efficiency values indicate that the amounts of drug added to proniosomes was enough to saturate the proniosome formulations. This will ensure equal thermodynamic activity among the tested proniosomes. This is necessary to ensure that any difference in transdermal drug delivery efficiency will depend on the composition of proniosomes. The high entrapment efficiency of nisoldipine in plain proniosomes (P) is expected for such a lipophilic drug. Similar entrapment efficiency patterns were recorded for lipophilic drugs like levonorgesterel, estradiol and tenoxicam (Vora et al., 1998; Fang et al., 2001; Ammar et al., 2011). Incorporation of oleic acid or IPM into proniosomes can add to the lipophilic domains of proniosomes. Taking this into consideration with the high solubility of nisoldipine in oleic acid and IPM (data not shown), the increase in entrapment efficiency of the drug in proniosomes can be explained. Incorporation of lecithin resulted in a reduction of the entrapment efficiency of the drug into proniosomes. This may suggest low solubility of the drug in lecithin. Another possible explanation can be based on positioning of lecithin and oleic acid into the vesicular structure. The former is known to intercalate in the lipid domains between Span and cholesterol but the later associates forming a pole within the vesicular structure (El Maghraby et al., 2004). Based on this arrangement lecithin can compete with the drug for a location within the vesicular membrane but the association of oleic acid molecules could provide islets into which more drug can be localised. It is important to note that the addition of propylene glycol with oleic acid reduced the entrapment efficiency to become comparable to that of the standard proniosomes.
The shear thinning flow behaviour of proniosomes (Fig. 2) is an advantage as it will allow easy application and spreading on the skin surface. The reduction in the viscosity of proniosomes after incorporation of penetration enhancers can be explained on the base of the fluidizing effect of the enhancers on the lamellar structure of proniosomes (El Maghraby et al., 2004).
The in vitro release of nisoldipine from various proniosomal formulations was studied at the same skin permeation experimental conditions but the skin was replaced with artificial semi-permeable membrane. This was conducted so as to correlate the release results with the skin permeation data (El Maghraby, 2010). The recorded linear release after a lag time (Fig. 3) is similar to previously recorded patterns for proniosomes containing tenoxicam which is a lipophilic drug like nisoldipine (Ammar et al., 2011). The recorded higher release rates of nisoldipine from proniosomes compared to saturated aqueous solution can be attributed to higher drug concentrations in proniosomal formulations compared with aqueous solution due to very poor solubility of such lipophilic drugs in water. This will in turn result in a higher concentration gradient in the case of proniosomes which is the main driving force in passive diffusion through semi-permeable membranes. With respect to the effect of composition of proniosomes on drug release, the recorded increase in the release rate after incorporation of various penetration enhancers can be attributed to the fluidizing effect of the enhancers on the provesicular system. The release rate generally correlated with the viscosity data with the most viscous formulation producing the slowest release. This pattern was not the same in the presence of propylene glycol which resulted in a higher release rate compared to the oleic acid containing formulation which was less viscous (Table 2). It is important to highlight the inverse relationship between the entrapment efficiency and the drug release. Taking the entrapment efficiency as a measure of the formulation ability to retain the drug, high entrapment efficiency should theoretically suggest slower release rate (El-Laithy et al., 2011).
Ethical requirements and lack of good human skin samples forced researchers to use other skin models (El Maghraby et al., 2008). Accordingly, full thickness skin obtained from the inner side of freshly excised rabbit ears was utilised in the current study. This skin has been successfully used in various transdermal permeation studies of lipophilic drugs like nisoldipine (Touitou et al., 2000; El Maghraby et al., 2009). The receptor fluid was 40% (v/v) ethanol in water to ensure sink conditions. Such a receptor has been successfully employed as a receptor to monitor skin delivery of a lipophilic drug from vesicular delivery systems (El Maghraby et al., 1999).
Successful transdermal delivery from proniosomes has been reported in the literature with proniosomes comprising Spans with cholesterol and ethanol. Those authors investigated the potential skin delivery of flurbiprofen from proniosomes and their corresponding vesicles. They investigated the effect of the type of Span and revealed a dependence of the release rate and diffusion rate of the drug on the type of such surfactant (Mokhtar et al., 2008; Ibrahim et al., 2008). Other authors included lecithin into proniosomes and recorded successful transdermal delivery of a variety of drugs from these proniosomes. These investigations revealed the success of these systems in enhanced transdermal delivery of levonorgesterol, estradiol and piroxicam (Vora et al., 1998; Fang et al., 2001; Chandra and Sharma, 2008). However, none of these articles provided direct comparison between lecithin free and lecithin containing proniosomes using the same drug. Alternative mechanisms have been suggested for the enhanced transdermal drug delivery from proniosomes. The earliest one depended on the possibility that proniosomes can form niosomes after mixing with skin secretion in vivo or mixing with the back diffusing receptor fluid in vitro (Vora et al., 1998). Accepting this hypothesis, the mechanism of action of proniosomes can be similar to niosomes which can enhance the drug by adsorption on or adhesion to the skin surface creating high thermodynamic activity with subsequent drug transfer. Another possible reason for niosome enhanced transdermal delivery can depend on the penetration enhancing effect of the vesicle components (Schreier and Bouwstra, 1994; Uchegbu and Vyas, 1998). Later on, the hypothesis of transformation of proniosomes to niosomes after topical application was rejected (Fang et al., 2001). This rejection was based on recording the drug in the receptor in the first 30 min after application. Authors considered this as a short time for niosome formation and subsequent drug transfer. In addition the hydrated proniosomes (niosomes) were less efficient compared to the corresponding proniosomes. Authors suggested direct drug transfer from proniosomes with a role for the penetration enhancing effect of the non-ionic surfactant component of such systems (Fang et al., 2001). However, studies comparing niosomes with proniosomes, tested dilute niosomes, a process which may not mimic the actual situation where proniosomes form niosomes after topical application. Accordingly, the potential of proniosomes transformation into niosomes after application to the skin cannot be rejected completely before constructing an experiment comparing different dilutions of proniosomes with respect to transdermal drug delivery.
The recorded superiority of lecithin containing proniosomes can be explained on the basis of the ability of lecithin to mix with skin lipids creating a more permeable environment and adding to the penetration enhancing effect of proniosome components. Mixing of lecithin with skin is believed to be better in the presence of ethanol which is a component of proniosomes (Junginger et al., 1991). Additionally, soya lecithin has been reported to contain unsaturated fatty acids like oleic acid and linoleic acid which have penetration enhancing properties (Rita and Lakshmi, 2012). Accordingly, the superiority of lecithin containing proniosomes can be explained on the basis of the fluidizing effect of these fatty acids on the vesicular membrane (El Maghraby et al., 2004). This explanation is suggested here taking into consideration that proniosomes have to form niosomes after mixing with skin secretions or back diffusing receptor fluid before delivering the drug into and through the skin. Another possible explanation for the superiority of lecithin containing proniosomes can depend on the direct skin penetration enhancing effect of the unsaturated fatty acid components of lecithin. Both explanations considered the importance of the fatty acid components of lecithin in enhanced transdermal delivery from proniosomes. To verify this importance other proniosomes formulations were devised with lecithin being replaced by pure skin penetration enhancers. Oleic acid was used for this purpose both in the presence and absence of propylene glycol which is known to provide a synergistic penetration enhancing effect with oleic acid (Barry, 1987; Larrucea et al., 2001;). In addition, proniosomes containing IPM, a well known fatty acid ester which can intervene with the lipid lamellae making them more permeable (Gorukanti et al., 1999; El Maghraby et al., 2009), were prepared and tested for the same purpose.
With respect to the effect of penetration enhancers, the efficacy of the oleic acid containing system was comparable to that of the lecithin based formulation (Table 2). This suggests that the enhancing effect of lecithin can be due to its fatty acid components. The efficacy of oleic acid based proniosomes was enhanced further after incorporation of propylene glycol into proniosomes. This finding implies a synergistic effect for propylene glycol with oleic acid even after incorporation into proniosomes. Oleic acid is believed to intercalate between the lipid bilayer of the intercellular lipid lamellae of the stratum corneum leading to a more loose structure. This effect is believed to be improved in the presence of propylene glycol which allows for easy penetration of oleic acid into the skin (Barry, 1987; Larrucea et al., 2001;). This highlights the direct penetration enhancing effect of the proniosome components as an important factor in the superiority of enhancer containing proniosomes compared with plain ones. However, further mechanistic investigations are required to confirm this hypothesis. Incorporation of IPM into proniosomes resulted in a similar trend with the resulting provesicles being superior to the corresponding plain proniosomes. However, the efficacy of IPM based system was lower than that of lecithin or oleic acid containing proniosomes. This implies the need for further optimisation of the IPM based system with respect to the enhancer concentration. Finally, it is important to consider the penetration enhancing effect of ethanol which was employed as an integral component of proniosomes. Ethanol is believed to increase the diffusivity of drugs through the stratum corneum (Barry, 1987). Great success was recorded for ethanol when incorporated in a similar vesicular system (ethothomes) and it was considered to act as skin enhancer by itself with a possible fluidizing effect on the vesicular component. The latter effect is considered important for vesicular transdermal drug delivery (Touitou et al., 2000). The success of ethanol was also recorded for enhanced transdermal drug delivery from microemulsions (El Maghraby, 2008).
5. Conclusion
Proniosomes are promising transdermal drug delivery systems with their efficacy being dependent on their composition. Proper selection of the proniosome components may lead to better utilisation of such formulations. Penetration enhancer containing proniosomes were better than the corresponding plain formulation. The mechanism(s) of enhanced transdermal delivery from proniosomes requires further investigations although the recorded data and the effect of composition suggested the possibility for combined mechanisms. The article thus provided a new composition for transdermal proniosomes and opened gates for future research on optimisation of the composition together with mechanistic investigations.
Footnotes
Peer review under responsibility of King Saud University.

Contributor Information
Gamal M. El Maghraby, Email: gmmelmag@yahoo.com.
Amal A. Ahmed, Email: amal_aahmed2009@yahoo.com.
Mohamed A. Osman, Email: mosman444@yahoo.com.
References
- Ammar H.O., Ghorab M., El-Nahhas S.A., Higazy I.M. Proniosomes as a carrier system for transdermal delivery of tenoxicam. Int. J. Pharm. 2011;405:142–152. doi: 10.1016/j.ijpharm.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Barry B.W. Mode of action of penetration enhancers in human skin. J. Control Rel. 1987;6:85–97. [Google Scholar]
- Cevc G., Blume G. Lipid vesicles penetrate into intact skin owing to the transdermal osmotic gradients and hydration force. Biochem. Biophys. Acta. 1992;1104:226–232. doi: 10.1016/0005-2736(92)90154-e. [DOI] [PubMed] [Google Scholar]
- Chandra A., Sharma P.K. Proniosome based drug delivery system of peroxicam. Afr. J. Pharm. Phamacol. 2008;2(9):184–190. [Google Scholar]
- Corbo M., Liu J.C., Chien Y.W. Bioavailability of propranolol following oral and transdermal administration in rabbits. J. Pharm. Sci. 1990;79(7):584–587. doi: 10.1002/jps.2600790707. [DOI] [PubMed] [Google Scholar]
- El Maghraby G.M. Transdermal delivery of hydrocortisone from eucalyptus oil microemulsion: effects of cosurfactant. Int. J. Pharm. 2008;355:285–292. doi: 10.1016/j.ijpharm.2007.12.022. [DOI] [PubMed] [Google Scholar]
- El Maghraby G.M. Self microemulsifying and microemulsion systems for transdermal delivery of indomethacin: effect of phase transition. Colloids Surf., B. 2010;75:595–600. doi: 10.1016/j.colsurfb.2009.10.003. [DOI] [PubMed] [Google Scholar]
- El Maghraby G.M. Occlusive and non-occlusive application of microemulsion for transdermal delivery of progesterone: mechanistic studies. Sci. Pharm. 2012;80:765–778. doi: 10.3797/scipharm.1201-01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Maghraby G.M., Elsergany R.N. Fast disintegrating tablets of nisoldipine for intra-oral administration. Pharm. Dev. Technol. 2014;19(6):641–650. doi: 10.3109/10837450.2013.813543. [DOI] [PubMed] [Google Scholar]
- El Maghraby G.M., Williams A.C. Vesicular systems for delivering conventional small organic molecules and larger macromolecules to and through human skin. Expert Opin. Drug Deliv. 2009;6(2):149–163. doi: 10.1517/17425240802691059. [DOI] [PubMed] [Google Scholar]
- El Maghraby G.M.M., Williams A.C., Barry B.W. Skin delivery of oestradiol from deformable and traditional liposomes: mechanistic studies. J. Pharm. Pharmacol. 1999;51:1123–1134. doi: 10.1211/0022357991776813. [DOI] [PubMed] [Google Scholar]
- El Maghraby G.M.M., Williams A.C., Barry B.W. Skin delivery of 5-fluorouracil from ultradeformable and traditional liposomes in vitro. J. Pharm. Pharmacol. 2001;53:1069–1077. doi: 10.1211/0022357011776450. [DOI] [PubMed] [Google Scholar]
- El Maghraby G.M.M., Williams A.C., Barry B.W. Interactions of surfactants (edge activators) and skin penetration enhancers with liposomes. Int. J. Pharm. 2004;276:143–161. doi: 10.1016/j.ijpharm.2004.02.024. [DOI] [PubMed] [Google Scholar]
- El Maghraby G.M., Barry B.W., Williams A.C. Liposomes and skin: from drug delivery to model membranes. Eur. J. Pharm. Sci. 2008;34:203–222. doi: 10.1016/j.ejps.2008.05.002. [DOI] [PubMed] [Google Scholar]
- El Maghraby G.M., Alanazi F.K., Alsarra I.A. Transdermal delivery of tadalafil. I. Effect of vehicles on skin permeation. Drug Dev. Ind. Pharm. 2009;35:329–336. doi: 10.1080/03639040802360494. [DOI] [PubMed] [Google Scholar]
- El-Laithy H.M., Shoukry O., Mahran L.G. Novel sugar esters proniosomes for transdermal delivery of vinpocetine: preclinical and clinical studies. Eur. J. Pharm. Biopharm. 2011;77:43–55. doi: 10.1016/j.ejpb.2010.10.011. [DOI] [PubMed] [Google Scholar]
- Fang J.Y., Yu S.Y., Wu P.C., Huang Y.B., Tsai Y.H. In vitro skin permeation of estradiol from various proniosome formulations. Int. J. Pharm. 2001;215:91–99. doi: 10.1016/s0378-5173(00)00669-4. [DOI] [PubMed] [Google Scholar]
- Foco A., Gasperlin M., Kristl J. Investigation of liposomes as carriers of sodium ascorbyl phosphate for cutaneous photoprotection. Int. J. Pharm. 2005;291:21–29. doi: 10.1016/j.ijpharm.2004.07.039. [DOI] [PubMed] [Google Scholar]
- Gorukanti S.R., Li L., Kim K.H. Transdermal delivery of antiparkinsonian agent, benzotropine. I. Effect of vehicles on skin permeation. Int. J. Pharm. 1999;192:159–172. doi: 10.1016/s0378-5173(99)00305-1. [DOI] [PubMed] [Google Scholar]
- Hu C., Rhodes D.G. Proniosomes: A novel drug carrier preparation. Int. J. Pharm. 2000;206:110–122. [PubMed] [Google Scholar]
- Ibrahim M.M., Sammour O.A., Hammad M.A., Megrab N.A. In vitro evaluation of proniosomes as a drug carrier for flurbiprofen. AAPS PharmSciTech. 2008;9:782–790. doi: 10.1208/s12249-008-9114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junginger H.E., Hofland H.E.J., Bouwstra J.A. Liposomes and niosomes: interaction with human skin. Cosmet. Toil. 1991;106:45–50. [Google Scholar]
- Larrucea E., Arellano A., Santoyo S., Ygartua P. Combined effect of oleic acid and propylene glycol on the percutaneous penetration of tenoxicam and its retention in the skin. Eur. J. Pharm. Biopharm. 2001;52:113–119. doi: 10.1016/s0939-6411(01)00158-8. [DOI] [PubMed] [Google Scholar]
- Maestrelli F., Gonzalez-Rodrıguez M.L., Rabasco A.M., Mura P. Preparation and characterisation of liposomes encapsulating ketoprofen–cyclodextrin complexes for transdermal drug delivery. Int. J. Pharm. 2005;298:55–67. doi: 10.1016/j.ijpharm.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Manosroi A., Jantrawut P., Manosroi J. Anti-inflammatory activity of gel containing novel elastic niosomes entrapped with diclofenac diethylammonium. Int. J. Pharm. 2008;360:156–163. doi: 10.1016/j.ijpharm.2008.04.033. [DOI] [PubMed] [Google Scholar]
- Marinkovic V.D., Agbaba D., Karljikovic-Rajic K., Vladimirov S., Nedeljkovic J.M. Photochemical degradation of solid-state nisoldipine monitored by HPLC. J. Pharm. Biomed. Anal. 2003;32:929–935. doi: 10.1016/s0731-7085(03)00194-8. [DOI] [PubMed] [Google Scholar]
- Mezei M., Gulasekharam V. Liposomes-a selective drug delivery system for topical route of administration. 1. Lotion dosage form. Life Sci. 1980;26:1473–1477. doi: 10.1016/0024-3205(80)90268-4. [DOI] [PubMed] [Google Scholar]
- Mokhtar M., Sammour O.A., Hammad M.A., Megarb N.A. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int. J. Pharm. 2008;361:104–111. doi: 10.1016/j.ijpharm.2008.05.031. [DOI] [PubMed] [Google Scholar]
- Papahadjopoulos D., Jacobson K., Nir S., Isac T. Phase transition in phospholipid vesicles: fluorescence polarization and permeability measurements concerning the effect of temperature and cholesterol. Biochim. Biophys. Acta. 1973;311:330–348. doi: 10.1016/0005-2736(73)90314-3. [DOI] [PubMed] [Google Scholar]
- Rita B., Lakshmi P.K. Preparation and evaluation of modified proniosomal gel for localised urticaria and optimisation by statistical method. J. App. Pharm. Sci. 2012;2(3):85–91. [Google Scholar]
- Schreier H., Bouwstra J. Liposomes and niosomes as topical drug carriers: dermal and transdermal drug delivery. J. Control. Rel. 1994;30:1–15. [Google Scholar]
- Sharma A., Sharma U.S. Liposomes in drug delivery: progress and limitations. Int. J. Pharm. 1997;154:123–140. [Google Scholar]
- Touitou E., Dayan N., Bergelson L., Godin B., Eliaz M. Ethosomes – novel vesicular carrier for enhanced delivery: characterization and skin penetration properties. J. Control. Rel. 2000;65:403–418. doi: 10.1016/s0168-3659(99)00222-9. [DOI] [PubMed] [Google Scholar]
- Trotta M., Peira E., Debernardi F., Gallarate M. Elastic liposomes for skin delivery of dipotassium glycyrrhizinate. Int. J. Pharm. 2002;241:319–327. doi: 10.1016/s0378-5173(02)00266-1. [DOI] [PubMed] [Google Scholar]
- Uchegbu I.F., Vyas S.P. Non-ionic surfactant based vesicles (niosomes) in drug delivery. Int. J. Pharm. 1998;172:33–70. [Google Scholar]
- Vora B., Khopade A.J., Jain N.K. Proniosome based transdermal delivery of levonorgesterol for effective contraception. J. Control. Rel. 1998;54:149–165. doi: 10.1016/s0168-3659(97)00100-4. [DOI] [PubMed] [Google Scholar]
- Zannad F. Clinical pharmacology of nisoldipine coat core. Am. J. Cardiol. 1995;75:41E–45E. doi: 10.1016/s0002-9149(99)80447-0. [DOI] [PubMed] [Google Scholar]