

Abstract
Wilms tumor (WT) blastema retains gene expression profiles characteristic of the multipotent nephron progenitor pool, or cap mesenchyme (CM), in the developing kidney. As a result, WT blastema and the CM are believed to represent contextual analogues of one another. Sine oculis homeobox 2 (SIX2) is a transcription factor expressed specifically in the CM, provides a critical mechanism for CM self-renewal, and remains persistently active in WT blastema, although its purpose in this childhood malignancy remains unclear. We hypothesized that SIX2, analogous to its function in development, confers a survival pathway to blastema, the putative WT stem cell. To test its functional significance in WT biology, wild-type SIX2 was overexpressed in the human WT cell line, WiT49. After validating this model, SIX2 effects on anchorage-independent growth, proliferation, invasiveness, canonical WNT pathway signaling, and gene expression of specific WNT pathway participants were evaluated. Relative to controls, WiT49 cells overexpressing SIX2 showed significantly enhanced anchorage-independent growth and early-passage proliferation representing surrogates of cell survival. Interestingly, overexpression of SIX2 generally repressed TCF/LEF-dependent canonical WNT signaling, which activates and coordinates both differentiation and stem pathways, but significantly heightened canonical WNT signaling through the survivin promoter, a mechanism that exclusively maintains the stem state. In summary, when overexpressed in a human WT cell line, SIX2 enhances cell survival and appears to shift the balance in WNT/β-catenin signaling away from a differentiation path and toward a stem cell survival path.
Introduction
Wilms tumor (WT), the most common childhood kidney cancer, retains gene expression profiles and histologic elements characteristic of the embryonic kidney and so is classified among embryonal tumors [1], [2], [3]. Typically, WTs show a triphasic pattern of cellular features, comprised principally of 1) blastema, its putative cancer stem cell and the malignant analogue of nephron progenitors, 2) epithelia, which appears more differentiated as primitive tubules and glomeruli but lacks physiologic function and tissue architecture, and 3) stroma, which consists mostly of connective tissue fibroblasts but can show smooth or skeletal muscle, and even cartilaginous, differentiation [4], [5]. A predominant pattern of blastema, particularly if persisting after neoadjuvant therapy, represents a histologic marker of treatment resistance and has been shown to portend a worse prognosis [6], [7]. The 2013 Children’s Oncology Group blueprint for renal tumors therefore challenges investigators to identify the mechanisms that maintain blastema interminably and confer treatment resistance to WT as targets for more efficacious drugs, answers to which likely rest in the mysteries of cancer stem cell self-renewal and evasion of standard therapies [8].
Sine oculis homeobox 2 (SIX2) gene encodes a transcription factor that provides a necessary self-renewal mechanism to nephron progenitors residing within the cap mesenchyme (CM) of the embryonic kidney and that protects against canonical WNT pathway signals directing epithelial differentiation [9], [10], [11]. Interestingly, coordination of asymmetric stem cell division in the developing kidney, which spawns one daughter cell fated to become nephronic epithelia and the other to remain in the stem state, depends in part on integrity and balance of canonical WNT signaling. More specifically, this β-catenin–dependent cell fate decision, either to differentiate or to remain stem, relies on its interaction with two co-activators, CREB-binding protein (CBP), which directly promotes stemness, or p300, which directs epithelial conversion [12], [13], [14]. If the balance in this tightly regulated binary branch point shifts to a greater symmetric course of cell division, whereby two daughter stem cells are spawned, a state of interminable self-perpetuation is created. CBP/β-catenin–dependent symmetric cell division, which sets up a perpetual loop, has been proposed as a self-maintenance mechanism of the cancer stem cell [15]. How SIX2 and β-catenin interact in the highly regulated coordination of CM asymmetric cell division is incompletely understood; moreover, how this balance shifts to symmetric cell division in the cancer stem cell remains more elusive yet represents a candidate target of new therapies [16], [17], [18].
To coordinate the critical balance between maintaining a sufficient nephron progenitor pool while simultaneously spawning committed epithelial cells within the CM of the murine embryonic kidney, SIX2 and β-catenin have been shown to share regulatory gene networks, and a tight interplay has been observed between SIX2 and Wnt9b [11], [19]. Curiously, both SIX2 and β-catenin are broadly activated in WT, which represents an ideal paradigm to study self-renewal of an embryonal cancer stem cell and its potential for epithelial conversion, given the typical appearance of WT blastema immediately adjacent to or surrounding a variety of epithelial structures [20], [21], [22]. Although much has been revealed regarding the function of SIX2 and canonical WNT signaling in the coordinated process of nephron development, insufficient evidence has been uncovered regarding these transcriptional regulators in blastema self-renewal and maintenance of the WT cancer stem cell. Analogous to its functional significance in the CM, we hypothesized therefore that exuberant SIX2 expression confers a survival mechanism to WT and preferentially drives β-catenin toward the CBP-dependent arm of the canonical WNT pathway to maintain the stem state. These studies were designed to test SIX2 as a survival mechanism and a modifier of the canonical WNT pathway in the WT context.
Methods
SIX2 Cellular Distribution in WT
WT clinical specimens
To evaluate SIX2 as a marker of the putative WT stem cell, or blastema, we conducted a comprehensive immunohistochemical (IHC) analysis of its cellular distribution in relation to neural cell adhesion molecule (NCAM, a cell surface marker of the WT stem cell) [23], [24]. Because SIX2 provides self-renewal to the CM, we also examined SIX2 as a marker of proliferation by evaluating its association with proliferating cell nuclear antigen (PCNA, a marker of proliferating cells) expression across a large sampling of WT specimens. Briefly, using formalin-fixed paraffin-embedded renal tumor and adjacent kidney specimens collected prospectively and archived in our Institutional Review Board (IRB)-approved laboratory embryonal tumor repository, we created three tissue microarrays (TMA) comprised of 223 total punches (~ 1 mm in diameter each). Two hundred and fourteen of these punches were derived from 43 consecutive childhood renal tumors (41 WTs, 1 clear cell sarcoma, and 1 mesoblastic nephroma), and 9 punches originated from 9 discarded human fetal kidney specimens acquired from therapeutic abortuses. To ensure the highest punch quality and an adequate sampling of a given kidney tumor specimen, multiple punches were selected and taken from different blocks having histologic regions of interest. Serial 5-μm sections of these three TMAs were included for the IHC analysis, which was concentrated on the 41 WT specimens (39 favorable and 2 unfavorable histology).
Immunohistochemistry
Using our previously described protocol for peroxidase-based IHC analysis, serial sections of the three TMAs were probed, respectively, for SIX2, NCAM, and PCNA [21]. Briefly, the paraffin blocks containing each TMA were cut in 5-μm sections, were subjected to heat-induced epitope retrieval in 10 mM citrate buffer, and were incubated in affinity-purified rabbit anti-SIX2 (1:20 dilution; US Biological Corp, Marblehead, MA), mouse anti-NCAM (1:100 dilution; Cell Signaling Technology, Danvers, MA), or mouse anti-PCNA (1:100 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) antibodies overnight at 4°C. Anti-rabbit or anti-mouse labeled HRP polymer was applied to tissue sections at room temperature for 30 minutes, and antigens were visualized with a DAKO Envision kit (DakoCytomation, Carpinteria, CA). Additional sections of whole WT specimens were incubated with affinity-purified rabbit anti-CITED1 antibody (1:50; Lab Vision/Neomarkers, Fremont, CA) and developed also using the DAKO Envision kit, as described [20].
Immunofluorescence to Label the WT Stem Cell
In vivo immunofluorescence
To determine the spatial distribution of blastema (i.e., the putative WT stem cell) and more differentiated epithelial structures and patterns of protein co-expression, human WT was co-labeled with rabbit anti-SIX2 (1:1000) and mouse anti-NCAM (1:1000) antibodies. Importantly, NCAM has been shown recently to label both nephron progenitors in the embryonic kidney and a putative WT stem cell [24], [25], [26], [27]. Given that SIX2 is a fundamental marker of nephron progenitors in the CM and WT blastema, we questioned what overlap it and NCAM would show in this malignant context; we hypothesized that SIX2+/NCAM+ would co-label the most undifferentiated WT blastema. The immunofluorescence protocol followed for this co-labeling has been described elsewhere, but briefly, after antigen retrieval and overnight incubation with these two primary antibodies, DAKO Envision (DakoCytomation) anti-rabbit HRP polymer and tyramide signal amplification (PerkinElmer, Waltham, MA) with the fluorescein isothiocyanate fluorophore (SIX2 labels green) were added to the slides for 10 minutes [20]. After quenching and washing, DAKO Envision anti-mouse HRP polymer and tyramide signal amplification with the Cy3 fluorophore (NCAM labels red) were added, respectively. Tissue sections were then counterstained with 4′,6-diamidino-2-phenylindole,dihydrochloride (DAPI) (1:50,000; Invitrogen, Carlsbad, CA), which labels all nuclei blue.
Model of SIX2 Overexpression in WT
WT cell lines
To mimic the observed overexpression of SIX2 in clinical WT specimens, the human WT cell line, WiT49, was cultured as described and then transfected with the pcDNA3.1 plasmid containing the Venus green fluorescent protein (vGFP) gene only (control cell line) or containing both the vGFP and wild-type SIX2 genes (experimental cell line) [28]. This latter transgene was designed uniquely to produce a vGFP-2A-SIX2 protein, in which the intervening 19 amino acid 2A peptide is cleaved immediately after synthesis, thereby liberating the GFP marker from the amino terminus of SIX2 and eliminating any physical constraints of this epitope on transcriptional function [29], [30]. Briefly, separate six-well plates of WiT49 cells grown to 80% confluence were transfected with either transgene using the XtremeGene Transfection System according to the manufacturer’s protocol (Roche, Indianapolis, IN) and using 2 μg of DNA and 6 μl of transfection reagent in a 1:3 ratio. The neomycin analog, G418, was added subsequently at a concentration of 1 mg/ml to select the successfully transfected cells, as the pcDNA3.1 plasmid contains a neomycin resistance cassette. After 5 weeks of G418 selection, inducing stable integration of the transgenes, both the WiT49-GFP-2A-SIX2 and WiT49-GFP cell lines were flow sorted (FACS) for viability and the vGFP signal to enrich a population of high-expressing, GFP-positive cells in each line. Specifically, five million cells were stained with 7-AAD (BD Biosciences, San Jose, CA) for 20 minutes at 4°C. After washing with 1 × phosphate-buffered saline (PBS) containing 2% FBS, cells were sorted in the Vanderbilt Flow Cytometry Shared Resource for viability and high expression of vGFP. Cells were collected in medium containing antibiotics and were placed immediately in culture conditions for expansion. All validation and functional assays outlined below were performed using samples from flow-sorted populations of high GFP-expressing cells.
Model Validation Assays
In vitro immunofluorescence
To validate expression of both transgenes in the WiT49 cells using detection of vGFP as the surrogate marker, glass coverslips were coated with 50 μg/ml poly-d-Lysine for 1 hour at room temperature and rinsed two times with 1 × PBS. Cells from the respective lines were plated onto coverslips and, after reaching 80% confluency, were fixed with 4% paraformaldehyde in PBS at 37°C for 30 minutes. Cells were washed twice with 1 × PBS, and coverslips were applied with fluorescent mounting media. Images were captured on a Zeiss Axioflor fluorescent microscope.
Western blot
To validate further the integrity of transgene expression in both cell lines, and specifically to quantitate the differences in SIX2 expression between control and experimental WiT49 cell lines, total protein was extracted from whole-cell lysates, as described [20]. Equal concentrations of protein from passages 2, 3, and 4 of WiT49-GFP-2A-SIX2 and WiT49-GFP cells were separated on 10% bis-Tris sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels; protein samples extracted from wild-type WiT49 cells (SIX2 protein not detected typically at standard loading concentrations) and a human WT specimen that expresses abundant SIX2 served as controls. After protein separation and transfer, cellulose blots were probed using anti-SIX2 primary antibody (1:1000; US Biological Corp) overnight at 4°C. Blots were washed thrice and then probed with an anti-rabbit IgG secondary antibody (1:5000; Jackson ImmunoResearch, West Grove, PA) at room temperature for 1 hour. SIX2 bands were visualized using Pierce ECL Western Blotting Substrate (Thermo Scientific, Waltham, MA). The same protocol was followed to detect vGFP expression (Venus GFP primary antibody, 1:3000; Aves and an anti-chicken IgY secondary antibody, 1:5000; Jackson ImmunoResearch). Both the SIX2 and vGFP blots were stripped and reprobed for β-actin (primary antibody, 1:5000; Sigma, St. Louis, MO, and an anti-mouse IgG secondary antibody at 1:3000, KPL, Gaithersburg, MD).
Functional Assays
Soft agar tumorigenesis assay
To measure SIX2 effects on anchorage-independent growth of WiT49 cells, the bottom agar was prepared with 10 ml of 1.6% agar (0.72 g of SeaPlaque agar, 45 ml of sterile ddH2O), 1 ml of FBS, 0.2 ml of penicillin/streptomycin, and 8.8 ml of Dulbecco's modified Eagle's medium (DMEM) [28]; 1.5 ml of bottom agar mix was added to each well of a six-well plate and allowed to solidify for 1 hour at room temperature. The top agar layer was prepared with 5 ml of 1.6% agar, 1 ml of FBS, 0.2 ml of penicillin/streptomycin, and 13.8 ml of DMEM. Three milliliters of top agar was added to separate conical tubes, followed by the addition of WiT49-GFP-2A-SIX2 cells or WiT49-GFP cells to each tube; 0.75 ml of top agar, containing 12,000 cells per well (n = 5 technical replicates; n = 3 biologic replicates—passages 2, 3, and 5), was added to each well of the six-well plate. The agar was allowed to solidify for 30 minutes at room temperature and was placed in a 37°C incubator with 5% CO2 for 3 weeks. The plates were read with a GelCount instrument (Oxford Optronix, Oxfordshire, United Kingdom), housed within the Epithelial Biology Shared Resource of Vanderbilt University Medical Center, to count colony numbers and to measure colony diameter.
Cellular proliferation
To determine SIX2 effects on WT cellular proliferation, the CellTiter 96 non-radioactive kit was used (Promega, Madison, WI), as described [28]. Briefly, 5000 WiT49-GFP-2A-SIX2 cells or WiT49-GFP cells were plated onto a 96-well flat-bottom plate in media containing 0.2% FBS. Cells were grown for 24, 48, or 72 hours, and 15 μl of dye reagent was added to each well according to the manufacturer’s instructions. After 4 hours of incubation with the dye reagent, 100 μl of stop solution was added. The plates were read in a SpectraMax spectrophotometer (Molecular Devices, Sunnyvale, CA). Three biologic replicates (passages 2, 3, and 5; n = 4 technical replicates per passage) of WiT49-GFP-2A-SIX2 cells and WiT49-GFP cells were analyzed.
Invasion
To assay effects of SIX2 on WT invasiveness, the QCM Fluorometric 24-Well Cell Invasion Assay was used (Millipore, Billerica, MA). Cells from passage numbers 3, 4, and 5 of both WiT49-GFP-2A-SIX2 and WiT49-GFP were serum-starved (0.2% FBS) for 18 hours. After harvesting and washing, cell pellets were resuspended in 1 ml of serum-free medium, counted, and brought to a volume to give 1 × 106 cells per ml. Invasion assay inserts were handled with sterile forceps; 300 μl of serum-free DMEM was added to the interior of each insert and allowed to rehydrate the insert for 15 minutes at room temperature. After rehydration, 250 μl of media was removed from each insert; 250 μl of a cell suspension containing 1 × 106 cells per ml in chemoattractant-free media was added to each insert; 500 μl of media containing 15% FBS was added to the outer chamber. Plates were covered and incubated for 24 hours at 37°C in a CO2 incubator. The remaining cell suspension was pipetted out of each insert, and the invasion chambers were inserted into clean wells containing 225 μl of cell detachment solution. Invasion chambers were incubated for 30 minutes at 37°C. Cells were dislodged from the underside of the chamber by tilting during incubation, and inserts were removed from the wells; 75 μl of lysis buffer/dye solution (CyQuant GR Dye diluted 1:75 with 4 × lysis buffer) were added to each well containing 225 μl of cell detachment solution with the cells that invaded through the membrane. The wells were incubated for 15 minutes at room temperature; 200 μl of the mixture was transferred to a 96-well plate and read with a fluorescence plate reader using 480/520 nm filter set.
Canonical WNT Signaling
To investigate the effects of SIX2 on canonical WNT pathway activation, the TOPFlash and FOPFlash (gifts of Ethan Lee, Vanderbilt) and survivin (gift of Michael Khan, University of Southern California) reporter plasmids were transfected into the experimental cell line WiT49-GFP-2A-SIX2 and the control cell line WiT49-GFP [28], [31]. Cells were transfected using the X-tremeGENE HP DNA Transfection Reagent (Roche) following the manufacturer’s protocol. Twenty four hours after transfection, WNT3a conditioned media was added to cultures to activate reporter plasmids. The Luciferase Assay System (Promega) was used to measure WNT signaling activation. Growth media were removed from the TOPFlash and FOPFlash transfected cells, and cells were rinsed with PBS; 200 μl of 1 × lysis buffer was added to the cells, and one freeze-thaw cycle was performed to ensure cell lysis. Cells were scraped from the culture plates and transferred to microcentrifuge tubes. Tubes were vortexed for 10 seconds and centrifuged at 12,000g for 2 minutes at 4°C. The supernatant, containing the cell lysate, was transferred to a new tube, and 20 μl of cell lysate per sample was added to a 96-well plate; 100 μl of luciferase assay reagent was added per well, and the light produced was measured immediately in a SpectraMax spectrophotometer (Molecular Devices).
Real-Time Polymerase Chain Reaction
To evaluate effects of SIX2 on WNT pathway signaling, reverse transcription was performed with 1 μg of RNA using the RT2 First Strand cDNA synthesis kit (SA Biosciences, Valencia, CA). The RT2 Profiler PCR arrays specific for the WNT signaling pathway (PAHS-043YA) was purchased from SA Biosciences and used according to the manufacturer's instructions [28], [32]. Changes in WNT pathway genes were evaluated on a BioRad iCycler for passages 2, 3, and 4 of each cell line (one plate per passage). This array provides quantitative analysis of 84 WNT pathway components and target genes with expression normalized to a panel of five housekeeping genes on a 96-well plate format.
Statistics
Analysis of variance (ANOVA) was used to compare differences in experimental and control cell lines for colony number and size, WNT reporter assays, and invasion. Passage effects were not statistically significant for these assays (all P > .1). However, due to important interactions observed between group and passage for the proliferation assay, proliferation was analyzed separately by passage using a two-way ANOVA for group and time and their interaction. Model-based least square means were used to compare pairwise group differences, differences between time points, and group differences by time point, as warranted, adjusting for multiple comparisons using the Tukey-Kramer method. Differences in WNT pathway gene expression between cell lines were evaluated using statistical software available through the SA Biosciences website and were specific to these quantitative polymerase chain reaction (PCR) analyses. Statistical significance for all comparisons was set at P < .05. ANOVA and Multilevel Multivariate Analysis of Variance (MMANOVA) were conducted in SAS 9.2. Data were represented graphically using GraphPad Prism 6 software.
Results
Co-Expression of SIX2 and NCAM Specifies the Multipotent CM of the Embryonic Kidney
Serial sections of human fetal kidney specimens ranging between 16 and 20 weeks gestation age were probed for SIX2 and NCAM antigens and showed overlapping yet disparate expression domains of these two nephron progenitor markers (Figure 1). As expected, both SIX2 and NCAM co-labeled most robustly the condensed CM, whereas SIX2 appeared to label the loosely aggregated metanephric mesenchyme (MM) more diffusely than NCAM, where the latter was detected minimally if at all. Interestingly, NCAM appeared to label a larger population of the CM (up to four cell layers) than SIX2, which was visualized robustly in only one or two cell layers immediately surrounding the ureteric bud (UB). UB structures were negative for both antigens. Among the early epithelial structures differentiated from the CM, NCAM appeared to label through the comma-shaped body and no further, and SIX2 labeled at most through the pre-tubular aggregate.
Figure 1.
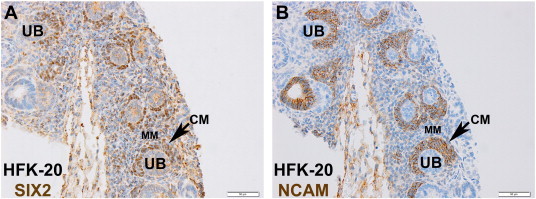
Immunoperoxidase staining (brown) for SIX2 (A) and NCAM (B) in serial sections of a 20-week gestation human fetal kidney included in one TMA. (A) SIX2 shows rich nuclear expression in one or two cell layers of the CM (arrow) immediately adjacent to the UB, which is negative for this protein. The intervening MM shows positive SIX2 expression mixed between the cytosol and nuclear compartments. (B) In contrast to SIX2, NCAM is detected exclusively in the CM but in two or more cell layers and is also negative in the UB. All images are captured at 400 × (bar = 50 μm). The sectioning artifact in both serial sections should be regarded as non-specific staining.
Co-Expression of SIX2 and NCAM Principally Labels Undifferentiated and Proliferating WT Blastema
Of the 41 WT specimens included in the three TMAs, 36 were primary lesions untreated before resection (4 had been pretreated and 1 was obtained from a metastatic deposit only). Metastatic specimens accompanied 5 of these 36 (13.9%) primary WTs. Overall, SIX2 was expressed in 35 (97.2%), and NCAM in 33 (91.7%), of these primary untreated WTs (Figure 2). Expression of both SIX2 and NCAM within any cell type (i.e., either blastema or epithelia) was visualized in 29 of the total 36 (80.6%) primary untreated WT specimens.
Figure 2.
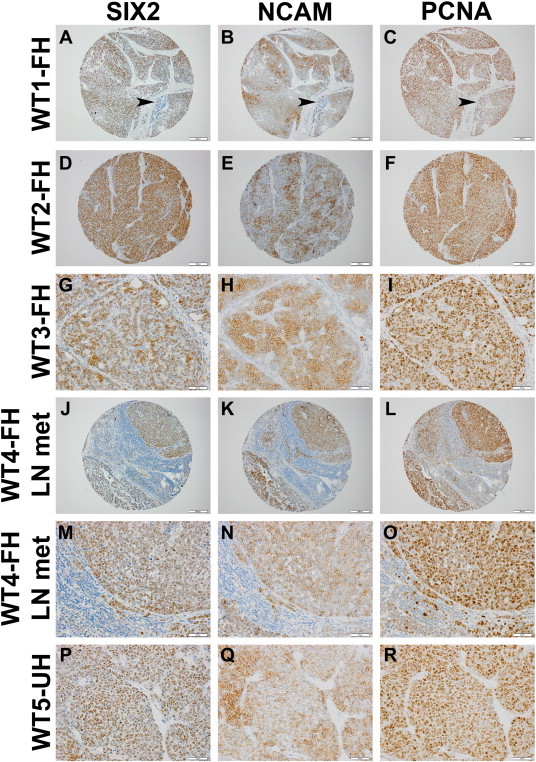
Representative images of immunoperoxidase staining (brown) for SIX2, NCAM, and the proliferation marker PCNA as captured from serial sections of the three TMAs. (A–C) A blastemal predominant but triphasic favorable histology WT (WT1-FH) shows the typical expression domain for each protein. Note that significant regions also show overlapping expression yet discrete individual areas of detection. Arrowheads show a pocket of epithelia (blue only) that do not express either SIX2 or NCAM but are proliferative. The intervening stroma (white regions) does not express any of these three proteins. (D–F) A second FH WT (WT2-FH) that is blastemal predominant shows a similar expression domain for each protein. Note again that the stroma is negative and that PCNA staining more closely parallels SIX2 than NCAM. Punch images are captured at 100 × (bar = 200 μm). (G–I) A third FH WT (WT3-FH) also shows overlapping yet unique expression domain for each protein. Intervening stroma again is negative. Images are captured at 400 × (bar = 50 μm). (J–L) Low and (M–O) high powered images (scales as above) of a metastatic deposit in a lymph node (LN) obtained from a fourth FH WT (WT4-FH). Note that the metastatic WT cells are strongly positive for each protein without significant variability in expression domains. (P–R) For comparison, expression patterns of these three proteins are provided within an unfavorable histology WT (WT5-UH) specimen (400 ×; bar = 50 μm). As with the FH specimens above, considerable overlap, yet with areas of distinctive expression domains, was observed again. However, it appeared that SIX2 more closely paralleled the expression pattern of PCNA. Stroma remained negative for these three proteins in the UH context.
When evaluating specific cell types for expression of either or both proteins, blastema was present in 30 of these 36 primary untreated WT specimens, and 29 (96.7%) of these undifferentiated cellular compartments showed highly specific co-expression of SIX2 and NCAM (Figure 2). Acknowledging that blastema occurred far more abundantly than epithelia in this cohort of consecutive WTs, only 11 of 27 (40.7%) specimens having epithelia for evaluation co-expressed both SIX2 and NCAM within this differentiated cellular compartment. While NCAM is almost exclusively detected at the cell membrane in either blastema or epithelia (i.e., its subcellular localization does not appear to change in WT), unexpectedly, 7 (38.9%) of the 18 primary untreated WTs having epithelial detection of SIX2 showed cytosolic compartmentalization, which is an unusual subcellular location for this transcription factor in either undifferentiated blastema or the multipotent CM. In general, SIX2 and NCAM detection using immunoperoxidase techniques largely overlapped in the same regions of specific cell types (i.e., blastema and epithelia) within a given WT; however, stroma showed no detection of either protein.
Contained within these TMAs were five specimens acquired from metastatic deposits (lung, liver, nephrectomy bed, and lymph nodes). All five metastases showed NCAM expression, and four showed SIX2 expression. As expected, neither the malignant (i.e., clear cell sarcoma) nor the benign (i.e., mesoblastic nephroma) childhood renal tumors serving as controls showed expression of the SIX2 or NCAM proteins, suggesting a different cell of origin for these tumor types other than the CM and its epithelial derivatives.
To determine if SIX2 and/or NCAM labeled proliferative cells in these WT specimens, immunostaining for the proliferation marker, PCNA, was performed on a third serial section as well (Figure 2). WTs are notoriously proliferative cancers, and as expected, PCNA was visualized in a highly specific and diffuse pattern within these WT specimens; although the clear cell sarcoma specimen showed detection of PCNA, the mesoblastic nephroma benign control tumor was entirely devoid of this proliferation marker. As depicted, PCNA showed an expression domain that closely paralleled both proteins but subtly appeared to associate more with SIX2 (Figure 2). The conclusion of these extensive IHC experiments is that regions of highly proliferative blastema (and epithelia more rarely) express both NCAM and SIX2 robustly and that SIX2 more specifically labels individual proliferating cells.
Overall, from this IHC analysis of the three TMAs, cellular regions within each WT punch composed of a specific cell type (e.g., blastema or epithelia) often expressed both SIX2 and NCAM, although individual cells within these regions appeared to express variably both, one or the other protein. To examine the cellular and subcellular co-expression domains of SIX2 and NCAM more closely in WT, we performed co-immunofluorescence of additional WT specimens not included in the TMAs. As shown, co-expression of SIX2 (green) and NCAM (red) in a clinical WT specimen labels a population of loosely aggregated blastema that appears furthest from the more differentiated elements (DAPI blue only) in various zones of epithelial transition (Figure 3). Analogous to compartmentalization observed within the CM of the embryonic kidney, in which progenitor cells are coursing through the mesenchymal-to-epithelial transition, a spatial gradient in degree of differentiation also appears in this malignant context, passing from the most undifferentiated cell type, SIX2+/NCAM+, to the most differentiated, i.e., SIX2−/NCAM− (DAPI only), with an intermediate cell type SIX2−/NCAM+ existing between these cellular compartments. Lending further evidence that co-expression of SIX2 and NCAM designate a blastema population likely containing the WT cancer stem cell, serial sections of WT show tight blastemal co-detection of NCAM and CITED1, as we have shown previously that the latter transcriptional regulator is a marker of proliferating WT cells and overlaps considerably, but not exclusively, with the SIX2 expression domain in WT (Figure 4) [20], [28].
Figure 3.
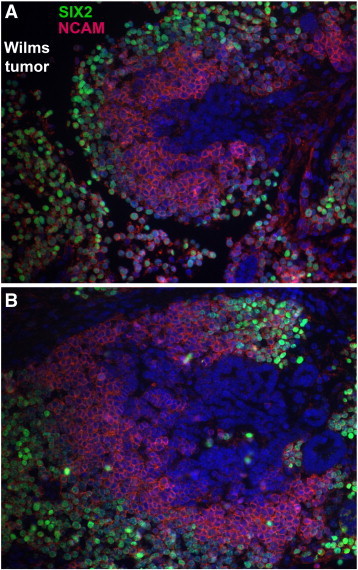
(A and B) Immunofluorescent co-localization of SIX2 (green) and NCAM (red) in two distinct “zones of differentiation” within an FH WT specimen. The loosely aggregated and most undifferentiated blastema stains positively for both SIX2 and NCAM, although this region also contains more rare SIX2+/NCAM− cells. As cells move toward the more differentiated epithelial state (SIX2−/NCAM−/DAPI+; blue only) showing tubule formation, the intermediate group of aggregating cells is SIX2−/NCAM+ (red only). Although not absolute, the majority of differentiating or differentiated WT cells appear to lose SIX2 expression, as shown in these two images (400 ×).
Figure 4.
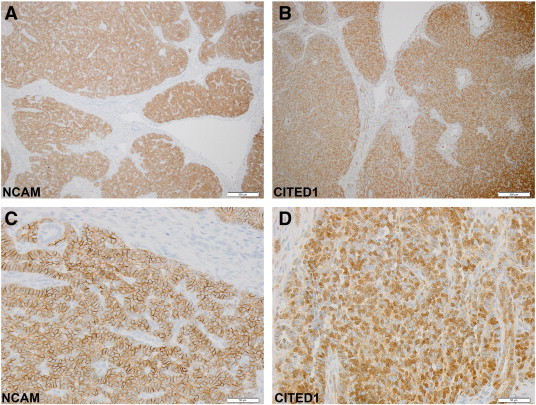
Serial sections of a human WT specimen immunostained for NCAM and CITED1 (B and D appear rotated 90° counterclockwise relative to A and C because of orientation from tissue sectioning). (A and B) Low power photomicrographs of a WT specimen immunostained for NCAM and CITED1, respectively (100 ×; bar = 200 μm). The blastemal compartments show nearly exact overlap in detection of both antigens with differences only appearing in the more mature tubular elements (arrowhead) that do not express NCAM but variably express CITED1. (C and D) High power photomicrographs (400 ×; bar = 50 μm) show differences in subcellular compartment detection of both antigens but also the tight overlap between blastemal detection.
SIX2 Overexpression in a WT Cell Line—Model Validation
To mimic the clinically observed overexpression of SIX2 in WT and to test its biologic effects and mechanism of action, we engineered a model using the human WT cell line, WiT49, which forms as wild-type cells predominantly differentiated (stromal and epithelial compartments with rare differentiating blastema) xenograft tumors and is a low expresser of SIX2 (e.g., detected by PCR only) [28], [33]. After transfection of WiT49 cells with either the control (vGFP only) or experimental (vGFP-2A-SIX2) plasmids in our model, stable integration of each transgene was achieved within 5 weeks of neomycin pressure selection. To enrich each cell line for high expression of the plasmid, as determined from intensity of vGFP detection, viable cells were sorted according to fluorescent signal, and high expressers were collected for expansion in culture (Figure 5, A and B). Using this approach, the percent of vGFP-positive cells for both cell lines increased from 80% at baseline to nearly 100%.
Figure 5.

Model validation studies in the human WT cell line, WiT49. (A and B) Results of FACS for viability and high expression of vGFP. (A) Control cell line: vGFP only. (B) Experimental cell line expressing vGFP-2A-SIX2 transgene. (C and D) Direct immunofluorescence of the two cell lines shows high vGFP detection in the nucleus and weak vGFP detection in the cytosol. (E) Western blot to confirm transgene expression and protein translation in the two cell lines. SIX2 is detected at its appropriate weight in a positive-control clinical WT specimen and in three consecutive passages of the WiT49-vGFP-2A-SIX2 cell line (expression continues similarly to passage 6—data not shown). Importantly, SIX2 protein is not detected in wild-type WiT49 cells nor in the control vGFP-only cells. vGFP was detected in all transfected cells but importantly not in the clinical WT specimen or wild-type WiT49 cells. β-Actin loading controls are shown for each Western blot.
To validate the model, direct fluorescence microscopy of cells in vitro showed rich visualization of predominantly nuclear vGFP signal in both cell lines (Figure 5, C and D). To ensure that the vGFP marker and SIX2 were indeed liberated from one another by inserting the intervening 2A cleavage peptide, Western blot analysis of protein collected from multiple passages (2-4) of each cell line showed that both vGFP and SIX2 traveled at the standard weights (Figure 5).
SIX2 Effects on WiT49 Biology
Having validated this model as continually overexpressing SIX2 across multiple passages of WiT49 cells (p5) and without the potential for physical constraints of a large epitope tagged onto this transcription factor, we next proceeded to test the effects of persistent SIX2 activation on WT cell survival, given its established role in the maintenance of, and repression of WNT-dependent differentiation in, the CM of the embryonic kidney. Using our established protocol for anchorage-independent growth in soft agar, we evaluated the effects of SIX2 overexpression on WiT49 cell survival (Figure 6, A–D). SIX2-overexpressing cells showed a significantly greater colony count (median count, 3429; interquartile range: 540-9704) than vGFP control cells (median 160; interquartile range: 150-186; P = .027; Figure 6). Colony diameter was also significantly greater for SIX2-overexpressing cells relative to controls (median diameter 90.8 vs 67.6, respectively; P = .0009; Figure 6).
Figure 6.
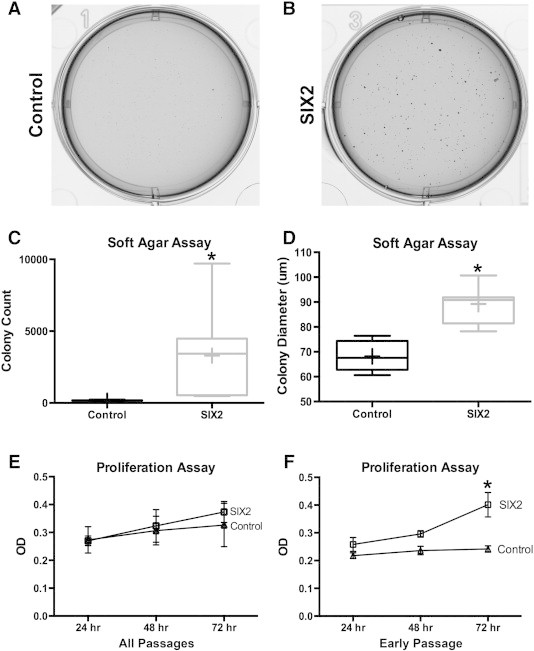
SIX2 effects on WiT49 cell survival. (A and B) Images captured from a representative soft agar well showing grossly differences in colony number and size between control and experimental cell lines. (C) Colony count was significantly increased for WiT49 cells transfected with the vGFP-2A-SIX2 transgene (interquartile box shown, with median bar and mean + sign). (D) Colony size was also greater in the SIX2-expressing WiT49 cells. (E) Although SIX2-expressing cells appeared to have enhanced proliferative response when analyzing all passages and time points simultaneously, an insufficient number of observations were performed to conclude statistically significant differences. However, when separating out individual passages, early passages of SIX2-expressing WiT49 cells were noted to be significantly more proliferative than vGFP-only cells (F). This effect of SIX2 persisted although statistical significance was mitigated as control cells became more proliferative in later passages too; 72-hour time points showed significantly more proliferative cells than 24-hour time points.
We next evaluated SIX2 effects on proliferative response of WiT49 cells as another in vitro measure of enhanced cell survival. Using our established model of WiT49 proliferation, we detected an overall trend that SIX2 enhanced proliferation and separated from controls over time, although statistical significance was not met when comparing all passages together (Figure 6). However, the effect of SIX2 to enhance proliferation appeared to be dependent both on time point evaluated (e.g., 72 hours vs 24 hours, P = .047) and passage number after transfection, as early passage achieved the highest statistical significance between cell lines (passage 2; P < .0001; Figure 6). In later passages, SIX2 continued to show the same enhanced proliferative response in WiT49 cells across the three time points evaluated (as in earlier passages), although the control cells in later passages also showed an increased proliferative response, which mitigated statistically significant differences between cell lines when all passages were analyzed together.
Regarding SIX2 effects on invasiveness of WiT49 cells, we had an insufficient number of observations to conclude firmly any change in this biologic parameter, although a tendency toward repression was detected (data not shown).
SIX2 Effects on Canonical WNT Pathway Reporter Assays
Because SIX2 is known to regulate self-renewal of nephron progenitors and to counterbalance WNT-dependent differentiation signals in the CM, we next sought to clarify its effects on canonical WNT signaling in the WT context. First, to gain perspective in the embryonic context, using our previously published model on WNT signaling in HEK-293 cells that have been stably transfected with a TOPFlash reporter, we evaluated the effects of transiently overexpressing SIX2 in this immortalized but non-malignant cell line [28]. In HEK-293 cells, we observed that SIX2 had no effect on global canonical WNT signaling when testing the TCF/LEF-dependent TOPFlash reporter assay (Figure 7A). Interestingly, when this reporter was transfected into the two WiT49 cell lines, we observed across 11 biologic replicates a consistent effect of SIX2 to repress canonical WNT signaling, although variability in the magnitude of repression between experiments mitigated statistical significance when comparing mean values between groups (Figure 7B). Specifically, 8 of the 11 (72.7%) biologic replicates tested showed that SIX2 overexpression repressed TOPFlash activation in WiT49 cells. The variability in absolute magnitude of TOPFlash activation is likely explained by variations in concentration of WNT3a collected from supernatant of transfected production cells and used to activate the reporter between different experiments (a standard technique to generate WNT proteins for TOPFlash assays). Taken together, these observations using a TOPFlash reporter assay likely reflect a balanced effect in the embryonic context of SIX2 to repress differentiation and to activate stem cell survival through interactions with specific components of the canonical WNT pathway but that becomes deregulated and unbalanced in malignancy.
Figure 7.
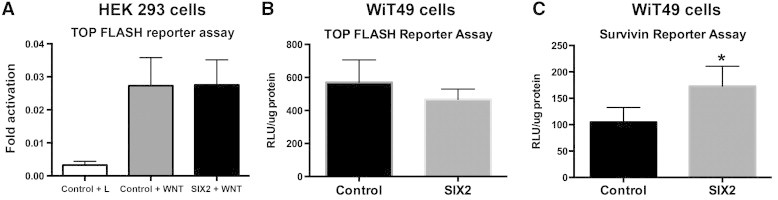
SIX2 effects on canonical WNT reporter assay activation. (A) TOPFlash activation in HEK-293 cells stably transfected with this reporter plasmid. Baseline control HEK-293 cells having the TOPFlash reporter but not transfected with either vGFP or vGFP-2A-SIX2 plasmid and not stimulated with WNT3a (L) show low activation. When transfected with either control or SIX2 plasmid, and stimulated with WNT3a, canonical WNT activation appears balanced. (B) After transfection of the TOPFlash reporter into the respective model cell lines, an insufficient number of observations were made to conclude statistically significant differences, although 8 of 11 (72.7%) experiments showed repression of reporter activation versus 3 of 11 (27.3%) that showed enhanced activation. (C) To examine whether SIX2 promoted enhanced WiT49 cell survival through CBP/β-catenin–dependent interaction, a survivin reporter assay was transfected into the model cell lines. Notably, SIX2 significantly enhanced canonical WNT pathway activation through the survivin promoter, which represents a pathway to maintain stemness and cell survival.
Because canonical WNT signaling operates through β-catenin interaction with either CBP, leading to maintenance of stemness, or p300, resulting in commitment to epithelial differentiation, we sought next to distinguish what effect SIX2 has on the former pro-stem pathway and if this transcription factor tips the balance toward a cell survival pathway in the malignant context. To test SIX2 effects on CBP/β-catenin–dependent signaling, we transfected a survivin reporter, which is activated specifically through the interaction of β-catenin with CBP, into the two WiT49 cells lines established in this model. Interestingly, SIX2 was observed to enhance significantly survivin-dependent signaling in the WT context (P = .034; Figure 7C). Although we are unaware of a reporter assay that is specific to β-catenin interaction with p300, we suspect that the relative balance observed with the TOPFlash reporter potentially originates from repression of this differentiation pathway, given the observed up-regulation of the pro-stem CBP/β-catenin signaling.
SIX2 Effects on WNT Gene Expression in WiT49 Cells
Because we detected differential effects of SIX2 on canonical WNT signaling, we questioned next what WNT pathway interactors would be altered in this WT model across three passages (i.e., biologic replicates). Using quantitative PCR to analyze expression changes in 84 WNT pathway genes having known functions, we detected statistically significant effects of SIX2 on 35 genes (41.7%), 31 (36.9%) of which were repressed (Table 1).
Table 1.
SIX2 Effects on WNT Pathway Gene Expression.
| Symbol | Fold Regulation | P Value |
|---|---|---|
| MT1A | 23.464 | .02682 |
| CCND2 | 7.057 | .000623 |
| WNT1 | 1.8909 | .040953 |
| CXADR | 1.7642 | .04628 |
| BTRC | − 1.1336 | .043991 |
| RUVBL1 | − 1.3022 | .010522 |
| GSK3B | − 1.3326 | .031276 |
| CTNNB1 | − 1.4283 | .016683 |
| CTNNBIP1 | − 1.4958 | .024324 |
| DVL1 | − 1.4958 | .020268 |
| DVL2 | − 1.5308 | .03886 |
| CSNK1A1 | − 1.7183 | .006969 |
| FBXW11 | − 1.7183 | .001276 |
| VANGL2 | − 1.7183 | .023559 |
| PITX2 | − 1.7584 | .007196 |
| AXIN2 | − 1.7995 | .045722 |
| PRICKLE1 | − 1.8846 | .008141 |
| CALM1 | − 1.8846 | .000792 |
| LEF1 | − 2.1154 | .014944 |
| CTBP1 | − 2.1649 | .005634 |
| WNT3 | − 2.2673 | .013814 |
| DAB2 | − 2.5449 | .004921 |
| SFRP4 | − 2.6653 | .040353 |
| FZD9 | − 2.7276 | .015464 |
| WNT6 | − 2.8566 | .016166 |
| WISP1 | − 2.9233 | .037553 |
| NFATC1 | − 3.2813 | .025631 |
| SKP2 | − 3.358 | .000671 |
| WNT11 | − 3.4365 | .004348 |
| WNT9A | − 3.5991 | .010208 |
| KREMEN1 | − 3.8574 | .001497 |
| CYP4V2 | − 4.0398 | .005576 |
| SFRP1 | − 4.9736 | .000086 |
| DKK1 | − 7.1981 | .022828 |
| NKD1 | − 325.7498 | .002323 |
Discussion
This study showed foremost that SIX2 overexpression in a human WT cell line confers a survival predisposition to this embryonal tumor of childhood analogous to its documented functions in the developing kidney. Moreover, these studies showed that SIX2 appears to direct β-catenin interaction preferentially with the co-activator, CBP, which is a recognized mechanism of stem cells, whether normal or malignant, to maintain self-renewal and is currently being evaluated as a therapeutic target of PRI-724 in several adult solid cancers.
We found in previous studies that SIX2 is expressed aberrantly across a broad spectrum of WTs and is principally a marker of undifferentiated blastema, its putative cancer stem cell, yet an understanding of its functional significance in the malignant context until now has remained an extrapolation from development [20], [21]. Importantly, in the embryonic kidney, SIX2 has been established as a critical regulator and self-renewal mechanism of the nephron progenitor pool within the CM [9], [10], [11], [19]. As a result of those prior studies, we postulated that SIX2 likely would be hijacked for a similar gain of perpetuity in the WT context, albeit in an unchecked manner. Indeed, using a validated model of SIX2 overexpression in a WT cell line, we have observed significantly enhanced cell survival in soft agar and an early effect to increase cell proliferation, consistent with a previous report [22].
In the developing kidney, significant “cross-talk” occurs between the UB structures, which will form the collecting tubules of the distal nephron, and the condensing MM (or the CM), which will form the epithelial elements of the proximal nephron, including glomeruli [34]. To establish proper nephron endowment of the fully developed and mature kidney, this “cross-talk” must be tightly regulated and coordinated between the UB and the CM, which together direct reciprocal interactions that lead to growth and development of the progenitor pool and committed epithelial structures. Asymmetric cell division is at the cornerstone of this coordinated maintenance to spawn an equal balance of both stem and epithelial-committed daughter cells. Indeed, in the murine embryonic kidney, Wnt9b, secreted from the UB, has been shown to target SIX2 as a mechanism to induce epithelial conversion [11]. Furthermore, SIX2 and β-catenin have been shown to share gene regulatory networks in the embryonic kidney, providing additional evidence that these two transcriptional regulators tightly coordinate the mesenchymal-to-epithelial transition of the CM [19]. By evaluating differentially two canonical WNT pathway reporter assays in our WT model, it appears that SIX2 may direct β-catenin away from interaction with p300, which induces epithelial commitment within stem cells, and toward interaction with CBP that drives progenitor maintenance. Any imbalance in this tightly coordinated binary branch point for WNT-dependent cell fate decisions could result in symmetric cell division, whereby both daughter cells remain in a stem state. Effectively, in the malignant context of WT, SIX2 may promote β-catenin interaction with CBP, fueling perpetuity of a WT stem cell. Interestingly, in this current WT model, SIX2 was found to repress significantly an abundance of WNT pathway genes that could collectively shift the balance to maintaining a perpetual loop for WT survival, although admittedly our studies were not designed to test the effects on WT biology of each of these altered WNT genes.
Much emphasis in cancer biology has been directed recently toward defining the biologic properties of the mysterious cancer stem cell [35], [36]. Embryonal tumors of childhood by definition arise from rogue progenitor cells that somehow escape terminal differentiation and retain the hallmark feature of a “small round blue” cell having a high nuclear-to-cytosolic ratio. In this current study, we have shown that NCAM, a critical cell surface marker of a putative WT stem cell, largely associates with expression of SIX2 in clinical specimens [23], [24], [26]. Curiously, although SIX2 and NCAM appear to be co-expressed in a majority of individual blastema, and more rarely in a minority of epithelia, other cells within these two compartments express one or the other protein, but not both. The functional significance of these different expression patterns remains unclear, although we expect on the basis of our observations that co-expression of SIX2 and NCAM labels a less differentiated WT cell, likely one that is fixed in a perpetual loop of self-renewal and maintaining stemness.
We acknowledge several limitations of this study. First, we have evaluated only WiT49 cells showing high expression of vGFP as a surrogate for SIX2, given that the transgene we designed included a cleavage peptide and should yield equal protein translation (i.e., if vGFP detection is high then so should be SIX2). Importantly, however, a dose effect of SIX2 overexpression on WT biology was not evaluated in this model, as low vGFP-emitting cells were not collected for culture expansion, and the reason being that, within clinical WT specimens, blastema (and rarely epithelia) appears typically to express a consistent and strong content of SIX2. Second, we have evaluated the effects of SIX2 overexpression in only a single WT cell line, as no others are commercially available at the present time, and those primary WT lines that our laboratory has initiated have not yet been validated as tumorigenic in a xenograft model. Third, comparison of biologic effects between SIX2 and control plasmid was only conducted in detail for this WiT49 cell line and were not designed to test differential effects in the embryonic context (e.g., HEK-293 cells) beyond the TOPFlash reporter assay. We only evaluated overexpression of SIX2 and the control plasmid in this embryonic kidney cell line to establish baseline context of WNT/β-catenin signaling and to reveal more clearly whether deregulated pathway activation in the experimentally manipulated WiT49 cells occurred. Fourth, although we observed a consistent effect of SIX2 to repress TOPFlash activation across the majority of multiple experiments, statistical significance was not achieved when comparing mean values between cell lines given the variability in magnitude of activation between observations. Nevertheless, we believe that this repressive effect is real and that the variability is explained by differences in WNT3a concentration used between experiments, which is challenging to measure when generating this activating protein from transfected cell cultures. Finally, our studies were not designed to determine whether SIX2 is a driver or passenger in WT survival, which is the focus of ongoing studies.
In summary, we developed and validated an in vitro model of SIX2 overexpression in a human WT cell line to mimic observations of its expression domain in clinical specimens and to test SIX2 effects on WT biology. It appears from these studies that SIX2, especially when co-expressed with NCAM, labels the putative WT stem cell in clinical specimens and promotes WT cell survival in this model. Our studies introduce an early clue to the mechanism through which SIX2 operates as a means to confer WT cell survival by activating survivin-dependent signaling, a path that requires β-catenin interaction with the CBP co-activator. Future studies are warranted to dissect further the mechanism as to how deregulated SIX2 activity shifts the balance of WNT/β-catenin signaling toward symmetrical maintenance of a WT stem cell. Such work will likely reveal a targetable mechanism in WT stem cell perpetuity that is currently being evaluated in various adult solid tumors.
Acknowledgments
The authors thank Ethan Lee for providing the resources to complete the TOPFlash analyses and Michael Kahn for providing the survivin reporter assay. The authors would like to recognize the expertise within the Vanderbilt Translational Pathology Shared Resource to assist with TMA development and histologic processing of all clinical specimens that comprise our laboratory tissue repository and within the Vanderbilt University Flow Cytometry Shared Resource. Both of these Shared Resources are supported by the Vanderbilt Ingram Cancer Center (grant P30 CA68485).
Footnotes
This work was supported by funding generously provided through the National Cancer Institute [5R00CA135695-05 (H.N.L. and J.P.) and T32CA106183 (A.J.M.)].
References
- 1.Kalapurakal JA, Dome JS, Perlman EJ, Malogolowkin M, Haase GM, Grundy P, Coppes MJ. Management of Wilms' tumour: current practice and future goals. Lancet Oncol. 2004;5:37–46. doi: 10.1016/s1470-2045(03)01322-6. [DOI] [PubMed] [Google Scholar]
- 2.Beckwith JB. Nephrogenic rests and the pathogenesis of Wilms tumor: developmental and clinical considerations. Am J Med Genet. 1998;79:268–273. doi: 10.1002/(sici)1096-8628(19981002)79:4<268::aid-ajmg7>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 3.Li CM, Guo M, Borczuk A, Powell CA, Wei M, Thaker HM, Friedman R, Klein U, Tycko B. Gene expression in Wilms' tumor mimics the earliest committed stage in the metanephric mesenchymal-epithelial transition. Am J Pathol. 2002;160:2181–2190. doi: 10.1016/S0002-9440(10)61166-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beckwith JB, Kiviat NB, Bonadio JF. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms' tumor. Pediatr Pathol. 1990;10:1–36. doi: 10.3109/15513819009067094. [DOI] [PubMed] [Google Scholar]
- 5.Beckwith JB. National Wilms Tumor Study: an update for pathologists. Pediatr Dev Pathol. 1998;1:79–84. doi: 10.1007/s100249900010. [DOI] [PubMed] [Google Scholar]
- 6.Kinoshita Y, Suminoe A, Inada H, Yagi M, Yanai F, Zaizen Y, Nishi M, Inomata Y, Kawakami K, Matsufuji H. The prognostic significance of blastemal predominant histology in initially resected Wilms' tumors: a report from the Study Group for Pediatric Solid Tumors in the Kyushu Area, Japan. J Pediatr Surg. 2012;47:2205–2209. doi: 10.1016/j.jpedsurg.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 7.Graf N, van Tinteren H, Bergeron C, Pein F, van den Heuvel-Eibrink MM, Sandstedt B, Schenk JP, Godzinski J, Oldenburger F, Furtwangler R. Characteristics and outcome of stage II and III non-anaplastic Wilms' tumour treated according to the SIOP trial and study 93-01. Eur J Cancer. 2012;48:3240–3248. doi: 10.1016/j.ejca.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Dome JS, Fernandez CV, Mullen EA, Kalapurakal JA, Geller JI, Huff V, Gratias EJ, Dix DB, Ehrlich PF, Khanna G. Children's Oncology Group's 2013 blueprint for research: renal tumors. Pediatr Blood Cancer. 2012;60:994–1000. doi: 10.1002/pbc.24419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 2006;25:5214–5228. doi: 10.1038/sj.emboj.7601381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3:169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karner CM, Das A, Ma Z, Self M, Chen C, Lum L, Oliver G, Carroll TJ. Canonical Wnt9b signaling balances progenitor cell expansion and differentiation during kidney development. Development. 2011;138:1247–1257. doi: 10.1242/dev.057646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kahn M. Symmetric division versus asymmetric division: a tale of two coactivators. Future Med Chem. 2011;3:1745–1763. doi: 10.4155/fmc.11.126. [DOI] [PubMed] [Google Scholar]
- 13.Teo JL, Kahn M. The Wnt signaling pathway in cellular proliferation and differentiation: a tale of two coactivators. Adv Drug Deliv Rev. 2010;62:1149–1155. doi: 10.1016/j.addr.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 14.Miki T, Yasuda SY, Kahn M. Wnt/β-catenin signaling in embryonic stem cell self-renewal and somatic cell reprogramming. Stem Cell Rev. 2011;7:836–846. doi: 10.1007/s12015-011-9275-1. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–3162. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- 16.Nusse R. Cancer. Converging on beta-catenin in Wilms tumor. Science. 2007;316:988–989. doi: 10.1126/science.1143337. [DOI] [PubMed] [Google Scholar]
- 17.Perotti D, Hohenstein P, Bongarzone I, Maschietto M, Weeks M, Radice P, Pritchard-Jones K. Is Wilms tumor a candidate neoplasia for treatment with WNT/β-catenin pathway modulators?—A report from the renal tumors biology-driven drug development workshop. Mol Cancer Ther. 2013;12:2619–2627. doi: 10.1158/1535-7163.MCT-13-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pode-Shakked N, Harari-Steinberg O, Haberman-Ziv Y, Rom-Gross E, Bahar S, Omer D, Metsuyanim S, Buzhor E, Jacob-Hirsch J, Goldstein RS. Resistance or sensitivity of Wilms' tumor to anti-FZD7 antibody highlights the Wnt pathway as a possible therapeutic target. Oncogene. 2011;30:1664–1680. doi: 10.1038/onc.2010.549. [DOI] [PubMed] [Google Scholar]
- 19.Park JS, Ma W, O'Brien LL, Chung E, Guo JJ, Cheng JG, Valerius MT, McMahon JA, Wong WH, McMahon AP. Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev Cell. 2012;23:637–651. doi: 10.1016/j.devcel.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murphy AJ, Pierce J, de Caestecker C, Taylor C, Anderson JR, Perantoni AO, de Caestecker MP, Lovvorn HN. SIX2 and CITED1, markers of nephronic progenitor self-renewal, remain active in primitive elements of Wilms' tumor. J Pediatr Surg. 2012;47:1239–1249. doi: 10.1016/j.jpedsurg.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy AJ, Axt JR, de Caestecker C, Pierce J, Correa H, Seeley EH, Caprioli RM, Newton MW, de Caestecker MP, Lovvorn HN. Molecular characterization of Wilms' tumor from a resource-constrained region of sub-Saharan Africa. Int J Cancer. 2012;131:E983–E994. doi: 10.1002/ijc.27544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Senanayake U, Koller K, Pichler M, Leuschner I, Strohmaler H, Hadler U, Das S, Hoefler G, Guertl B. The pluripotent renal stem cell regulator SIX2 is activated in renal neoplasms and influences cellular proliferation and migration. Hum Pathol. 2013;44:336–345. doi: 10.1016/j.humpath.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 23.Pode-Shakked N, Metsuyanim S, Rom-Gross E. Developmental tumourigenesis: NCAM as a putative marker for the malignant renal stem/progenitor cell population. J Cell Mol Med. 2009;13:1792–1808. doi: 10.1111/j.1582-4934.2008.00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pode-Shakked N, Shukrun R, Mark-Danieli M, Tsvetkov P, Bahar S, Pri-Chen S, Goldstein RS, Rom-Gross E, Mor Y, Fridman E. The isolation and characterization of renal cancer initiating cells from human Wilms' tumour xenografts unveils new therapeutic targets. EMBO Mol Med. 2013;5:18–37. doi: 10.1002/emmm.201201516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pode-Shakked N, Dekel B. Wilms tumor—a renal stem cell malignancy? Pediatr Nephrol. 2011;26:1535–1543. doi: 10.1007/s00467-011-1858-1. [DOI] [PubMed] [Google Scholar]
- 26.Hohenstein P. The stem and roots of Wilms' tumours. EMBO Mol Med. 2013;5:4–6. doi: 10.1002/emmm.201202173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buzhor E, Omer D, Harari-Steinberg O, Dotan Z, Vax E, Pri-Chen S, Metsuyanim S, Pleniceanu O, Goldstein RS, Dekel B. Reactivation of NCAM1 defines a subpopulation of human adult kidney epithelial cells with clonogenic and stem/progenitor properties. Am J Pathol. 2013;183:1621–1633. doi: 10.1016/j.ajpath.2013.07.034. [DOI] [PubMed] [Google Scholar]
- 28.Murphy AJ, Pierce J, de Caestecker C, Ayers GD, Zhao A, Krebs JR, Saito-Diaz VK, Lee E, Perantoni AO, de Caestecker MP. CITED1 confers stemness to Wilms tumor and enhances tumorigenic responses when enriched in the nucleus. Oncotarget. 2014;5:386–402. doi: 10.18632/oncotarget.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osborn MJ, Panoskaltsis-Mortari A, McElmurry RT, Bell SK, Vignali DA, Ryan MD, Wilber AC, McIvor RS, Tolar J, Blazar BR. A picornaviral 2A-like sequence-based tricistronic vector allowing for high-level therapeutic gene expression coupled to a dual-reporter system. Mol Ther. 2005;12:569–574. doi: 10.1016/j.ymthe.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 30.Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, Kim MK, Shin BA, Choi SY. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One. 2011;6:e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/β-catenin-mediated survivin gene expression. Oncogene. 2005;24:3619–3631. doi: 10.1038/sj.onc.1208433. [DOI] [PubMed] [Google Scholar]
- 32.Murphy AJ, de Caestecker C, Pierce J, Boyle SC, Ayers GD, Zhao Z, Libes JM, Correa H, Walter T, Huppert SS. CITED1 expression in liver development and hepatoblastoma. Neoplasia. 2012;14:1153–1163. doi: 10.1593/neo.12958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li MH, Yamase H, Ferrer F. Characterization of a WiT49 cell line derived orthotopic model of Wilms tumor. Pediatr Blood Cancer. 2010;54:316–318. doi: 10.1002/pbc.22205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown AC, Muthukrishnan SD, Guay JA, Adams DC, Schafer DA, Fetting JL, Oxburgh L. Role for compartmentalization in nephron progenitor differentiation. Proc Natl Acad Sci U S A. 2013;110:4640–4645. doi: 10.1073/pnas.1213971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 36.Singh SR. Cancer stem cells: recent developments and future prospects. Cancer Lett. 2013;338:1–2. doi: 10.1016/j.canlet.2013.03.036. [DOI] [PubMed] [Google Scholar]