

Abstract
Several microRNAs (miRNAs) are associated with the molecular pathogenesis of hepatocellular carcinoma (HCC). However, previous studies analyzing the dysregulation of miRNAs in HCC show heterogeneous results. We hypothesized that part of this heterogeneity might be attributable to variations of miRNA expression deriving from the HCC capsule or the fibrotic septa within the peritumoral tissue used as controls. Tissue from surgically resected hepatitis C–associated HCC from six well-matched patients was microdissected using laser microdissection and pressure catapulting technique. Four distinct histologic compartments were isolated: tumor parenchyma (TP), fibrous capsule of the tumor (TC), tumor-adjacent liver parenchyma (LP), and cirrhotic septa of the tumor-adjacent liver (LC). MiRNA expression profiling analysis of 1105 mature miRNAs and precursors was performed using miRNA microarray. Principal component analysis and consecutive pairwise supervised comparisons demonstrated distinct patterns of expressed miRNAs not only for TP versus LP (e.g., intratumoral down-regulation of miR-214, miR-199a, miR-146a, and miR-125a; P< .05) but also for TC versus LC (including down-regulation within TC of miR-126, miR-99a/100, miR-26a, and miR-125b; P< .05). The tumor capsule therefore demonstrates a tumor-like phenotype with down-regulation of well-known tumor-suppressive miRNAs. Variations of co-analyzed fibrotic tissue within the tumor or in controls may have profound influence on miRNA expression analyses in HCC. Several miRNAs, which are proposed to be HCC specific, may indeed be rather associated to the tumor capsule. As miRNAs evolve to be important biomarkers in liver tumors, the presented data have important translational implications on diagnostics and treatment in patients with HCC.
Introduction
Hepatocellular carcinoma (HCC) is a major complication of end-stage liver disease. The worldwide annual incidence of HCC is estimated to be 600,000 [1], [2], [3], [4]. In Western countries and Japan, the incidence of hepatitis C virus (HCV)–related liver cirrhosis and liver cancer is rising because of the HCV epidemic [5], [6]. A further increase is assumed, because obesity and nonalcoholic steatohepatitis have been identified as risk factors of HCC especially in Western countries [7].
MicroRNAs (miRNAs) are a class of endogenous small regulatory RNA molecules that regulate mRNA expression by either translational repression or mRNA degradation [8]. MiRNAs are involved in the regulation of genes associated with different biologic processes, such as development, cell proliferation, apoptosis, and tumorigenesis [9]. Various studies have uncovered dysregulation of different miRNAs in HCC, with implication for prognosis and as possible targets for future therapy [10], [11], [12]. Unfortunately, data published on miRNA expression in HCC are highly heterogeneous and partly conflicting. Of note, only a small number of specific miRNAs were found consistently upregulated or downregulated in different studies [13]. Part of this heterogeneity of expression data may be attributable to HCC being a heterogeneous disease. Expression patterns of miRNAs correlate with activation of different well-known oncogenic pathways in HCC (e.g., β-catenin, mTOR, or interferon response–related pathways) [14]. Moreover, some miRNAs correlate with early stages of liver carcinogenesis, while at later HCC stages a global miRNA down-regulation has been found [15]. Apart from cancer cell–specific factors, miRNA expression in HCC is influenced by non–tumor-associated factors such as differences in ethnicity, etiology of liver disease, or progression of underlying liver fibrosis [16], [17]. Early studies investigating miRNA expression in HCC compared HCC tissue to healthy liver tissue of different patient cohorts [18]. More recently, tumor-adjacent non-tumorous tissue (TAT) of the same HCC patient has been preferred as control [19], [20]. Throughout most of the studies, whole tissue sections from either fresh tissue or formalin-fixed paraffin-embedded (FFPE) tissue have been used. When HCC whole tissue sections are used, the extracted RNA originates from all cell types comprising the tumor, among which the fibrous compartment may contribute a particular source of variation, especially as the HCC capsule not only surrounds the macroscopic tumor nodule but often shows a radial distribution throughout the tumor section. In addition, tissue sections of TAT are a similar mixture of liver cells and cirrhotic septa. Both capsule tissues consist of fibrous connective tissue and different cellular components including fibroblasts, lymphocytes, and granulocytes. We therefore speculated that quality and quantity of co-analyzed non-tumorous tissue could contribute to the heterogeneity of miRNA expression data in HCC.
To study the influence of non–tumor-associated factors on HCC miRNA expression in more detail, we isolated different tissue compartments of the tumor-infiltrated liver and studied patterns of miRNAs in their respective microenvironment.
Material and Methods
Patient Selection
The current study is a retrospective analysis, which was approved by the local Ethics Committee of the University Hospital Frankfurt, Germany (Ethik-Kommission des Fachbereichs Medizin der Johann Wolfgang Goethe-Universität). Written informed consent was obtained from all patients. The screening group of six patients was carefully selected for matching clinical characteristics to eliminate bias by ethnicity, tumor stage, liver function, or type of underlying liver disease. All patients underwent primary surgical resection of HCV-associated HCC [all Barcelona Clinic Liver Cancer Staging System (BCLC) stage 0/A; see Table 1]. The validation group consists of 20 patients, all HCV-positive and subject to either primary resection or liver transplantation (Table 2).
Table 1.
Patient Cohort Used for Microarray Experiments (Screening Cohort)⁎
| Age—mean (min, max) | 65 (54–80) | (years) |
| Gender—no. (%) | ||
| Female | 4 | (66) |
| Male | 2 | (33) |
| Ancestry region—no. | ||
| Europe | 6 | (100) |
| Underlying liver cirrhosis | 6 | (100) |
| ISHAK fibrosis score = 6 | 6 | (100) |
| METAVIR fibrosis score = F4 | 6 | (100) |
| Cause of liver cirrhosis—no. (%) | ||
| HCV | 6 | (100) |
| HCV genotype—no. (%) | ||
| Genotype 1 | 5 | (83) |
| Genotype 3 | 1 | (17) |
| Body mass index—mean ± SD** | 31.5 ± 4.2 | (kg/m2) |
| ECOG performance status—no. (%)** | ||
| 0 | 5 | (83) |
| 1 | 1 | (17) |
| Child-Pugh class—no. (%)** | ||
| A | 6 | (100) |
| MELD score—median (min, max)** | 9.33 (7, 11) | |
| AFP serum level—median (min, max)** | 15 (2.9, 58) | ng/ml |
| BCLC stage—no. (%)** | ||
| 0 | 5 | (83) |
| A | 1 | (17) |
| Histologic grading (G)—no. (%)* | ||
| G1 | 2 | (11) |
| G2 | 4 | (79) |
*Screening microarray experiments were performed for seven patients. One technical outlier sample was removed from the analysis. **At the time point of HCC resection.
Table 2.
Patient Cohort Used for Quantitative PCR Experiments (Validation Cohort)
| Age—mean (min, max) | 58 (42–79) | (years) |
| Gender—no. (%) | ||
| Female | 5 | (33) |
| Male | 15 | (66) |
| Ancestry region—no. | ||
| Europe | 18 | (90) |
| Asia | 1 | (5) |
| Africa | 1 | (5) |
| Underlying liver cirrhosis | 20 | (100) |
| ISHAK fibrosis score = 6 | 20 | (100) |
| METAVIR fibrosis score = F4 | 20 | (100) |
| Cause of liver cirrhosis—no. (%) | ||
| HCV | 20 | (100) |
| HCV genotype—no. (%) | ||
| Genotype 1 | 11 | (55) |
| Genotype 2 | 1 | (5) |
| Genotype 3 | 2 | (10) |
| Unknown | 6 | (30) |
| Child-Pugh class—no. (%)⁎ | ||
| A | 16 | (80) |
| B | 3 | (15) |
| C | 1 | (5) |
| MELD score—median (min, max)⁎ | 11.6 (6, 29) | |
| AFP serum level—median (min, max)⁎ | 15 (1.9, 84,798) | ng/ml |
| BCLC stage—no. (%)⁎ | ||
| 0 | 8 | (40) |
| A | 11 | (55) |
| B | 1 | (5) |
| Histologic grading (G)—no. (%)⁎ | ||
| G1 | 3 | (15) |
| G2 | 13 | (65) |
| G3 | 3 | (15) |
| G4 | 1 | (5) |
| Therapy type—no. (%) | ||
| Surgical resection | 11 | (55) |
| Liver transplantation | 9 | (45) |
| TACE prior to resection or transplantation—no. (%) | 12 | (60) |
At the time point of HCC resection. MELD, Model for End-Stage Liver Disease.
Histologic Assessment of Liver Cancer
Diagnosis of HCC was confirmed by an experienced pathologist. Tumors were classified according to BCLC criteria, and staging and grading were assessed following current TNM classification of malignant tumors guidelines [3], [21]. All patients featured underlying liver cirrhosis. Two observers independently evaluated the percentage of each tissue compartment within the whole tissue sections (Tables S1 and S2 [48]).
Isolation and Expression Profiling Analysis of miRNA
FFPE tissue from surgically resected HCC was microdissected using laser microdissection and pressure catapulting technique with a UV laser beam (PALM). Four distinct histologic compartments were isolated: tumor parenchyma (TP), fibrinous capsule of the tumor (TC), tumor-adjacent liver parenchyma (LP), and cirrhotic septa of the tumor-adjacent liver (LC; Figure S1). Total RNA was extracted from tumor FFPE specimen using an RNeasy FFPE Kit (Qiagen, Hilden, Germany) following the manufacturer's protocol. RNA quantification was performed using a Nanodrop 2000 spectrometer (Thermo Fisher Scientific, Wilmington, USA) and 50 ng of total RNA was used for further analysis. We performed a large-scale miRNA expression profiling analysis of 1105 miRNAs using Affymetrix GeneChip miRNA Array v2.0 in a total of 28 tissue compartments. The probe set composition of the arrays is available at www.affymetrix.com. Microarray experiments were performed following the manufacturer's protocol with exception to the hybridization duration. Hybridization time was extended from 18 to 40 hours to compensate for low amount RNA input, following the recommendation of the manufacturer. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus [22] and are accessible through Gene Expression Omnibus Series accession number GSE54537 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE54537).
Real-Time Polymerase Chain Reaction miRNA Expression Analysis
TaqMan MicroRNA Q-RT-PCR assays (Applied Biosystems, Darmstadt, Germany) were used to quantify miRNAs according to the manufacturer’s protocol. Expression was analyzed for three miRNAs (hsa-miR-146a, hsa-miR-126, and hsa-miR-150) and one endogenous control (U6). Samples were analyzed in triplicate, and ΔCt values were calculated using the endogenous control.
MiRNA In Situ Hybridization
miRNA in situ hybridization (miRNA-ISH) was carried out as reported elsewhere [23]. Briefly, 11 digoxigenin-labeled locked nucleic acid probes antisense to miR-150 or let-7a (Exiqon, Vedbaek, Denmark) were used for overnight staining on tissue sections at 61°C. Detection was accomplished with anti-digoxigenin alkaline phosphatase-conjugated Fab fragment followed by nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate color development (Roche, Basel, Switzerland).
Bioinformatic and Statistical Analyses
Clinical and biochemical characteristics of patients were expressed as mean ± SD or median and range as appropriate. Unless indicated otherwise, all tests were two tailed and P values < .05 were considered significant. Unpaired or paired Student’s t test was applied as appropriate. Bioinformatic analysis of miRNA profiles was performed using the statistical computing environment R. Additional software packages (HTqPCR, geneplotter, and gplots) were taken from the Bioconductor project. Unsupervised hierarchical clustering was performed for miRNAs with an SD ≥ 1.5 across all samples using the Manhattan distance method and the average linkage method. For the supervised analysis, expression intensity and variance filters were used to reduce the dimension of the microarray data. Data were filtered by an intensity filter (gene intensity is > 100 in at least 25% of the samples, if the group size is equal) as well as a variance filter (the interquartile range of log2 intensities is > 0.5, if the group size is equal). After application of paired t test for comparisons of compartments of each patient sample, average P values were calculated to identify differentially expressed miRNAs between two compartment groups. For multiple testing problems, a false discovery rate was used [24]. In addition, fold change (FC) between the two groups was calculated for each gene. DIANA miRPath v.2.0 was used as a computational predictive model to calculate potentially targeted genes and pathways using microT-CDS database (P value threshold of .05, MicroT threshold of 0.8, false discovery rate correction, and conservative stats were applied) [25]. Depicted pathways were derived from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [26].
Results
Using Affymetrix GeneChipR miRNA microarray, miRNA expression profiles were generated from six HCC samples that each had been laser-microdissected into four subcompartments. After global filtering (only miRNAs with an SD ≥ 1.5 across all samples were used), a panel of 301 miRNAs was used to perform unsupervised hierarchical clustering analysis (UCA) from 24 subcompartments. UCA did not show a tissue-wise grouping of the four isolated histologic sectors but rather showed a patient-dependent clustering of subcompartments indicating a high interindividual variance of miRNA expression patterns (Figure 1).
Figure 1.
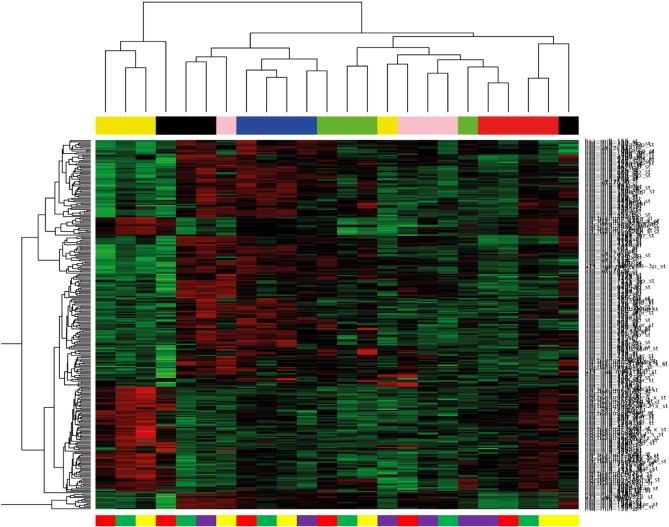
Illustration of UCA of miRNA expression profiles generated from a total of 24 subcompartments. Heat map of profiles deriving from microdissected FFPE tissue of six surgically resected HCC specimens (all HCV positive). The dendrogram is based on the semiquantitative expression data after global filtering for miRNA probe sets with SD > 1.5. The color code on top of the illustration represents the different patient samples. Subcompartments are displayed as color code below (tumor cells, yellow bars; liver cells, green bars; tumor fibrinous capsule, red bars; liver fibrinous capsule, light purple bars). High expression in red and low expression in green.
To increase robustness against the high global variation of miRNA expression between the samples, a principal component analysis (PCA) was calculated for each of the four subcompartments. Here, the non-tumorous tissue compartments (TC and LC) show a closer relationship, while liver tissue and tumor tissue remain widely separated (Figure 2). Nevertheless, miRNA expression in TC and LC, despite being morphologically similar, shows a clear separation in the PCA.
Figure 2.
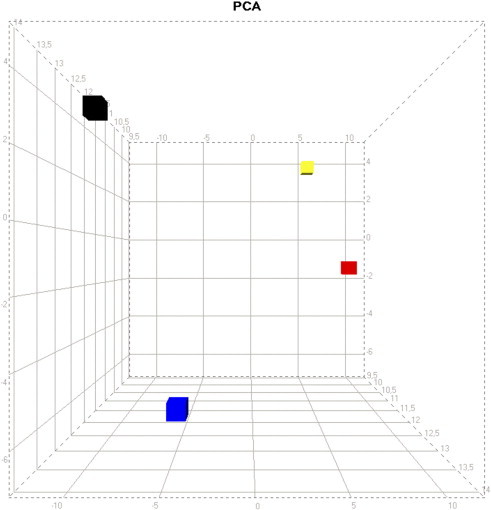
PCA of miRNA expression profiles. Profiles derived from microdissected FFPE tissue of six surgically resected HCC specimens (all HCV positive). The plot is based on the semiquantitative expression data after global filtering for miRNA probe sets with SD > 1.5. The first principal component (x-axis) has the largest possible variance, and each succeeding component (y-axis, z-axis) has the highest variance possible while being uncorrelated with the preceding components. Subcompartment color code: tumor cells, black; liver cells, blue; liver fibrinous capsule, red; tumor fibrinous capsule, yellow.
To correct for the high interindividual variation observed in the UCA, the supervised analysis was adapted to first calculate intraindividual pairwise comparisons of subcompartments, while the comparisons between different HCC samples were analyzed in a second step. Pairwise comparisons of subcompartments showed distinct patterns of expressed miRNAs for LC versus LP, TC versus TP, LP versus TP, and LC versus TC (Table 3). Of note, LC and LP show a number of differentially expressed miRNAs, supporting the hypothesis that the degree of liver cirrhosis may significantly influence the results of the miRNA expression analyses from patients with HCC. This may be of special importance, when whole tissue sections (comprising tumor tissue as well as TAT) are used for miRNA profiling. For validation purposes, miRNAs differentially expressed between LC and LP in this cohort were screened for in the expression data from healthy non-cirrhotic livers deriving from one of our previous works [27]. No dysregulation was found for these miRNAs in healthy livers. Moreover, though LC and TC show identical histomorphology and have a similar composition, there is a pronounced difference in miRNA expression.
Table 3.
Supervised Analysis—Differentially Expressed miRNAs between Subcompartments
| Liver Capsule versus Liver Parenchyma |
Tumor Capsule versus TP |
Liver Parenchyma versus TP |
Liver Capsule versus Tumor Capsule |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| miRNA | FC | P Value | miRNA | FC | P Value | miRNA | FC | P Value | miRNA | FC | P Value |
| miR-19b | − 2.3 | .03 | miR-548c-3p | 3.6 | .02 | miR-214 | 3.9 | 3.1E−6 | miR-126 | 2.8 | .002 |
| let-7a | 5.3 | .03 | miR-3201 | − 2.1 | .03 | miR-199a-5p | 2.8 | 2.7E−4 | miR-99a | 2.0 | .003 |
| miR-16-2* | − 3.1 | .04 | p-miR-1979* | − 1.8 | .03 | miR-199a-3p | 3.8 | 2.9E−4 | miR-146a | 4.6 | .003 |
| miR-150 | 4.2 | .05 | p-miR-520 h | − 1.9 | .04 | miR-199b-3p | 3.4 | 4.9E−4 | let-7 g | 3.4 | .004 |
| miR-548a-3p | − 2.5 | .05 | miR-150 | 3.4 | .04 | miR-125a-5p | 2.7 | .002 | miR-27a | 2.8 | .01 |
| miR-425 | 1.7 | .05 | miR-3128 | − 2.3 | .04 | miR-342-3p | 2.7 | .003 | miR-26a | 2.5 | .01 |
| p-miR-522 | − 1.9 | .04 | miR-99b | 1.9 | .01 | miR-150 | 3.7 | .02 | |||
| miR-335 | − 2.1 | .04 | miR-145 | 2.6 | .01 | miR-125b | 2.8 | .02 | |||
| miR-548e | 6.0 | .05 | let-7e | 2.7 | .02 | let-7i | 3.5 | .02 | |||
| let-7a | 4.9 | .05 | miR-27a | 2.6 | .02 | miR-100 | 2.2 | .02 | |||
| p-miR-548i | 2.4 | .05 | miR-195 | 2.9 | .02 | miR-155 | 5.0 | .02 | |||
| miR-548a-3p | 1.8 | .05 | miR-143 | − 2.9 | .02 | miR-199a-5p | 3.0 | .03 | |||
| p-miR-520g | − 1.6 | .05 | miR-23a | 2.8 | .02 | let-7c | 2.8 | .03 | |||
| miR-3148 | − 2.6 | .05 | miR-150 | − 2.8 | .03 | miR-768-5p | 2.0 | .03 | |||
| miR-146a | 3.9 | .03 | miR-143 | 4.8 | .03 | ||||||
| miR-155 | 2.8 | .04 | miR-23a | 3.3 | .03 | ||||||
| let-7a | 3.1 | .04 | |||||||||
| miR-195 | 3.8 | .04 | |||||||||
| let-7f | 3.4 | .05 | |||||||||
| miR-199a-3p | 5.7 | .05 | |||||||||
| miR-342-3p | 2.7 | .05 | |||||||||
MiRNA expression was compared case-wise for two compartments, respectively. Semiquantitative FC of miRNA expression and P value (paired t test) were calculated. miRNAs with FC ≥ 2 and P≤ .05 are displayed. MiRNA that were chosen for validation experiments are printed in bold. *miR-1979 is rather a fragment of Y RNA and no miRNA. p-mir, precursor-miRNA.
The results show a global down-regulation of miRNAs in TC compared to LC, including miR-126, miR-125b, let-7 group, or miR-26a. DIANA miRPath v2.0 enrichment analysis of predicted miRNA target genes was used to identify pathways influenced by the found miRNA profiles [25]. Interestingly, the TC profile was highly enriched for several well-known oncogenic signaling pathways (e.g., Wnt, MAPK, and PI3K/Akt pathways) as well as profiles of focal adhesion and extracellular matrix signaling (P< 1E−10; Table W1). Wnt and PI3K/Akt pathways with potentially affected genes marked are shown in Figure S2.
The version of the Affymetrix miRNA Array (v2.0) used in the present study allows screening for not only 1105 mature miRNAs but also for their respective precursor (pre)-miRNA. The comparison of TC and TP revealed several differentially expressed miRNAs, which have only recently been added to the miRBase database (e.g., miR-3201, miR-3128, and miR-3148). However, the majority of genes we found dysregulated between compartments include previously studied miRNAs. While five pre-miRNAs (pre-miR-1979, pre-miR-520h, pre-miR-522, pre-miR-548i, and pre-miR-520g) were found downregulated within the TC compartment as compared to TP, all other comparisons of subcompartments did not feature differentially expressed pre-miRNAs.
To simulate the comparison of whole sections of liver versus tumor tissue, pairs of subcompartments (LP and LC vs TP and TC) were pooled and consecutively compared for miRNA expression differences. As the individual compartments comprise different area fractions within the tissue samples, every case was reviewed to estimate percentages for each subcompartment within the whole tissue area. When the miRNA expression data were reanalyzed as pooled data of TC and TP versus LC and LP, taking into account the different percentages of tissue amounts, we found no differently expressed miRNAs (Table S3).
Results from the microarray experiments were validated in a larger independent cohort of patients with HCC (n= 20) using real-time polymerase chain reaction (PCR) for four exemplary miRNAs (miR-150, miR-126, and miR-146a). Differential regulation of miRNAs discovered in the screening population was confirmed in the validation cohort (Figure 3). The quantitative measurement of expression correlated well with semiquantitative results from the array experiments.
Figure 3.
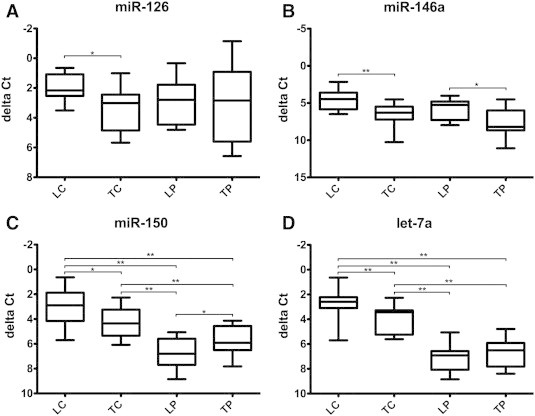
Validation by real-time RT-PCR for miRNAs miR-126, miR-146a, miR-150, and let-7a. Four representative miRNAs were selected for validation by quantitative RT-PCR in an independent cohort of 20 HCV-positive HCC specimens. Samples were analyzed in triplicate and ΔCt values were calculated using an endogenous control (U6). The analysis confirmed the dysregulation of selected miRNAs in the different subcompartments with comparable results to the microarray analysis (Table 3). ΔCt values are depicted on the y-axis with inverted scale. Values are displayed as box plots with mean, whiskers min to max (*P< .05; **P< .01).
Furthermore, miRNA-ISH was performed as an additional step of validation of two differentially expressed miRNAs (miR-150 and let-7a). In line with the microarray data and real-time PCR results, both miRNAs show markedly higher expression in the fibrous tissue compartments and decreased expression in TC compared to LC (Figure 4).
Figure 4.
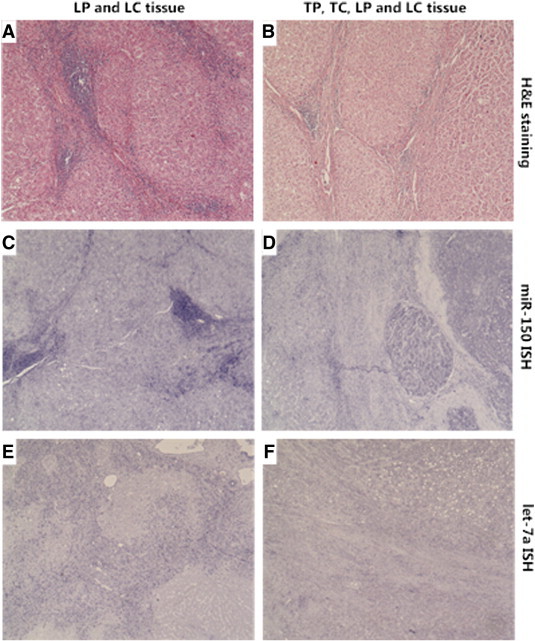
miRNA-ISH of miR-150 and let-7a. Representative microscopic pictures (40-fold magnification) of H&E staining and miRNA-ISH on paraffin-embedded tissue sections (miRNA is stained blue). Left column (A, C, E): non-tumorous liver cells (LP) and surrounding fibrotic tissue (LC); right column (B, D, F): non-tumorous liver (LP) with fibrotic tissue (LC) and HCC (TC) with surrounding tumor capsule (TC). (A) H&E staining of LP and LC; (B) H&E staining of TP and TC; (C) miRNA-ISH for miR-150 demonstrating high expression in LC compared to LP; (D) miRNA-ISH for miR-150 demonstrating high expression in LC and TC and higher expression in TP compared to LP; (E) miRNA-ISH for let-7a demonstrating strongly increased expression in LC compared to LP; (F) miRNA-ISH for let-7a demonstrating high general expression in the two fibrotic tissue compartments with stronger expression in LC versus TC and no difference between LP and TP. Microscope: Nikon Eclipse E1000. Camera: Nikon DS-Ri 1.
Twelve of 20 patients within the validation group received transarterial chemoembolization (TACE). The samples from the validation cohort were analyzed for impact of TACE therapy on the expression of the validated miRNAs. There were no significant differences between the TACE and non-TACE groups for the expression of miR-150, miR-126, miR-146a, or let-7a (Figure S3).
Discussion
HCC is an increasing burden in the world and is the second leading cause of cancer-related mortality [4]. Most patients are diagnosed at advanced stages, and thus, overall therapeutic options are limited and the prognosis is poor [21]. Current treatment strategies for advanced disease offer only limited efficacy [28], [29]. MiRNAs are promising candidates to act as future biomarkers in HCC as various studies associated miRNA expression with early detection, prognosis, or recurrence of HCC after resection [14], [30], [31]. Moreover, influencing oncogenic or tumor-suppressive miRNAs may be a part of potent future treatment strategies in HCC [32]. The development of clinically reliable biomarkers or new drug targets is hampered by the high versatility of the HCC phenotype.
Data published on miRNA expression in HCC are heterogeneous and partly conflicting. Only few specific miRNAs were found upregulated or downregulated in a consistent manner in more than two studies [13]. The majority of patients with HCC suffer from severe underlying liver disease. Despite efforts to correct for histologic staging of liver fibrosis or etiology of liver disease, significant interindividual differences in tissue gene expression likely remain. This is also reflected by the UCA analysis in the current study. Previous approaches compared miRNA expression of HCC tissue to either TAT of the same patient or normal liver tissue deriving from cohorts without liver disease. TAT is the preferred reference, as HCC typically originates from severely altered fibrotic or cirrhotic liver tissue. Still, even when TAT is used as reference, the comparison involves not only HCC cells and liver cells but also includes the tumor-surrounding cell compartment as well as fibrinous and inflammatory tissue of the adjacent cirrhotic liver.
We hypothesized that the heterogeneity of data likely derives not only from different molecular HCC pathomechanisms but may also be attributable to variations in the tumor environment.
First, we found that the fibrotic tissue of the tumor adjacent non-neoplastic liver expresses distinctively different miRNA patterns than its neighboring liver tissue, including high semiquantitative expression differences for single miRNAs. The analysis of the combined expression of LP and LC versus TP and TC showed that the heterogeneity of pooled data probably prevented the detection of differentially expressed miRNAs in this small sample size. Clearly, this in silico approach of pooling of data has to be treated with caution as the array analysis is only a semiquantitative method. Nevertheless, the stage or distribution pattern of cirrhosis may induce a bias in studies comparing miRNA expression from whole tissue sections of HCC and adjacent non-tumorous liver tissue. This effect would be partly compensated by well-matched patient cohorts or could average out with increasing sample size. Indeed, the miRNA expression patterns of microdissected HCC tissue compared to microdissected liver tissue in our small cohort is most similar to the larger studies investigating miRNA expression in HCC [13].
Second, this study is the first to describe the global miRNA expression of HCC capsule tissue. The TC compartment demonstrated an oncogenic phenotype. We observed that the fibrinous tissue of the tumor capsule within and around the tumor has a distinctively different miRNA expression pattern in comparison to the fibrinous tissue within the tumor adjacent non-tumorous liver, although both tissue types are morphologically indistinguishable and feature comparable composition of cell types. The differentially expressed genes compared to LC included known tumor-suppressive miRNAs such as miR-126, miR-99/100, miR-26a, or let-7 group [33], [34], [35], [36], [37]. The miRNAs downregulated in TC to some extent resemble those also downregulated in the corresponding HCC cell compartment (e.g., miR-146a, miR-27a, miR-155, or miR-342-3p). However, we found several miRNAs dysregulated exclusively in the TC compartment, which have been evaluated as promising prognostic biomarkers or implicated in HCC pathogenesis (miR-126, miR-99a, miR-100, miR-26a, and miR-125b) [32], [38], [39], [40], [41], [42]. Moreover, well-known oncogenic pathways were found highly enriched in the MiRPath analysis supporting the suggestion of a premalignant phenotype of the TC. Laser microdissection on a cellular level was used to ensure that results of the TC compartment truly represent measurements from the fibrinous tumor capsule and not only of the leading tumor edge. Furthermore, the miRNA-ISH technique was used to confirm and visualize miRNA dysregulation by the respective compartment, although it has to be taken into the account that the comparability to quantitative methods such as quantitative PCR is limited.
Prognostic biomarkers and novel therapeutic concepts in HCC are urgently needed. The tumor microenvironment likely plays a decisive role in HCC pathogenesis. Indeed, patterns of miRNAs expressed within the tumor adjacent liver tissue of patients with HCC are associated with tumor recurrence after surgical resection [43], [44]. Moreover, the TC may result in a reduced accessibility of solid tumors for systemic chemotherapies [45], [46]. Therefore, the TC may be a potential drug target to improve outcome in HCC. Therapeutic miRNA delivery has been successfully applied to suppress tumorigenesis in a murine liver cancer model [32]. Consequently, specific tissue compartments might be of high relevance in future therapeutic studies aiming to modulate miRNAs in HCC. Thus, targeting strategies for the different cell types involved in the tumorigenesis may be quite different. With our analysis, we provide additional insight into potential oncogenic and tumor-suppressive mechanisms within the tumor-surrounding tissue compartments and offer possible vantage points for new treatment approaches.
Several limitations of our study have to be acknowledged. Our analysis is limited by a rather small case number, as we aimed to match included patients meticulously for any confounding factors, such as tumor stage, etiology of liver disease, or extent of fibrosis. In addition, we focused on early-stage HCC (BCLC stage 0/A), so the possibility to translate our findings to more advanced stages of HCC is clearly limited. Nevertheless, the validity of our data could be demonstrated by application of complimentary methods of miRNA quantification in an independent patient cohort. Through microscopic visualization during and after microdissection a cross-contamination between tissue compartments can be effectively excluded. However, there is still a mixture of cell types within the isolated compartments, e.g., the tumor capsule compartment, among others, comprises fibroblasts, lymphocytes, and granulocytes. Analysis of miRNA signatures from single-cell fractions within our HCC compartments may address this issue in future studies. Furthermore, the percentage of female patients with 66% in the screening cohort is high compared to the typical population of patients with HCC [47]. Moreover, in this study, only HCV-associated HCC was investigated. It would be valuable to study the miRNA expression of the tumor microenvironment in HCC patients with other etiologies of underlying liver disease.
In conclusion, our data support the hypothesis that the heterogeneity of published data on miRNA expression in HCC may, in part, be attributable to variations of co-analyzed fibrotic tissue in the tumor and in controls. The use of TAT as control is of high importance, but analyses may still be subject of significant bias through variations in quantity and quality of fibrinous or cirrhotic tissue within the control tissue. The tumor capsule demonstrates a tumor-like phenotype with down-regulation of well-known tumor-suppressive miRNAs. Several miRNAs that are proposed to be HCC specific might indeed be rather associated to the tumor capsule. The finding that miRNA dysregulation varies within tumor compartments can be important for the development of future treatment strategies in HCC.
Acknowledgment
We thank Ekaterini Hadzoglou, Sabine Albrecht, and Janine Bronckhorst for excellent technical assistance.
Footnotes
This article refers to supplementary materials, which are designated by Tables S1 to S3 and Figures S1 to S3 and are available online at www.transonc.com.
This work was supported by the “Patenschaftsmodell” grant from the medical faculty of the J.W. Goethe University Hospital and by the Deutsche Forschungsgemeinschaft (KFO129).
Conflict of interest statement: The authors have no conflict of interest to declare in the context of this manuscript.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.tranon.2014.09.003.
Appendix A. Supplementary Materials
Hematoxylin and eosin (H&E) staining of HCC tissue undergoing microdissection. Colored lines represent borders for the PALM laser to cut the tissue (TP in red, TC in yellow, LP in green, and LC in blue). Slides before (left) and after (right) removal of tissue compartments are shown.
DIANA miRPath v2.0 enrichment analysis of predicted miRNA target pathways. The gene-to-miRNA interaction is calculated using the DIANA microT-CDS prediction software. Pathways deriving from KEGG database [26] are marked for genes with potential interaction with miRNAs dysregulated in the TC compartment (genes with one miRNA match are depicted in yellow and those with more than two miRNAs are depicted in orange). (A) PI3K/AKT signaling pathway. (B) WNT signaling pathway.
Comparison of miRNA expression between patients who received TACE (n = 12) and non-TACE patients (n = 8). Results of real-time reverse transcription (RT)–PCR for miRNAs miR-126, miR-146a, miR-150, and let-7a are shown. Samples were analyzed in triplicate and ΔCt values were calculated using an endogenous control (U6). ΔCt values are depicted on the y-axis with inverted scale. Values are displayed as box plots with mean, whiskers min to max (*P < .05; **P < .01).
Table S1. Percentages of Tissue Area for Each Subcompartment of the Screening Group
Table S2. Signaling Pathways Enriched in the TC Compartment
Pooled miRNA Expression Data of TC and TP versus LC and LP
References
- 1.Cabibbo G., Enea M., Attanasio M., Bruix J., Craxì A., Cammà C. A meta-analysis of survival rates of untreated patients in randomized clinical trials of hepatocellular carcinoma. Hepatology. 2010;51:1274–1283. doi: 10.1002/hep.23485. [DOI] [PubMed] [Google Scholar]
- 2.El Serag H.B., Rudolph K.L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 3.Llovet J.M., Burroughs A., Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 5.El Serag H.B., Mason A.C. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340:745–750. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- 6.Taura N., Yatsuhashi H., Hamasaki K. Increasing hepatitis C virus-associated hepatocellular carcinoma mortality and aging: long term trends in Japan. Hepatol Res. 2006;34:130–134. doi: 10.1016/j.hepres.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Ekstedt M., Franzen L.E., Mathiesen U.L. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 8.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 9.Esquela-Kerscher A., Slack F.J. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 10.Coulouarn C., Factor V.M., Andersen J.B., Durkin M.E., Thorgeirsson S.S. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28:3526–3536. doi: 10.1038/onc.2009.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ji J., Zhao L., Budhu A. Let-7g targets collagen type I α2 and inhibits cell migration in hepatocellular carcinoma. J Hepatol. 2010;52:690–697. doi: 10.1016/j.jhep.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong Y., Fang J.H., Yun J.P. Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology. 2010;51:836–845. doi: 10.1002/hep.23380. [DOI] [PubMed] [Google Scholar]
- 13.Borel F., Konstantinova P., Jansen P.L. Diagnostic and therapeutic potential of miRNA signatures in patients with hepatocellular carcinoma. J Hepatol. 2012;56:1371–1383. doi: 10.1016/j.jhep.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 14.Toffanin S., Hoshida Y., Lachenmayer A. MicroRNA-based classification of hepatocellular carcinoma and oncogenic role of miR-517a. Gastroenterology. 2011;140:1618–1628. doi: 10.1053/j.gastro.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong C.M., Wong C.C., Lee J.M., Fan D.N., Au S.L., Ng I.O. Sequential alterations of microRNA expression in hepatocellular carcinoma development and venous metastasis. Hepatology. 2012;55:1453–1461. doi: 10.1002/hep.25512. [DOI] [PubMed] [Google Scholar]
- 16.Ladeiro Y., Couchy G., Balabaud C. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47:1955–1963. doi: 10.1002/hep.22256. [DOI] [PubMed] [Google Scholar]
- 17.Varnholt H., Drebber U., Schulze F. MicroRNA gene expression profile of hepatitis C virus-associated hepatocellular carcinoma. Hepatology. 2008;47:1223–1232. doi: 10.1002/hep.22158. [DOI] [PubMed] [Google Scholar]
- 18.Meng F., Henson R., Wehbe-Janek H., Ghoshal K., Jacob S.T., Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–658. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Su H., Yang J.R., Xu T. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009;69:1135–1142. doi: 10.1158/0008-5472.CAN-08-2886. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y., Lee A.T., Ma J.Z. Profiling microRNA expression in hepatocellular carcinoma reveals microRNA-224 up-regulation and apoptosis inhibitor-5 as a microRNA-224-specific target. J Biol Chem. 2008;283:13205–13215. doi: 10.1074/jbc.M707629200. [DOI] [PubMed] [Google Scholar]
- 21.Llovet J.M., Bru C., Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis. 1999;19:329–338. doi: 10.1055/s-2007-1007122. [DOI] [PubMed] [Google Scholar]
- 22.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan L.P., Wang M., Robertus J.L. miRNA profiling of B-cell subsets: specific miRNA profile for germinal center B cells with variation between centroblasts and centrocytes. Lab Invest. 2009;89:708–716. doi: 10.1038/labinvest.2009.26. [DOI] [PubMed] [Google Scholar]
- 24.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 25.Vlachos I.S., Kostoulas N., Vergoulis T. DIANA miRPath v. 2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012;40:W498–W504. doi: 10.1093/nar/gks494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peveling-Oberhag J., Doring C., Hartmann S. Feasibility of global microRNA analysis from fine-needle biopsy FFPE material in patients with hepatocellular carcinoma treated with sorafenib. Clin Sci(Lond) 2015;128:29–37. doi: 10.1042/CS20140007. [DOI] [PubMed] [Google Scholar]
- 28.Cheng A.L., Kang Y.K., Chen Z. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 29.Llovet J.M., Ricci S., Mazzaferro V. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 30.Murakami Y., Yasuda T., Saigo K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2005;25:2537–2545. doi: 10.1038/sj.onc.1209283. [DOI] [PubMed] [Google Scholar]
- 31.Sato F., Hatano E., Kitamura K. MicroRNA profile predicts recurrence after resection in patients with hepatocellular carcinoma within the Milan Criteria. PLoS One. 2011;6:e16435. doi: 10.1371/journal.pone.0016435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kota J., Chivukula R.R., O'Donnell K.A. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen G., Umelo I.A., Lv S. miR-146a inhibits cell growth, cell migration and induces apoptosis in non-small cell lung cancer cells. PLoS One. 2013;8:e60317. doi: 10.1371/journal.pone.0060317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Felli N., Felicetti F., Lustri A.M. miR-126&126* restored expressions play a tumor suppressor role by directly regulating ADAM9 and MMP7 in melanoma. PLoS One. 2013;8:e56824. doi: 10.1371/journal.pone.0056824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jusufovic E., Rijavec M., Keser D. let-7b and miR-126 are down-regulated in tumor tissue and correlate with microvessel density and survival outcomes in non–small-cell lung cancer. PLoS One. 2012;7:e45577. doi: 10.1371/journal.pone.0045577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun D., Lee Y.S., Malhotra A. miR-99 family of MicroRNAs suppresses the expression of prostate-specific antigen and prostate cancer cell proliferation. Cancer Res. 2011;71:1313–1324. doi: 10.1158/0008-5472.CAN-10-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J., Han C., Wu T. MicroRNA-26a promotes cholangiocarcinoma growth by activating β-catenin. Gastroenterology. 2012;143:246–256.e8. doi: 10.1053/j.gastro.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen H., Miao R., Fan J. Decreased expression of miR-126 correlates with metastatic recurrence of hepatocellular carcinoma. Clin Exp Metastasis. 2013;30:651–658. doi: 10.1007/s10585-013-9569-6. [DOI] [PubMed] [Google Scholar]
- 39.Gong J., Zhang J.P., Li B. MicroRNA-125b promotes apoptosis by regulating the expression of Mcl-1, Bcl-w and IL-6R. Oncogene. 2013;32:3071–3079. doi: 10.1038/onc.2012.318. [DOI] [PubMed] [Google Scholar]
- 40.Li D., Liu X., Lin L. MicroRNA-99a inhibits hepatocellular carcinoma growth and correlates with prognosis of patients with hepatocellular carcinoma. J Biol Chem. 2011;286:36677–36685. doi: 10.1074/jbc.M111.270561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petrelli A., Perra A., Schernhuber K. Sequential analysis of multistage hepatocarcinogenesis reveals that miR-100 and PLK1 dysregulation is an early event maintained along tumor progression. Oncogene. 2012;31:4517–4526. doi: 10.1038/onc.2011.631. [DOI] [PubMed] [Google Scholar]
- 42.Zhu Y., Lu Y., Zhang Q. MicroRNA-26a/b and their host genes cooperate to inhibit the G1/S transition by activating the pRb protein. Nucleic Acids Res. 2012;40:4615–4625. doi: 10.1093/nar/gkr1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hernandez-Gea V., Toffanin S., Friedman S.L., Llovet J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144:512–527. doi: 10.1053/j.gastro.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Utsunomiya Y.T., Pérez O'Brien A.M., Sonstegard T.S. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS One. 2013;8:e64280. doi: 10.1371/journal.pone.0064280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chauhan V.P., Martin J.D., Liu H. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4:2516. doi: 10.1038/ncomms3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olive K.P., Jacobetz M.A., Davidson C.J. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El-Serag H.B. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 48.Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Hematoxylin and eosin (H&E) staining of HCC tissue undergoing microdissection. Colored lines represent borders for the PALM laser to cut the tissue (TP in red, TC in yellow, LP in green, and LC in blue). Slides before (left) and after (right) removal of tissue compartments are shown.
DIANA miRPath v2.0 enrichment analysis of predicted miRNA target pathways. The gene-to-miRNA interaction is calculated using the DIANA microT-CDS prediction software. Pathways deriving from KEGG database [26] are marked for genes with potential interaction with miRNAs dysregulated in the TC compartment (genes with one miRNA match are depicted in yellow and those with more than two miRNAs are depicted in orange). (A) PI3K/AKT signaling pathway. (B) WNT signaling pathway.
Comparison of miRNA expression between patients who received TACE (n = 12) and non-TACE patients (n = 8). Results of real-time reverse transcription (RT)–PCR for miRNAs miR-126, miR-146a, miR-150, and let-7a are shown. Samples were analyzed in triplicate and ΔCt values were calculated using an endogenous control (U6). ΔCt values are depicted on the y-axis with inverted scale. Values are displayed as box plots with mean, whiskers min to max (*P < .05; **P < .01).
Table S1. Percentages of Tissue Area for Each Subcompartment of the Screening Group
Table S2. Signaling Pathways Enriched in the TC Compartment
Pooled miRNA Expression Data of TC and TP versus LC and LP