

Abstract
INTRODUCTION: The rarity of dedifferentiated liposarcoma (DDLPS) and the lack of experimental DDLPS models limit the development of novel therapeutic strategies. Pazopanib (PAZ) is a tyrosine kinase inhibitor that is approved for the treatment of non-adipocytic advanced soft tissue sarcoma. The activity of this agent has not yet been properly explored in preclinical liposarcoma models nor in a randomized phase Ш clinical trial in this entity. The aim of the present study was to investigate whether PAZ had antitumor activity in DDLPS models in vivo. MATERIAL AND METHODS: We established two patient-derived DDLPS xenograft models (UZLX-STS3 and UZLX-STS5) through implantation of tumor material from sarcoma patients in athymic nude NMRI mice. An animal model of the SW872 liposarcoma cell line was also used. To investigate the efficacy of PAZ in vivo, mice bearing tumors were treated for 2 weeks with sterile water, doxorubicin (1.2 mg/kg, intraperitoneally, twice per week), PAZ [40 mg/kg, orally (p.o.), twice per day], or PAZ plus doxorubicin (same schedules as for single treatments). RESULTS: Patient-derived xenografts retained the histologic and molecular features of DDLPS. PAZ significantly delayed tumor growth by decreasing proliferation and inhibited angiogenesis in all models tested. Combining the angiogenesis inhibitor with an anthracycline did not show superior efficacy. CONCLUSION: These results suggest that PAZ has potential antitumor activity in DDLPS primarily through antiangiogenic effects and therefore should be explored in clinical trials.
Introduction
Liposarcomas arise from adipose tissue, representing the most frequent group of adult soft tissue sarcoma (STS). Liposarcomas are classified into four subtypes: atypical lipomatous tumor (ALT), dedifferentiated liposarcoma (DDLPS), myxoid liposarcoma, and pleomorphic liposarcoma. ALT and DDLPS, which are the most common subtypes, are cytogenetically characterized by chromosome 12q amplification leading to mouse double minute 2 homolog (MDM2) and/or cyclin-dependent kinase 4 gene amplification [1], [2], [3]. Surgical resection is the primary potentially curative treatment for localized liposarcomas, while patients with local advanced or metastatic disease qualify for palliative systemic therapy. Doxorubicin is the most commonly used first-line treatment for advanced liposarcomas but has only limited efficacy [3], [4]. Limitations for the development of innovative and effective treatments are the low incidence of liposarcomas, the heterogeneous clinical course of this disease, and the lack of experimental models.
Angiogenesis plays a fundamental role in the progression of cancers and is mainly activated by the vascular endothelial growth factor receptor (VEGFR) and the platelet-derived growth factor receptor (PDGFR) signaling pathways [5], [6]. Overexpression of VEGF and VEGFR is frequently found in patients bearing primary STS [6], [7], [8]. Serum VEGF levels in patients with different types of primary STS, including liposarcoma, are higher than those in healthy individuals [9].
Pazopanib (PAZ) is a small molecule multitargeted tyrosine kinase inhibitor, which mainly inhibits VEGFR, PDGFR, and KIT [10]. PAZ is approved for treatment of patients with renal cell carcinoma and for non-adipocytic advanced STS [11], [12], [13]. The liposarcoma stratum of a multi-sarcoma phase II study was closed early because it did not reach the predefined level of an antitumor efficacy [14]. However, after central pathologic review, two tumors registered for this trial as non-adipocytic STS were reclassified as liposarcoma, and these patients did benefit from PAZ treatment by achieving disease stabilization at 12 weeks of treatment [11], [14]. In retrospect, PAZ thus did show some clinical activity in adipocytic tumors, warranting further clinical exploration. At present, two phase II trials, testing PAZ in patients with liposarcoma, are revisiting this issue (ClinicalTrials.gov identifier: NCT01692496 and NCT01506596).
In our present preclinical study, we describe the development of two patient-derived DDLPS xenograft models and the exploration of the in vivo activity of PAZ in DDLPS, using these models. Our study provides additional support for the ongoing clinical trials testing PAZ in patients with advanced DDLPS.
Material and Methods
Drugs and Reagents
Doxorubicin hydrochloride (DOX; Sigma-Aldrich, St. Louis, Missouri) was dissolved in sterile 0.9% NaCl. PAZ (Sequoia Research Products, Pangbourne, United Kingdom) was dissolved in 0.5% hydroxypropyl methylcellulose (Sigma-Aldrich) with 1% Tween 80 (Sigma-Aldrich), pH 1.3 to 1.5. Before the administration, the solution was sonicated for at least 5 minutes. The following antibodies were used for Western blot analysis: VEGFR2 (55B11), phospho-VEGFR2 (15D2), AKT, phospho-AKT Ser473, p42/p44 mitogen-activated protein kinases (MAPK), phospho-T202/T204 MAPK, eukaryotic translation initiation factor 4E-binding protein 1, phospho–4E-binding protein 1, S6, phospho-S6, α-tubulin (all rabbit, 1:1000 dilution; Cell Signaling Technology, Danvers, Massachusetts). The secondary anti-rabbit antibody (1:2000 dilution) was purchased from DAKO (Glostrup, Denmark). Western blot analysis was performed using NuPAGE Bis-Tris gels (4-12%; Life Technologies, Carlsbad, California). For immunohistochemical (IHC) staining, antibodies against phospho-histone H3 (pHH3), cleaved caspase-3 (CC3), and VEGFR2 were purchased from Cell Signaling Technology. In addition, antibodies against Ki67 (Thermo Scientific, Rockford, Illinois), CD34 (Abcam, Cambridge, United Kingdom), and MDM2 (Life Technologies) were used. HRP polyclonal goat anti-rabbit Igs, anti-rabbit EnVision + System–HRP-labeled polymer, and 3′-diaminobenzidine-tetrahydrochloride were purchased from DAKO. For VEGFR2 immunostaining, SignalStain, the Boost IHC Detection Reagent (HRP, rabbit; Cell Signaling Technology), was used.
Establishment of Mouse DDLPS Xenografts
Female adult, partially immunodeficient, athymic NMRI nu/nu mice (JANVIER LABS, Saint Berthevin, France) were used for establishing xenograft models and for the in vivo experiments. Collection and usage of tumor samples from consenting patients were approved by the Medical Ethics Committee, University Hospitals Leuven. Animal experiments were approved by the Ethics Committee for Laboratory Animals, KU Leuven (Leuven, Belgium).
The SW872 liposarcoma cell line (Cell Lines Service, Eppelheim, Germany) was cultured in Dulbecco's modified Eagle's medium/F12 medium with 10% FBS (all from Life Technologies). The SW872 cell line has been previously studied in vitro and in vivo [15], [16]. The SW872 model was generated by subcutaneous, bilateral injection of 5 × 106 cells per mouse site. Patient-derived DDLPS xenografts (UZLX-STS3 and UZLX-STS5) were established by bilateral subcutaneous implantation of fresh surgically resected tumor specimens from patients with DDLPS. Tumor tissue was further re-transplanted from mouse to mouse at least twice. From each passage, tumor fragments were collected for histologic and molecular characterization.
Molecular Characterization of Xenografted Tumors
To assess MDM2 copy number, a dual-color interphase fluorescence in situ hybridization (FISH) using the LSI MDM2/CEP 12 FISH Probe Kit (Abbott Molecular, North Chicago, Illinois) was performed on paraffin sections. Hybridization and detection were carried out as previously described [17]. In addition, the copy number of MDM2 and cyclin-dependent kinase 4 was evaluated using quantitative polymerase chain reaction (qPCR), using HsRBP4 as a control gene (all primer sequences available on request). DNA was isolated from frozen tumor fragments, using QIAamp DNA Mini Kit (Qiagen, Venlo, Netherlands). qPCR analysis was performed using Light Cycler 480 (Roche Diagnostics, Basel, Switzerland).
In Vivo Experiments
For the in vivo studies, we used 18 mice with SW872, 18 with UZLX-STS3 (p.7), and 18 with UZLX-STS5 (p.6) xenografts. The average time between inoculation/transplantation and randomization was about 1 month (for SW872 and UZLX-STS3) or 2 months for UZLX-STS5. Mice were bearing tumors on both sides, with an average volume of ± 300 mm3 and were grouped as follows: 1) control mice treated with sterile water; 2) DOX treatment [1.2 mg/kg, intraperitoneally (i.p.), twice per week] [18]; 3) PAZ treatment (40 mg/kg, p.o., twice per day) [19]; 4) PAZ + DOX combination (same doses and schedules as for single treatments). The DOX schedule, with minimal toxicity, was used on the basis of the previous study [18]. Tumor volume was measured by digital caliper three times a week. Relative tumor volume (%) was calculated by comparing the tumor volumes of each group to those at the baseline (day 0). The body weight of the mice as well as their well-being was monitored daily. The treatment lasted 2 weeks, and afterward, mice were sacrificed 2 hours after administration of the last treatment. Tumor samples were immediately snap frozen in liquid nitrogen or fixed in 4% buffered formalin solution for further analysis (for details about the in vivo study design, see Table S1).
Western Blot Analysis
Tumor lysates were prepared from frozen samples and were used for Western blot analysis as previously described [20]. A lysate of the angiosarcoma cell line ASM with VEGFR2 activation was used as a positive control. Chemiluminescence levels (Western Lighting from PerkinElmer, Waltham, Massachusetts) were captured by the Fuji Mini-LAS3000-Plus Imaging System (Fuji, Tokyo, Japan).
Histologic Assessment
Formalin-fixed paraffin-embedded tissue blocks were cut (4 μm) for hematoxylin and eosin (H&E) and IHC stainings. Mitotic and apoptotic activity and Ki67 index were assessed as previously described [20]. Scoring of pHH3 and CC3 was performed to assess, respectively, the proliferation and apoptosis of tumor cells by counting positive nuclei or apoptotic bodies in 10 high power fields (HPFs). All assessments were carried out under 400-fold magnification (× 400) with an LH-30M microscope (Olympus, Tokyo, Japan). Microvascular density (MVD) was calculated as the average number of CD34- or VEGFR2-positive vessels in the area of the highest vascular density in five HPFs under 200-fold magnification (× 200). The total vascular area (TVA) of vessels was calculated as the average area (μm2) of CD34-positive vessels in five HPFs under × 200 using Cell D imaging software (Olympus), and images were captured with a Color View digital camera (Olympus).
Statistics
The comparisons between different groups for nonparametric values (relative tumor volume, histologic assessment) were performed using the Mann-Whitney U test. The value of P < .05 was considered as statistically significant. STATISTICA software (StatSoft, version 12.0, Tulsa, Oklahoma) was used for all calculations.
Results
Characterization of Patient-Derived Models
UZLX-STS3 and UZLX-STS5 models were established from tumors from patients diagnosed with DDLPS (Table S2). Tumors reached ± 100 mm3 within 2 to 4 months in early passages (p.0 to p.3). Afterward, the growth rates decreased and then remained stable (1-2 months). H&E staining of subsequent xenograft passages from both models confirmed the presence of histologic features of the original tumors. MDM2 immunopositivity was observed in both models (Figure 1). MDM2 amplification was observed by FISH analysis in both UZLX-STS3 and UZLX-STS5 (Figure 1) and was confirmed by qPCR (data not shown).
Figure 1.
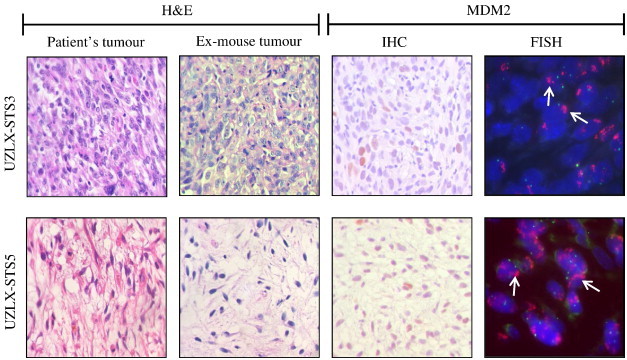
Representative images of H&E staining, MDM2 immunostaining, and FISH in UZLX-STS3 (p.7) and UZLX-STS5 (p.6) tumors, including H&E-stained images from the original patient samples. On FISH images, green signals identify chromosome 12 centromere, while the red cluster of signals (arrow) represents the amplified MDM2.
Tumor Volume Assessment
At the end of the 14-day treatment, the tumor volumes in the control group increased nearly five times in UZLX-STS3, four times in UZLX-STS5, and more than two times in SW872. Although DOX treatment slightly delayed the tumor growth in UZLX-STS3 and UZLX-STS5, the difference in tumor volume compared to untreated controls was significant only in the latter model (P < .005; Table 1 and Figure 2).
Table 1.
Relative Tumor Volume Assessment in DDLPS Models after Treatment. Mean (%) ± SD (%) was shown. Relative Tumor Volume Was Calculated by Comparing to the Tumor Volume at the Baseline (Day 0)
| Xenograft Model | Control | DOX | PAZ | PAZ + DOX |
|---|---|---|---|---|
| All models | 379 ± 165 | 303 ± 114⁎ | 189 ± 80⁎⁎## | 153 ± 48⁎⁎## |
| SW872 | 279 ± 65 | 271 ± 56 | 128 ± 22⁎⁎## | 125 ± 32⁎⁎## |
| UZLX-STS3 | 479 ± 263 | 384 ± 175 | 177 ± 39⁎⁎## | 175 ± 54⁎⁎## |
| UZLX-STS5 | 416 ± 55 | 274 ± 71⁎⁎ | 270 ± 86⁎ | 157 ± 42⁎⁎##$$ |
Statistical significance was calculated using Mann-Whitney U test.
Compared to control: *P < .05, **P < .005; compared to DOX: ##P < .005; compared to PAZ: $$P < .005.
Figure 2.
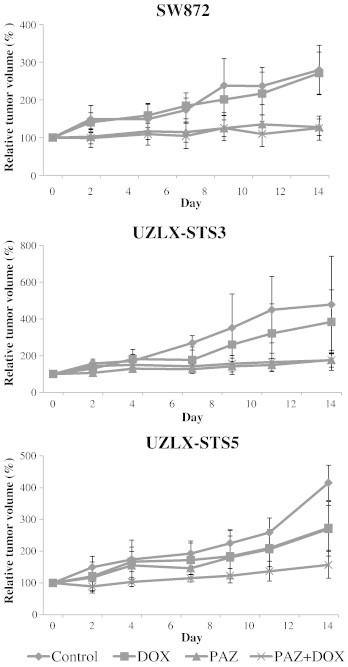
Tumor volume evaluation in SW872, UZLX-STS3, and UZLX-STS5 models. Tumor volume was measured three times per week and is presented as a relative tumor volume (%) compared to day 0. All data points are shown as mean ± SD of at least six tumors per treatment group.
In all models, at the end of the PAZ administration, the tumor volumes were one to two times higher than those at the baseline. Treatment with PAZ as a single agent significantly delayed the tumor growth compared with control animals in all models (P < .005 in SW872 and UZLX-STS3; P < .05 in UZLX-STS5) and DOX-treated groups in UZLX-STS3 and SW872 (P < .005), although it did not cause tumor shrinkage (Table 1 and Figure 2).
The PAZ + DOX combination treatment inhibited tumor growth compared to control animals and DOX-treated groups in all models tested (P < .005). Only in the UZLX-STS5 the combination treatment showed better tumor volume inhibition in comparison to single PAZ treatment (P < .05; Table 1 and Figure 2).
Histologic Assessment
Regardless of the model, tumors in the control group showed the highest level of mitotic activity among all treatment arms. PAZ alone significantly reduced mitotic activity in all the models in comparison with either control or DOX-treated cohorts, but the reduction was most pronounced in SW872 and UZLX-STS3 (P < .005, compared to untreated tumors). The combination of PAZ + DOX reduced antiproliferative activity by about two-fold in all models (P < .05, compared to control and DOX groups). These observations were confirmed by Ki67 and pHH3 immunostainings (Table 2).
Table 2.
Histologic Assessment of Proliferative and Apoptotic Activity of Tumor Cells. Histologic Assessment Was Performed for the Tumors Collected at the End of the Treatment. Results Are Shown as Fold Changes of Mean for Each Group in Comparison with Control. Arrows Indicate an Increase (Arrow Up) or Decrease (Arrow Down) of Proliferative or Apoptotic Activity in Treated Tumors versus Respective Controls
| All Models | SW872 | UZLX-STS3 | UZLX-STS5 | |
|---|---|---|---|---|
| Proliferative activity | ||||
| H&E | ||||
| DOX | ↓1.3⁎ | ↓1.3⁎ | ↓1.7⁎ | 1.0 |
| PAZ | ↓2.2⁎⁎## | ↓2.1⁎⁎## | ↓2.5⁎⁎# | ↓1.7⁎# |
| PAZ + DOX | ↓2.0⁎⁎## | ↓1.8⁎⁎## | ↓2.3⁎⁎# | ↓2.4⁎## |
| pHH3 | ||||
| DOX | ↓1.2 | ↑1.1 | ↓1.5⁎ | ↓1.2⁎ |
| PAZ | ↓1.9⁎⁎# | ↓2.0⁎⁎## | ↓1.8⁎ | ↓1.6⁎ |
| PAZ + DOX | ↓1.9⁎# | ↓2.0⁎⁎## | ↓1.9⁎ | ↓1.2 |
| Ki67 | ||||
| DOX | 1.0 | 1.0 | ↑1.2 | 1.0 |
| PAZ | ↓1.2⁎# | ↓1.2⁎ | ↓1.2# | ↓1.2 |
| PAZ + DOX | ↓1.5⁎⁎## | ↓1.3⁎⁎# | ↓2.7⁎⁎## | 1.0 |
| Apoptotic activity | ||||
| H&E | ||||
| DOX | ↑1.1 | 1.0 | ↑1.7⁎ | 1.0 |
| PAZ | ↑1.4 | ↑1.4⁎ | ↑2.2⁎⁎ | 1.0 |
| PAZ + DOX | ↑1.6⁎ | ↑1.3 | ↑2.2⁎⁎ | ↑3.0⁎ |
| CC3 | ||||
| DOX | ↑1.1 | 1.0 | ↑1.3 | ↑2.0 |
| PAZ | ↑1.3 | ↑1.1 | ↑1.7⁎⁎ | 1.0 |
| PAZ + DOX | ↑1.3 | 1.0 | ↑1.6⁎⁎ | ↑4.0⁎ |
Statistical significance was calculated using Mann-Whitney U test.
Compared to control: *P < .05, **P < .005; compared to DOX: #P < .05, ##P < .005.
H&E, hematoxylin and eosin staining; pHH3, phospho-histone H3 immunostaining; CC3, cleaved caspase-3 immunostaining.
In comparison with control, pro-apoptotic activity was observed in PAZ-treated UZLX-STS3 and SW872 tumors (P < .05). PAZ + DOX combination also induced a significant increase in apoptotic activity in UZLX-STS3 and UZLX-STS5 (P < .05). The results of apoptotic count assessment were confirmed by CC3 immunostaining (Table 2). There was no superior effect observed on cell proliferation or apoptosis in combination treatment group compared with PAZ alone.
Assessment of Angiogenesis
The antiangiogenic effect of PAZ in xenografted liposarcomas was evaluated by calculating MVD and TVA on the basis of CD34 and VEGFR2 immunostainings. As indicated by CD34 staining, after 2-week treatment, the tumors in control or in DOX treatment groups showed the highest vascular density in all models (Table 3 and Figure 3). Both PAZ-based treatments significantly reduced MVD by about two-fold and TVA by nearly three-fold in PAZ and PAZ + DOX treated tumors in comparison with control and DOX-treated tumors (P < .05; Table 3 and Figure 3). In addition, results of VEGFR2 immunostaining confirmed results obtained with CD34 staining (Table 3).
Table 3.
Assessment of MVD and TVA. Assessment Was Done for the Tumors after Treatment. Results Are Shown as Fold Changes of Mean for Each Group in Comparison with Control. Arrows Indicate an Increase (Arrow Up) or Decrease (Arrow Down) of MVD or TVA in Treated Tumors versus Respective Controls
| All Models | SW872 | UZLX-STS3 | UZLX-STS5 | |
|---|---|---|---|---|
| MVD | ||||
| CD34 | ||||
| DOX | 1.0 | ↑1.3⁎ | ↓1.1 | ↓1.2 |
| PAZ | ↓2.3⁎⁎## | ↓2.0⁎⁎## | ↓2.4⁎⁎## | ↓3.3⁎# |
| PAZ + DOX | ↓2.3⁎⁎## | ↓2.0⁎⁎## | ↓2.4⁎⁎## | ↓3.3⁎# |
| VEGFR2 | ||||
| DOX | 1.0 | 1.0 | 1.0 | ↓1.2 |
| PAZ | ↓2.0 ⁎⁎## | ↓2.0⁎⁎## | ↓2.2⁎⁎## | ↓2.0⁎# |
| PAZ + DOX | ↓2.0 ⁎⁎## | ↓2.0⁎⁎## | ↓2.2⁎⁎## | ↓2.0⁎⁎## |
| TVA | ||||
| CD34 | ||||
| DOX | ↓1.2 | ↑1.3 | ↓1.7 | ↓1.9 |
| PAZ | ↓2.6⁎⁎## | ↓1.7⁎# | ↓3.4⁎# | ↓4.2⁎# |
| PAZ + DOX | ↓2.9⁎⁎## | ↓1.7⁎## | ↓3.9⁎⁎## | ↓4.7⁎# |
Statistical significance was calculated using Mann-Whitney U test.
Compared to control: *P < .05, **P < .005; compared to DOX: #P < .05, ##P < .005.
Figure 3.
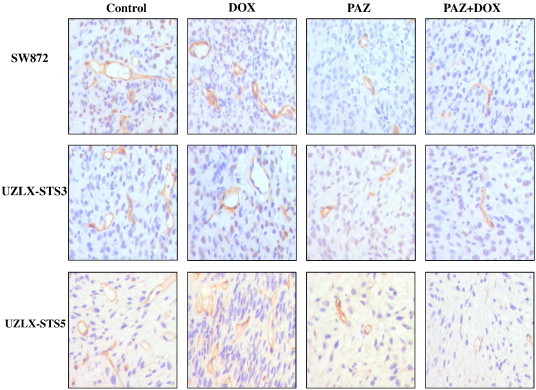
Representative images of tumor vascularity using CD34 immunostaining in SW872, UZLX-STS3, and UZLX-STS5 models. Images were captured under 400-fold magnification.
Evaluation of Oncogenic Signaling Pathways
Western blot analysis showed that AKT and MAPK were activated in all models tested, but substantial expression of VEGFR2 was only detected in SW872. No evident inhibition in either AKT or MAPK signaling pathways was observed in the treatment groups in any of the models tested (Figure S1).
Discussion
The present in vivo study reports two new patient-derived DDLPS xenografts UZLX-STS3 and UZLX-STS5 and their use for in vivo testing of antiangiogenic and cytotoxic compounds. Both established DDLPS xenograft models retained histologic and molecular features of the respective original tumor. Interestingly, it was noticed that their growth rate increased after several passages without affecting the tumors' histopathologic features. This phenomenon has been described before in a study using myxoid liposarcoma xenografts [21]. Recent studies revealed that genetic alternations associated with stromal microenvironment occurred during passages of xenografts [22], [23]. Therefore, it was hypothesized by some researchers that acquired growth advantage due to genetic alternations correlated to mouse stromal compartment during engraftment may contribute to the change in the growth rate of the xenograft [24].
We have previously shown that patient-derived xenografts from gastrointestinal stromal tumors can be successfully used for in vivo preclinical drug testing [20], [25]. In the current study, we present the activity of PAZ alone and in combination with DOX in patient- and cell line–derived DDLPS xenografts.
In the present study, DOX did not display significant antitumor activity in the three DDLPS models when compared with the control groups. Such result was not unexpected, since DOX usually only has very limited cytotoxic effects in DDLPS in the clinic. The lack of response may have also been due to the relatively low dose and i.p. administration of DOX as used in our experiments (1.2 mg/kg i.p., twice per week). We used this schedule based on published in vivo experience of other groups with this well-tolerated scheme [18], [26], [27] to minimize the potential toxicity of PAZ + DOX combination in mice.
PAZ delayed tumor growth, although there was no tumor shrinkage observed in any model. In the clinic, the primary goal of palliative treatment of locally advanced or metastatic STS is to prolong time to progression [28]. Accordingly, the objective response rate to experimental treatments in STS is not used as a primary endpoint of early clinical trials in this setting anymore [29]. Of note, the pivotal registration trial of PAZ in non-adipocytic sarcomas also showed that the rate of objective responses (all partial responses) was < 10% and the drug mainly induced disease stabilization (67%) [12]. Tumor growth delay rather than tumor shrinkage was also observed in synovial sarcoma and rhabdomyosarcoma models treated with PAZ [30], [31]. Therefore, a delayed tumor growth in our PAZ-treated tumors during the period of treatment implies a promising effect of this drug, which is currently not used for the treatment of liposarcomas outside of clinical trials.
Furthermore, emerging evidence indicates that PAZ does not only inhibit VEGF-induced endothelial cell proliferation in vitro but also blocks angiogenesis in vivo and in patients with STS [19], [32], [33]. In the present study, a remarkable reduction in MVD and TVA was observed in animals exposed to PAZ or PAZ-based combination treatment, regardless of the models tested. These changes suggested that PAZ treatment had significant antiangiogenic effect and decreased the blood flow to/in the tumor. Nevertheless, we did not observe any synergistic effect between PAZ and the anthracycline chemotherapeutic agent used in our experiments. The reason for the lack of synergy may be the disturbance of the balance between antiangiogenesis and vascular normalization as hypothesized by some researchers [34]. Such compounds can initially normalize the tumor vasculature, but continuous aggressive antiangiogenic therapy may eventually remove these vessels. Consequently, the vascular environment of tumor can become resistant to subsequent treatments and the use of the antiangiogenic compounds may even limit the delivery of other cytotoxic drugs; however, this hypothesis should be tested in the experimental setting.
To investigate whether PAZ exhibited a direct effect on oncogenic signaling pathways of tumor cells, Western blot analysis was conducted. VEGFR2 was hardly detected by Western blot analysis in the samples obtained from UZLX-STS3 and UZLX-STS5 in contrast to SW872, in which cells also showed VEGFR2 expression in vitro (data not shown). As only a fragment of tumor samples instead of whole tumors was used for lysing, it was expected that majority of proteins detected in the sample were from tumor cells and not from vessels. This may partially explain why VEGFR2 expression was remarkably higher in SW872 than those in UZLX-STS3 and UZLX-STS5, in which VEGFR2 may mostly originate from vessels rather than from tumor cells. However, AKT and MAPK pathways were both activated in the tumors of all the models, suggesting that VEGFR2 may not be primarily responsible for the activation of AKT and MAPK pathways in the DDLPS models. It has been shown that the activation of AKT is involved in the oncogenesis of DDLPS and ALT [35] and the activation of the AKT pathway in synovial cell lines can be inhibited by PAZ [30]. However, in our study, the inhibitory effect of PAZ on either AKT or MAPK pathway was not evident, suggesting that PAZ may not have a direct effect on oncogenic signaling pathways of tumor cells in the DDLPS xenografts. Taken all above together, we hypothesized that the antitumor activity of PAZ in DDLPS was mainly a result of antiangiogenic effect on tumor vessels rather than inhibition in cell signaling pathways of tumor cells.
Moreover, we observed a significant suppression of cell proliferation in PAZ-treated tumors. PAZ as a single agent or combined with DOX, however, did not enhance the pro-apoptotic activity in comparison with DOX. However, data from previous studies concerning whether PAZ had direct proliferative-inhibitory efficacy on STS cells in vitro were controversial. In synovial sarcoma cells, PAZ inhibited cell proliferation in vitro through inducing G1 arrest [30]. However, PAZ did not cause any effect on cell viability in vitro in rhabdomyosarcoma and bone sarcoma cell lines, although tumor growth delay and inhibition of angiogenesis were observed in vivo [31]. In the study of synovial sarcoma cell lines, activation of PDGFR and AKT pathways was also suppressed in the synovial cell lines with high level of PDGFR expression, implying that molecular factors of cell signaling may have an impact on the response of tumor cells to PAZ. Given these evidences above, it was rational to deduce that the direct effect of PAZ on tumor cells depended on the status of dominant tyrosine kinases of signaling pathways in tumor cells. However, as we discussed above, the evident inhibition in both AKT and MAPK pathways was not observed in the present study, which suggested that PAZ may not have a direct effect on oncogenic signaling pathways of tumor cells in the DDLPS models. However, since cancer cell survival and proliferation relies on neovascular vessels to supply oxygen and nutrients, angiogenesis blockade can also lead to inhibition of cell proliferation [36]. Thus, we hypothesized that proliferation arrest of tumor cells was mainly caused by angiogenesis inhibition in the PAZ-treated tumors in our DDLPS models.
It was already reported that DDLPS xenograft models exhibit a variable response to targeted therapies [37], which was also noticed in our study. Single agent PAZ treatment did not cause a significant difference in tumor volume compared with DOX treatment in UZLX-STS5 as it did in SW872 and UZLX-STS3, but combination treatment showed better efficacy than either single PAZ or DOX treatment in UZLX-STS5. Considering DDLPS as a highly heterogeneous tumor type, such distinct response was not unexpected, as observed also in a clinical setting. The difference in response may be due to the diverse histology of the DDLPS tumors.
In the phase II study with PAZ in multiple subtypes of STS, which was based on a two-stage design, the liposarcoma stratum was closed after stage 1 because the stratum did not meet the predefined level of antitumor activity [14]. Patient entry into this trial and the go/no go decision to proceed to stage 2 was based on the local histopathologic diagnosis. All diagnoses were reviewed by independent pathologists while the trial was proceeding. On the basis of this review, a number of treated cases were revised, some patients entered into non-liposarcoma stratum of the trial were reclassified as having DDLPS, and they did benefit from PAZ treatment [11], [14]. The fact that liposarcomas were excluded from the consecutive phase III trial is thus to be regarded as a methodological artifact. Hence, two phase II trials are currently readdressing this issue (ClinicalTrials.gov identifier: NCT01692496 and NCT01506596). In the present preclinical study, we clearly demonstrated the antitumor potential of PAZ in liposarcoma models, and we were able to show that the antitumor efficacy was mainly due to antiangiogenic effects of PAZ. We strongly believe that PAZ deserves prospective clinical testing in patients with adipocytic tumors.
In conclusion, our newly established DDLPS xenograft models, which recapitulate the histologic and molecular features of DDLPS in the clinic, provide a very useful platform for investigation of the efficacy of novel therapies in DDLPS. More importantly, we show for the first time that PAZ has antitumor activity in DDLPS xenografts, mainly through angiogenesis inhibition. Our results provide supportive evidence for the exploration of single agent PAZ in clinical trials of patients with DDLPS.
The following is the Supplementary data to this article.
Effects of PAZ and PAZ + DOX on phosphatidylinositide 3-kinase (PI3K)/AKT and MAPK signaling pathways in SW872, UZLX-STS3, and UZLX-STS5 models by Western blot analysis. The cropped images of three randomly selected tumors per treatment group are displayed. AMS (+): angiosarcoma cell line, stimulated with VEGF, used as a positive control.
In Vivo Study Design. All Mice Were Injected with SW872 Cells or Transplanted with Fresh Tumor Fragments (UZLX-STS3 or UZLX-STS5) at Both Sides.
Clinical Characteristics of Patients with DDLPS and Histopathologic Features of Xenografted Samples.
Acknowledgment
The AMS cell line was a kind gift from P. Ströbel (Universitätsmedizin Mannheim, Mannheim, Germany).
Footnotes
This article refers to supplementary materials, which are designated by Tables S1 and S2 and Figure S1 and are available online at www.transonc.com.
The experimental work was supported by research grants from the Fonds voor Wetenschappelijk Onderzoek Vlanderen (FWO grant GA01311N to P.S.) and the Chinese Scholarship Council (grant 2010601062 to H.L.). The sponsor did not influence the study design, analysis or data interpretation, or report writing and submission at any stage. Conflict of interest statement: P.S. did receive honoraria for advisory and consulting functions and educational activities from GlaxoSmithKline.
Equal contribution of the authors.
Contributor Information
Haifu Li, Email: haifu.li@med.kuleuven.be.
Agnieszka Wozniak, Email: agnieszka.wozniak@med.kuleuven.be.
Raf Sciot, Email: raf.sciot@uzleuven.be.
Jasmien Cornillie, Email: jasmien.cornillie@med.kuleuven.be.
Jasmien Wellens, Email: jasmien.wellens@med.kuleuven.be.
Thomas Van Looy, Email: thomas.vanlooy@med.kuleuven.be.
Ulla Vanleeuw, Email: ulla.vanleeuw@med.kuleuven.be.
Marguerite Stas, Email: marguerite.stas@uz.kuleuven.ac.be.
Daphne Hompes, Email: daphne.hompes@uzleuven.be.
Maria Debiec-Rychter, Email: maria.debiec-rychter@med.kuleuve.be.
Patrick Schöffski, Email: patrick.schoffski@uzleuven.be.
References
- 1.Dei Tos A.P., Pedeutour F., Marino-Enriquez A., Rossi S., Antonescu C.R., Coindre J.-M., Ladanyi M., Nielsen G.P., Mandahl M., Rosenberg A.E. Chapter: Adipocytic tumours. In: Fletcher C.D.M., Bridge J.A., Hogendoorm P.C.W., Mertens F., editors. WHO Classification of Tumours of Soft Tissue and Bone. International Agency for Research on Cancer; Lyon: 2013. pp. 33–43. [Google Scholar]
- 2.Dalal K.M., Antonescu C.R., Singer S. Diagnosis and management of lipomatous tumors. J Surg Oncol. 2008;97:298–313. doi: 10.1002/jso.20975. [DOI] [PubMed] [Google Scholar]
- 3.Jones R.L., Fisher C., Al-Muderis O., Judson I.R. Differential sensitivity of liposarcoma subtypes to chemotherapy. Eur J Cancer. 2005;41:2853–2860. doi: 10.1016/j.ejca.2005.07.023. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman A., Lazar A.J., Pollock R.E., Lev D. New frontiers in the treatment of liposarcoma, a therapeutically resistant malignant cohort. Drug Resist Updat. 2011;14:52–66. doi: 10.1016/j.drup.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Welti J., Loges S., Dimmeler S., Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–3200. doi: 10.1172/JCI70212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Potti A., Ganti A.K., Tendulkar K., Sholes K., Chitajallu S., Koch M., Kargas S. Determination of vascular endothelial growth factor (VEGF) overexpression in soft tissue sarcomas and the role of overexpression in leiomyosarcoma. J Cancer Res Clin Oncol. 2004;130:52–56. doi: 10.1007/s00432-003-0504-0. [DOI] [PubMed] [Google Scholar]
- 7.Kilvaer T.K., Valkov A., Sorbye S., Smeland E., Bremnes R.M., Busund L.T., Donnem T. Profiling of VEGFs and VEGFRs as prognostic factors in soft tissue sarcoma: VEGFR-3 is an independent predictor of poor prognosis. PLoS One. 2010;5:e15368. doi: 10.1371/journal.pone.0015368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L., Hannay J.A., Liu J., Das P., Zhan M., Nguyen T., Hicklin D.J., Yu D., Pollock R.E., Lev D. Vascular endothelial growth factor overexpression by soft tissue sarcoma cells: implications for tumor growth, metastasis, and chemoresistance. Cancer Res. 2006;66:8770–8778. doi: 10.1158/0008-5472.CAN-06-1217. [DOI] [PubMed] [Google Scholar]
- 9.Yoon S.S., Segal N.H., Park P.J., Detwiller K.Y., Fernando N.T., Ryeom S.W., Brennan M.F., Singer S. Angiogenic profile of soft tissue sarcomas based on analysis of circulating factors and microarray gene expression. J Surg Oncol. 2006;135:282–290. doi: 10.1016/j.jss.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 10.Hamberg P., Verweij J., Sleijfer S. (Pre-)clinical pharmacology and activity of pazopanib, a novel multikinase angiogenesis inhibitor. Oncologist. 2010;15:539–547. doi: 10.1634/theoncologist.2009-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schöffski P. Pazopanib in the treatment of soft tissue sarcoma. Expert Rev Anticancer Ther. 2012;12:711–723. doi: 10.1586/era.12.41. [DOI] [PubMed] [Google Scholar]
- 12.Van der Graaf W.T., Blay J.-Y., Chawla S.P., Kim D.W., Bui-Nguyen B., Casali P.G., Schöffski P., Aglietta M., Staddon A.P., Beppu Y. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379:1879–1886. doi: 10.1016/S0140-6736(12)60651-5. [DOI] [PubMed] [Google Scholar]
- 13.Sternberg C.N., Davis I.D., Mardiak J., Szczylik C., Lee E., Wagstaff J., Barrios C.H., Salman P., Gladkov O.A., Kavina A. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 14.Sleijfer S., Ray-Coquard I., Papai Z., Le Cesne A., Scurr M., Schoffski P., Collin F., Pandite L., Marreaud S., De Brauwer A. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043) J Clin Oncol. 2009;27:3126–3132. doi: 10.1200/JCO.2008.21.3223. [DOI] [PubMed] [Google Scholar]
- 15.Stratford E.W., Castro R., Daffinrud J., Skårn M., Lauvrak S., Munthe E., Myklebost O. Characterization of liposarcoma cell lines for preclinical and biological studies. Sarcoma. 2012;2012:148614. doi: 10.1155/2012/148614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang K., Chu K., Wu X., Gao H., Wang J., Yuan Y.C., Loera S., Ho K., Wang Y., Chow W. Amplification of FRS2 and activation of FGFR/FRS2 signaling pathway in high-grade liposarcoma. Cancer Res. 2013;73:1298–1307. doi: 10.1158/0008-5472.CAN-12-2086. [DOI] [PubMed] [Google Scholar]
- 17.Debiec-Rychter M., Cools J., Dumez H., Sciot R., Stul M., Mentens N., Vranckx H., Wasag B., Prenen H., Roesel J. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 18.Ren W., Korchin B., Zhu Q.S., Wei C., Dicker A., Heymach J., Lazar A., Pollock R.E., Lev D. Epidermal growth factor receptor blockade in combination with conventional chemotherapy inhibits soft tissue sarcoma cell growth in vitro and in vivo. Clin Cancer Res. 2008;14:2785–2795. doi: 10.1158/1078-0432.CCR-07-4471. [DOI] [PubMed] [Google Scholar]
- 19.Kumar R., Knick V.B., Rudolph S.K., Johnson J.H., Crosby R.M., Crouthamel M.C., Hopper T.M., Miller C.G., Harrington L.E., Onori J.A. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol Cancer Ther. 2007;6:2012–2021. doi: 10.1158/1535-7163.MCT-07-0193. [DOI] [PubMed] [Google Scholar]
- 20.Floris G., Debiec-Rychter M., Sciot R., Stefan C., Fieuws S., Machiels K., Atadja P., Wozniak A., Faa G., Schöffski P. High efficacy of panobinostat towards human gastrointestinal stromal tumors in a xenograft mouse model. Clin Cancer Res. 2009;15:4066–4076. doi: 10.1158/1078-0432.CCR-08-2588. [DOI] [PubMed] [Google Scholar]
- 21.Frapolli R., Tamborini E., Virdis E., Bello E., Tarantino E., Marchini S., Grosso F., Sanfilippo R., Gronchi A., Tercero J.C. Novel models of myxoid liposarcoma xenografts mimicking the biological and pharmacologic features of human tumors. Clin Cancer Res. 2010;16:4958–4967. doi: 10.1158/1078-0432.CCR-10-0317. [DOI] [PubMed] [Google Scholar]
- 22.Zhao X., Liu Z., Yu L., Zhang Y., Baxter P., Voicu H., Gurusiddappa S., Luan J., Su J.M., Leung H.C. Global gene expression profiling confirms the molecular fidelity of primary tumor-based orthotopic xenograft mouse models of medulloblastoma. Neuro Oncol. 2012;14:574–583. doi: 10.1093/neuonc/nos061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reyal F., Guyader C., Decraene C., Lucchesi C., Auger N., Assayag F., De Plater L., Gentien D., Poupon M.F., Cottu P. Molecular profiling of patient-derived breast cancer xenografts. Breast Cancer Res. 2012;14:R11. doi: 10.1186/bcr3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siolas D., Hannon G.J. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73:5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Floris G., Wozniak A., Sciot R., Li H., Friedman L., Van Looy T., Wellens J., Vermaelen P., Deroose C.M., Fletcher J.A. A potent combination of the novel PI3K Inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin Cancer Res. 2013;19:620–630. doi: 10.1158/1078-0432.CCR-12-2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang S., Ren W., Liu J., Lahat G., Torres K., Lopez G., Lazar A.J., Hayes-Jordan A., Liu K., Bankson J. TRAIL and doxorubicin combination induces proapoptotic and antiangiogenic effects in soft tissue sarcoma in vivo. Clin Cancer Res. 2010;16:2591–2604. doi: 10.1158/1078-0432.CCR-09-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez G., Liu J., Ren W., Wei W., Wang S., Lahat G., Zhu Q.S., Bornmann W.G., McConkey D.J., Pollock R.E. Combining PCI-24781, a novel histone deacetylase inhibitor, with chemotherapy for the treatment of soft tissue sarcoma. Clin Cancer Res. 2009;15:3472–3483. doi: 10.1158/1078-0432.CCR-08-2714. [DOI] [PubMed] [Google Scholar]
- 28.Grimer R., Judson I., Peake D., Seddon B. Guidelines for the management of soft tissue sarcomas. Sarcoma. 2010;2010:506182. doi: 10.1155/2010/506182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Glabbeke M., Verweij J., Judson I., Nielsen O.S., EORTC Soft Tissue and Bone Sarcoma Group Progression-free rate as the principal end-point for phase II trials in soft-tissue sarcomas. Eur J Cancer. 2002;38:543–549. doi: 10.1016/s0959-8049(01)00398-7. [DOI] [PubMed] [Google Scholar]
- 30.Hosaka S., Horiuchi K., Yoda M., Nakayama R., Tohmonda T., Susa M., Nakamura M., Chiba K., Toyama Y., Morioka H. A novel multi-kinase inhibitor pazopanib suppresses growth of synovial sarcoma cells through inhibition of the PI3K-AKT pathway. J Orthop Res. 2013;30:1493–1498. doi: 10.1002/jor.22091. [DOI] [PubMed] [Google Scholar]
- 31.Kumar S., Mokhtari R.B., Sheikh R., Wu B., Zhang L., Xu P., Man S., Oliveira I.D., Yeger H., Kerbel R.S. Metronomic oral topotecan with pazopanib is an active antiangiogenic regimen in mouse models of aggressive pediatric solid tumor. Clin Cancer Res. 2010;17:5656–5667. doi: 10.1158/1078-0432.CCR-11-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Podar K., Tonon G., Sattler M., Tai Y.T., Legouill S., Yasui H., Ishitsuka K., Kumar S., Kumar R., Pandite L.N. The small-molecule VEGF receptor inhibitor pazopanib (GW786034B) targets both tumor and endothelial cells in multiple myeloma. Proc Natl Acad Sci U S A. 2006;103:19478–19483. doi: 10.1073/pnas.0609329103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glade Bender J.L., Lee A., Reid J.M., Baruchel S., Roberts T., Voss S.D., Wu B., Ahern C.H., Ingle A.M., Harris P. Phase I pharmacokinetic and pharmacodynamic study of pazopanib in children with soft tissue sarcoma and other refractory solid tumors: a Children's Oncology Group Phase I consortium report. J Clin Oncol. 2013;31:3034–3043. doi: 10.1200/JCO.2012.47.0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jain R.K. Antiangiogenic therapy normalization of tumour vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 35.Gutierrez A., Snyder E.L., Marino-Enriquez A., Zhang Y.X., Sioletic S., Kozakewich E., Grebliunaite R., Ou W.B., Sicinska E., Raut C.P. Aberrant AKT activation drives well-differentiated liposarcoma. Proc Natl Acad Sci U S A. 2011;108:16386–16391. doi: 10.1073/pnas.1106127108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leite de Oliveira R., Hamm A., Mazzone M. Growing tumor vessels: more than one way to skin a cat—implications for angiogenesis targeted cancer therapies. Mol Aspects Med. 2011;32:71–87. doi: 10.1016/j.mam.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Smith K.B., Tran L.M., Tam B.M., Shurell E.M., Li Y., Braas D., Tap W.D., Christofk H.R., Dry S.M., Eilber F.C. Novel dedifferentiated liposarcoma xenograft models reveal PTEN down-regulation as a malignant signature and response to PI3K pathway inhibition. Am J Pathol. 2013;180:1400–1411. doi: 10.1016/j.ajpath.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of PAZ and PAZ + DOX on phosphatidylinositide 3-kinase (PI3K)/AKT and MAPK signaling pathways in SW872, UZLX-STS3, and UZLX-STS5 models by Western blot analysis. The cropped images of three randomly selected tumors per treatment group are displayed. AMS (+): angiosarcoma cell line, stimulated with VEGF, used as a positive control.
In Vivo Study Design. All Mice Were Injected with SW872 Cells or Transplanted with Fresh Tumor Fragments (UZLX-STS3 or UZLX-STS5) at Both Sides.
Clinical Characteristics of Patients with DDLPS and Histopathologic Features of Xenografted Samples.