

Abstract
Although rare in its idiopathic form, pulmonary arterial hypertension (PAH) is not uncommon in association with various associated medical conditions, most notably connective tissue disease (CTD). In particular, it develops in approximately 10% of patients with systemic sclerosis and so these patients are increasingly screened to enable early detection. The response of patients with systemic sclerosis to PAH-specific therapy appears to be worse than in other forms of PAH. Survival in systemic sclerosis-associated PAH is inferior to that observed in idiopathic PAH. Potential reasons for this include differences in age, the nature of the underlying pulmonary vasculopathy and the ability of the right ventricle to cope with increased afterload between patients with systemic sclerosis-associated PAH and idiopathic PAH, while coexisting cardiac and pulmonary disease is common in systemic sclerosis-associated PAH. Other forms of connective tissue-associated PAH have been less well studied, however PAH associated with systemic lupus erythematosus (SLE) has a better prognosis than systemic sclerosis-associated PAH and likely responds to immunosuppression.
Introduction
Pulmonary hypertension is defined as a mean pulmonary arterial pressure (mPAP) of ≥25 mmHg when measured at right heart catheterization [1]. Current classification describes five main groups with shared clinical and pathophysiological characteristics (Figure 1): group 1, PAH; group 2, pulmonary hypertension associated with left heart disease; group 3, pulmonary hypertension associated with lung disease; group 4, chronic thromboembolic pulmonary hypertension (CTEPH); and group 5, miscellaneous with unclear mechanisms [2]. Although PAH is rare in the general population (idiopathic PAH having an incidence of 1–2/million/year) [3] it is more common in several associated conditions, most noticeably CTD. As a consequence of the high prevalence of both left heart disease and interstitial lung disease in CTD, the accurate diagnosis of PAH is particularly challenging. Although PAH in other forms of CTD (CTD-PAH) will be briefly discussed, PAH is most commonly seen in association with systemic sclerosis and is the main focus of this review. In particular the differences between systemic sclerosis with PAH and idiopathic PAH will be explored.
Figure 1. Current PH classification system.

Patients with pulmonary hypertension (PH) in association with connective tissue disease may sit in group 1 (pulmonary arterial hypertension), group 2 (PH associated with left heart disease) or group 3 (PH associated with lung disease) while group 4 (chronic thromboembolic pulmonary hypertension) disease must also be excluded. Reproduced with permission [77]
Systemic sclerosis
Epidemiology
The prevalence of systemic sclerosis within the general population ranges from 80/million in the UK [4] to 240/million in the USA [5]. Prospective right heart catheter-based studies have observed a prevalence of PAH in patients with systemic sclerosis of 7.8–12% while a recent meta-analysis calculated the prevalence to be 9% [6,7]. The French Itiner-Air group observed an annual incidence of PAH in patients with systemic sclerosis of 0.61% [8].
Pathogenesis
Systemic sclerosis-PAH is characterized by intimal hyperplasia, medial hypertrophy and adventitial fibrosis as in other forms of PAH although, compared to idiopathic PAH, a lower number of plexiform lesions are observed [9]. Recent histological data suggest a surprisingly high involvement of pulmonary venules, although the proportion of patients with clinically overt pulmonary veno-occlusive disease (PVOD) is lower [7,9,10]. Mutations in bone morphogenetic receptor type 2 (BMPR-2), which are well described in idiopathic and heritable PAH, have not been demonstrated in systemic sclerosis-PAH. An increased frequency of a polymorphism in the gene coding for endoglin, a component of the transforming growth factor beta (TGF-B) receptor complex, which is associated with hereditary haemorrhagic telangectasia and has been identified in patients with idiopathic PAH [11], has been recently identified in patients with systemic sclerosis-PAH [12].
Systemic sclerosis is an autoimmune condition and, as such, it seems likely that autoimmunity and inflammation will play an important role in the development of PAH. In support of this hypothesis, lymphocytes, macrophages and leucocytes have been identified in pulmonary arterial vascular lesions in systemic sclerosis-PAH [10]. The association between several auto-antibodies and the presence of isolated systemic sclerosis are well recognized, including anticentromere, anti-topoisomerase-1 (SCl-70), anti-RNA-polymerase-III and anti-Th/To antibodies. However, their exact role in the pathogenesis of PAH is unclear [13]. Studies in systemic sclerosis patients without PAH have identified high levels of molecules associated with endothelial cell activation and apoptosis (VCAM) and angiogenesis (vascular endothelial growth factor [VEGF]), which would be consistent with the risk of subsequent PAH development [14,15]. Anti-fibroblast IgG from sera of systemic sclerosis patients has been shown to activate normal fibroblasts, and it is hypothesized that fibroblast activation may lead to the induction of collagen synthesis, which may promote subsequent vascular remodeling [16]. Becker et al. have recently reported elevated levels of stimulating anti-endothelin receptor type A antibodies and anti-angiotensin receptor type-1 antibodies in systemic sclerosis-PAH and CTD-PAH when compared with idiopathic PAH [17]. Serum levels of both antibodies were prognostic, despite a lack of correlation with pulmonary haemodynamics, and also predicted the development of PAH in a separate cohort of systemic sclerosis patients without PAH at baseline.
Risk factors
The classical systemic sclerosis patient considered to be at high-risk of developing PAH has limited cutaneous disease of >5 years duration, a low lung gas transfer (DLCO) and is anticentromere antibody positive [18–21]. More recent data have challenged some of these suppositions. A recent study of 78 consecutively diagnosed systemic sclerosis-PAH patients reported the onset of PAH <5 years from the onset of limited cutaneous systemic sclerosis in 55% of patients [22]. Similarly, more recent studies have suggested that PAH is almost as common in diffuse disease [17,23]. A reduced and falling gas transfer (DLCO) is common in patients with systemic sclerosis-PAH, with a mean DLCO of 52% predicted 4.5 years prior to the diagnosis of PAH, compared with a mean DLCO of 80% predicted in non-PAH systemic sclerosis patients [19,24]. Interestingly, patients with systemic sclerosis-PAH have lower DLCO than patients with idiopathic PAH, despite having less severe pulmonary haemodynamics at presentation (discussed below) [25]. As well as the recognized risk of PAH in patients with anticentromere antibody, associations between the presence of anti-RNA-polymerase-III and anti-Th/To antibodies and PAH have also been reported [13].
Prognosis
Several large registries have reported survival in systemic sclerosis-PAH over recent years (Table 1): the UK national registry enrolled 259 incident patients and observed 1- and 3-year survival of 78% and 47% [26]; the REVEAL registry enrolled 399 predominantly prevalent cases and observed a 1-year survival of 82% [20]; the ASPIRE registry enrolled 156 incident patients and observed a 3-year survival of 52% [25]; the French Registry enrolled 85 incident patients and observed 1- and 3-year survival of 90% and 56% [27]; and the PHAROS registry enrolled 131 incident patients (with a high proportion in World Health Organisation [WHO] Functional Class [FC] I and II) and observed 1- and 3-year survival of 93% and 75% [28]. The different outcomes reported are likely to be impacted by differences in patient characteristics (e.g., in the PHAROS study a high proportion were in WHO FC I and II, reflecting a higher proportion of patients identified by screening). Lefevre et al. recently performed a meta-analysis to adjust for these differences, and observed 1- and 3-year survival of 81% and 52% [29].
Table 1. Prognosis of SSc-PAH in selected major registries.
| Registry | Year | n | Age (yrs) | Incident cases (%) | WHO FC I&II/III/IV (%) | mPAP (mmHg) | PVR (dyns.s.cm5) | 1 yr survival (%) |
3 yr survival(%) |
|---|---|---|---|---|---|---|---|---|---|
| UK [26] | 2009 | 259 | 64 | 100 | 16/68/16 | 42 | 715 | 78 | 47 |
| REVEAL [20] | 2010 | 399 | 62 | 18 | 25/60/15 | 45 | 768 | 82 | n/a |
| ASPIRE [25] | 2012 | 156 | 66 | 100 | 19/67/14 | 43 | 678 | 82 | 52 |
| French [27] | 2013 | 85 | 65 | 100 | 21/67/12 | 41 | 680 | 90 | 56 |
| PHAROS [28] | 2014 | 131 | 60 | 100 | 56/38/6 | 36 | 448 | 93 | 75 |
Age and haemodynamic data presented as mean.
Abbreviaitons: mPAP, mean pulmonary arterial pressure; PAH, pulmonary arterial hypertension; PVR, pulmonary vascular resistance; SSc, systemic sclerosis; WHO FC, World Health Organisation Functional Class
Prognostic factors identified at multivariate analysis have also differed between studies, but have included age [26,28], male gender [26,28], DLCO [26,28], functional class [26,28], pulmonary vascular resistance [21], pulmonary capacitance [21], stroke volume index [21] and estimated glomerular filtration rate [21].
It is unclear whether survival in systemic sclerosis-PAH has improved with the introduction of PAH-specific therapies. Historical case series prior to the widespread availability of PAH-specific therapies reported median survival rates of around one year, but are limited by their extremely small size [30]. Rubenfire et al. recently reported no difference in survival in patients diagnosed before 2002 (when only prostacyclin was available) than in patients diagnosed from 2002 onwards (when oral therapies were made available), whereas survival in idiopathic PAH did improve over the same period [31]. Similarly, in their meta-analysis Lefevre et al. also found no relationship between survival and years of enrollment of each study, which may also suggest no progressive improvement in survival over the last decade [29].
Detection
Given the relatively high prevalence of PAH in systemic sclerosis, active screening of patients is advocated [32]. Humbert et al. demonstrated that patients identified with systemic sclerosis-PAH via a screening programme had milder disease and superior survival than those patients who presented with symptomatic disease [33]. Although patients with mildly symptomatic disease have been shown to benefit from PAH therapy, the contribution lead-time bias plays in the superior survival of screened patients is still unclear [34]. Current European Respiratory Society (ERS)/European Society of Cardiology (ESC) guidelines recommend annual screening with echocardiography in patients with systemic sclerosis [32]. Mukerjee et al. demonstrated a DLCO of <55% to have high sensitivity for pulmonary hypertension, but no threshold for either echocardiography-derived systolic pulmonary artery pressure or DLCO could be identified which could confidently exclude pulmonary hypertension [35]. The recently published multicentre DETECT study enrolled 466 systemic sclerosis patients enriched for an increased risk of PAH (>3 years from diagnosis of systemic sclerosis with DLCO <60%) and derived a score based on 6 non-echocardiographic variables (forced vital capacity [FVC]) % predicted/ DLCO % predicted, present/past telengectasia, anticentromere antibody, N-terminal pro-brain natriuretic peptide (NT-proBNP), serum urate and right axis deviation on electrocardiogram (ECG) to guide whether or not an echocardiogram is subsequently required [36]. A second score based on right atrial area and tricuspid regurgitant velocity is then added to the first score to produce a total score that determines whether right heart catheterisation is required. The DETECT protocol resulted in more patients in this enriched cohort being referred for right heart catheterisation but resulted in less false negatives than an approach using echocardiography alone (4% versus 29%). There are important limitations to the DETECT approach: the protocol has not been validated in a separate cohort, has not been studied in patients <3 years from diagnosis of systemic sclerosis or with DLCO >60% and importantly has not been compared with a more holistic approach to identifying patients with systemic sclerosis-PAH currently used in many centers, which incorporates echocardiography, DLCO and symptoms to identify patients requiring right heart catheterisation. Furthermore, it is not clear how practical the DETECT algorithm will be in widespread clinical practice, especially in terms of accurate measurement of right atrial area and availability of NT-proBNP assays.
A proportion of patients undergoing screening will be found to have borderline mean pulmonary arterial pressures of 21-24 mmHg at right heart catheterization. A recent study of 228 patients in the Royal Free Hospital, London, observed progression to PAH in 31% of such patients at subsequent right heart catheterization, suggesting that patients with borderline pressures require careful monitoring [37]. Bae et al. repeated right heart catheterisation in 11/28 patients with the same borderline pulmonary pressures and found that 6/11 had developed resting pulmonary hypertension after a mean period of 14 months [38]. Of note, associated interstitial lung disease and restrictive spirometry was more common in those patients with borderline pressure than in those with normal pressure.
Exercise may have a role in the earlier identification of patients with pulmonary vasculopathy in connective tissue disease, with differential patterns of gas exchange on non-invasive exercise testing distinguishing pulmonary vascular disease from left heart disease in systemic sclerosis [39]. Patients with elevation of mPAP >30 mmHg on exercise appear to be at risk of developing PAH with 19% of patients developing PAH at rest in the UK national CTD-PAH registry [26]. Additional evidence demonstrating improved haemodynamics and exercise capacity in response to the endothelin-1 receptor antagonist, ambrisentan, in patients with exercise-induced systemic sclerosis- pulmonary hypertension further suggests this is a precursor to PAH [40]. The normal response in mPAP at exercise in patients without PAH at rest is not known [41] and so routine exercise at right heart catheterization is not currently recommended [1].
Treatment
Current PAH-specific therapies target 3 pathways (prostacyclin, endothelin-1 and nitric oxide) [42]. Several subgroup analyses of CTD-PAH patients enrolled in the pivotal randomized controlled trials (RCTs) have been published which, apart from one exception [43], suggest a less marked response to PAH therapy in CTD-PAH. It is possible that this may be partly due to the fact that the six minute walk distance (6MWD), which has been used as the primary endpoint in the majority of RCTs of PAH-specific therapies, may particularly be affected by age and musculoskeletal pathology in patients with CTD.
Prostanoids
An unblinded RCT of continuous intravenous epoprostenol (which is administered by a portable pump via an indwelling central line), involving 111 patients with systemic sclerosis-PAH, observed a significant improvement in mean 6MWD of 46 metres following 12 weeks of treatment, while patients receiving placebo experienced a mean decrease of 48 metres (P<0.05) [43]. Due to its much longer half-life, the prostacyclin analogue treprostinil can be administered via continuous subcutaneous infusion, thus removing the risk of line infection. Ninety patients with CTD-PAH (including 45 patients with systemic sclerosis-PAH) from the original RCT (which also included 270 patients with idiopathic PAH and 109 patients with congenital heart disease) were analysed separately [44]. Unlike in the original study, the between treatment group's difference in median 6MWD did not reach statistical significance (+25 metres, P=0.06) in the CTD-PAH group. Furthermore, improvements in the secondary endpoints of Borg dyspnoea score, mean right atrial pressure, mean pulmonary artery pressure, and mixed venous oxygen saturation seen in the original study, were not observed in the subgroup analysis.
Endothelin-1 receptor antagonist
Bosentan, an endothelin-1 receptor antagonist, was studied in an RCT involving 213 PAH patients [45]. In the 150 idiopathic PAH patients, the 6MWD increased by 46 metres after 16 weeks of therapy, while it decreased by 5 metres in the placebo group (P<0.05). A different picture was seen in patients with systemic sclerosis-PAH; among the 14 patients in the placebo group there was a decline of 40 metres, while in the 33 patients receiving active therapy deterioration was prevented with an improvement of 3 metres (P=not significant). No RCT data specifically on CTD-PAH have been published to date for the other currently available endothelin-1 receptor antagonists, ambrisentan and macitentan.
Phosphodiesterase-5 inhibitors
A subgroup analysis of the 84 patients with CTD-PAH (including 50 patients with systemic sclerosis) who were included in the SUPER-1 trial of sildenafil was subsequently published [46,47]. In the original study of 278 patients, in which patients received 20 mg, 40 mg or 80 mg three times a day, the mean placebo-controlled treatment effects on 6MWD were +45 metres, +46 metres and +50 metres after 12 weeks. Direct comparison is difficult especially since patients with SLE may have a greater response to treatment. However, the treatment effect in the subgroup analysis tended to be smaller, with the 6MWD increasing by +42 metres, +36 metres and +15 metres in patients receiving the three different doses.
Comparison of systemic sclerosis-PAH with idiopathic PAH
Survival in systemic sclerosis-PAH is significantly worse than in idiopathic PAH, despite less severe pulmonary haemodynamics (Figure 2) [20,25,48–50]. There are several potential reasons for this disconnect between pulmonary hypertension severity and outcome (Table 2).
Figure 2. Survival of IPAH and SSc-PAH in the ASPIRE registry.
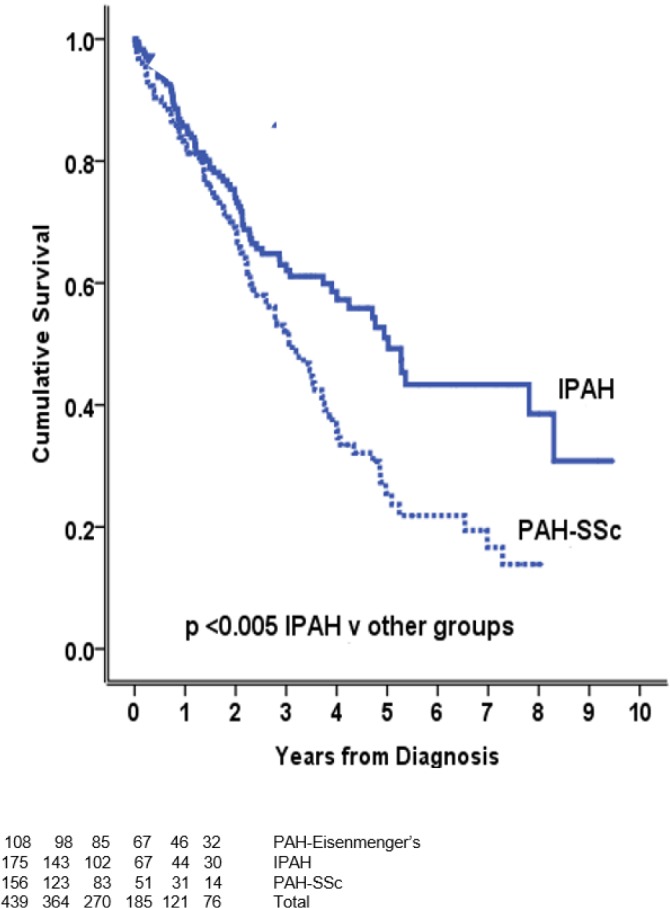
Adapted with permission [25]. Abbreviations: IPAH, idiopathic pulmonary arterial hypertension; PAH, pulmonary arterial hypertension; SSc, systemic sclerosis
Table 2. Potential reasons for poorer outcome in SSc-PAH than in IPAH.
| Factor | Evidence |
|---|---|
| Age | Mean age of diagnosis of SSc-PAH ≈10 yrs > IPAH [25]. Age was independent prognostic factor in combined group of SSc-PAH and IPAH [50]. |
| Pulmonary vasculopathy | Higher proportion of pulmonary venule involvement in SSc-PAH [10,51]. |
| Right ventricle (RV) | Reduced RV contractility assessed by pump function graph and pressure-volume loops in SSc-PAH [54,55]. Higher NT-proBNP in SSc-PAH despite less severe pulmonary haemodynamics [53]. |
| Left ventricle (LV) | High prevalence of LV systolic and diastolic dysfunction in SSc [50,57]. ≈1/3 of patients fulfilling haemodynamic criteria for SSc-PAH may have occult left heart disease based on response to fluid challenge and clinico-radiological characteristics [58]. |
| Interstitial lung disease (ILD) | ILD common in SSc. Different registries have included varying degrees of ILD in SSc-PAH cohorts [29]. Response to PH therapy in SSc in the presence of ILD appears poor [75]. |
| Multisystem disease | Coexisting renovascular and gastrointestinal disease including iron deficiency and malnutrition more common in SSc than in general population. |
| Antibodies | Exact role of SSc-associated autoantibodies (e.g. anticentromere) in pathogenesis of PAH and poorer prognosis compared with idiopathic PAH not clear [76]. Elevated stimulating anti-endothelin receptor type A antibodies and anti-angiotensin receptor type-1 antibodies in SSc-PAH compared with idiopathic PAH [17]. |
Abbreviations: IPAH, idiopathic pulmonary arterial hypertension; PAH, pulmonary arterial hypertension; PH, pulmonary hypertension; NT-proBNP, N-terminal pro-brain natriuretic peptide; SSc, systemic sclerosis
Age
Patients with systemic sclerosis-PAH are up to a decade older than patients with idiopathic PAH at diagnosis [25]. Age is an independent prognostic factor in systemic sclerosis-PAH [26] and was also found to be an independent predictor of mortality in a study involving 50 systemic sclerosis-PAH patients and 41 idiopathic PAH patients [50].
Pulmonary vasculopathy
Dorfmuller et al. studied lung tissue samples from 8 patients with various forms of CTD-PAH and compared them with 29 idiopathic PAH patients [10]. Obstructing lesions in pulmonary veins and venules were seen in 75% of the CTD-PAH patients and 17% of the idiopathic PAH patients (P<0.05). Overbeek et al. subsequently studied lung tissue samples from 8 systemic sclerosis-PAH and 11 idiopathic PAH patients [51]. They observed plexogenic lesions in all the idiopathic PAH patients and none of the systemic sclerosis-PAH patients. Furthermore, they observed pulmonary vein and venule fibrosis in all of the systemic sclerosis-PAH patients (with a pattern consistent with PVOD in 50%) and only 3 of the idiopathic PAH patients. Gunther et al. observed at least 2 radiographic signs consistent with PVOD on high resolution computerized tomography (CT) imaging (centrilobular ground glass abnormalities, septal lines and lymphadenopathy) in 16/26 patients with systemic sclerosis-PAH [52]. Furthermore, 8 of those 16 patients developed pulmonary oedema on commencement of PAH-specific therapy. Clinical experience in centres treating large numbers of patients with systemic sclerosis-PAH would suggest that frank PVOD is significantly less common than this. Nevertheless, idiopathic PVOD is associated with an exceedingly poor survival and, taken together, these data suggest that a PVOD-component in a proportion of patients may be responsible for some of the poorer outcomes in systemic sclerosis-PAH.
Right ventricular function
Despite pulmonary haemodynamics being more severe in the idiopathic PAH group, Mathai et al. observed significantly higher NT-proBNP levels in 55 systemic sclerosis-PAH patients when compared with 43 idiopathic PAH patients [53]. This difference persisted even when adjusting for renal function and age, and suggests a poorer ability of the right ventricle to cope with increased afterload. Overbeek et al. studied 13 systemic sclerosis-PAH and 17 idiopathic PAH patients using the pump function graph technique and demonstrated poorer right ventricle contractility in the systemic sclerosis-PAH group [54]. Tedford et al. performed pressure-volume loops in 5 idiopathic PAH, 7 systemic sclerosis-PAH and 7 systemic sclerosis non-pulmonary hypertension patients [55]. No significant difference in right ventricle afterload as assessed by pulmonary vascular resistance (PVR), compliance and arterial elastance (Ea) was identified between the idiopathic PAH and systemic sclerosis-PAH patients. Right ventricle contractility as measured using end-systolic elastance (Ees) and the coupling of right ventricle contractility with afterload (Ees:Ea) were, however, poorer in systemic sclerosis-PAH than in idiopathic PAH. Interestingly, Ees:Ea in systemic sclerosis patients without pulmonary hypertension was even more abnormal than in systemic sclerosis-PAH patients. The underlying reason for these observations is not clear. It is known that abnormal collagen deposition is high in both ventricles in patients with systemic sclerosis and no evidence of left heart disease or PAH [56]. However, when Overbeek et al. examined right ventricle samples for five systemic sclerosis-PAH and nine idiopathic PAH patients they found no difference in extent of interstitial fibrosis although there was a greater inflammatory infiltrate in the systemic sclerosis-PAH group [9].
Left heart disease
Hachulla et al. performed cardiac magnetic resonance imaging in 52 patients with systemic sclerosis and observed evidence of left ventricle systolic or diastolic dysfunction in approximately ⅓ of cases, raising the possibility that occult left heart disease may be responsible for a proportion of diagnosed systemic sclerosis-PAH [57]. Fisher et al. observed evidence of left heart diastolic dysfunction in approximately ⅓ of 50 patients fulfilling diagnostic criteria for systemic sclerosis-PAH (with a mean age of 59) but only in approximately 10% of patients diagnosed with idiopathic PAH (with a mean age of 48), although its presence was not predictive of higher mortality [49]. Fox et al. identified pulmonary hypertension in 53/107 systemic sclerosis patients who underwent left and right heart catheterization [58]. Originally, 29/53 patients were given a diagnosis of PAH, but after fluid challenge 11/29 (38%) were reclassified as pulmonary hypertension secondary to left heart disease, which was in keeping with radiological appearances of the left heart and risk factors for left heart disease. A subsequent study identified 207 patients fulfilling haemodynamic criteria for PAH, 49% of whom had underlying CTD [59]. Following fluid challenge, 46/207 (22%) were reclassified as pulmonary hypertension secondary to left heart disease. It is, however, well recognized that elderly patients fulfilling haemodynamic criteria for idiopathic PAH also have a high prevalence of risk factors for left heart disease and so it is unclear whether the potential misdiagnosis of pulmonary hypertension with left heart disease as PAH is more common in idiopathic PAH than in CTD-PAH [3]. Furthermore, the role of fluid challenge in the assessment of pulmonary hypertension is not well defined, with the positive predictive value for the presence of pulmonary hypertension secondary to left heart disease being unknown; hence it is not part of the diagnostic algorithm in the most recent world pulmonary hypertension guidelines [1].
Interstitial lung disease
Interstitial lung disease (ILD) is common in patients with systemic sclerosis and occurs more frequently in diffuse than in limited cutaneous disease [60]. Defining pulmonary hypertension in systemic sclerosis patients as being isolated PAH rather than ILD-associated pulmonary hypertension is not always straightforward. In the absence of significant pulmonary hypertension, an approach combining radiological assessment (limited disease when the extent of ILD <10% and extensive disease when the extent of disease >30%) and a spirometric threshold of FVC 70% (when the extent of ILD is estimated to be 10–30%) has been shown to effectively identify patients with systemic sclerosis and ILD with good and poor prognoses [46]. Whether this approach also robustly differentiates systemic sclerosis-PAH from ILD-associated pulmonary hypertension is unclear. Several thresholds involving FVC or total lung capacity (TLC) of 60 or 70% and/or varyingly-defined minor/mild versus moderate/severe ILD have been employed in different studies and it is very possible that cohorts of “systemic sclerosis-PAH” in the literature have been “contaminated” with a proportion of patients who actually have ILD-associated pulmonary hypertension [29]. Several groups have demonstrated that ILD-associated pulmonary hypertension in systemic sclerosis is associated with a significantly poorer survival than systemic sclerosis-PAH, despite similar pulmonary haemodynamics [26,61]. A retrospective study of 70 systemic sclerosis patients with ILD-associated pulmonary hypertension was unable to demonstrate any clear benefit from PAH-specific therapy. However, prospective data are required and many centres have a low threshold for PAH-specific therapy in this group of patients.
Iron deficiency
Iron deficiency has previously been shown to be common in idiopathic PAH, occurring in up to 61% of patients in the absence of overt anaemia in the majority of cases, and is associated with poorer outcome [62]. Inappropriate elevation in serum hepcidin levels with subsequent reduced iron gut absorption has been postulated as a possible mechanism. Iron deficiency has recently been reported as occurring in 46% of systemic sclerosis-PAH and 16% of systemic sclerosis non-pulmonary hypertension patients [63]. Both exercise capacity and survival were reduced in those systemic sclerosis-PAH patients with iron deficiency. Hepcidin levels were found to be elevated while underlying gastrointestinal angiodysplastic lesions may also predispose iron deficiency in systemic sclerosis patients. Although it is not clear whether iron deficiency is more severe in systemic sclerosis-PAH, it is interesting to note that haemoglobin levels were statistically significantly lower in a recent study comparing 228 systemic sclerosis-PAH patients and 279 idiopathic PAH patients [47].
Multisystem disease
Extra-cardiorespiratory manifestations of systemic sclerosis may impact on both morbidity and mortality. Scleroderma renal crisis was previously a major cause of mortality, although is now more treatable since the availability of ACE inhibitors. Gastrointestinal involvement can lead to severe nutritional deficiency.
Selected other CTD-PAH
The exact prevalence of pulmonary hypertension in patients with SLE is unknown and estimates have ranged from 2–43% depending on diagnostic methods used [64,65]. Echocardiography-based studies are associated with a significant risk of over-estimating pulmonary pressures. Despite the prevalence of SLE being at least 3 times that of systemic sclerosis in the UK [66], the number of SLE-associated PAH (SLE-PAH) patients in the UK national registry was only 11% of the number of systemic sclerosis-PAH patients, suggesting that the true prevalence of PAH in patients with SLE is likely to be <1% [26]. A recent prospective study of 245 patients with SLE demonstrated pulmonary hypertension associated with significant lung or left heart disease in 12 patients (5%), but no cases of PAH were identified [67]. Patients with SLE-PAH are younger than those with systemic sclerosis-PAH, with higher pulmonary pressures and lower DLCO [26,68]. Survival appears to be superior with 3-year survival in the UK national registry of 74%, compared to 47% in systemic sclerosis-PAH [26]. Significant improvements in PAH have been described in patients with SLE-PAH (as well as some patients with mixed connective tissue disease-associated PAH) treated with immunosuppression alone, although no good RCT data exist [69]. Our approach is to treat active lupus aggressively and ensure that all patients with SLE-PAH are adequately immunosuppressed. Antiphospholipid syndrome is a risk factor for the development of chronic thromboembolic pulmonary hypertension and so, if SLE-PAH is suspected, then particular attention to the exclusion of thromboembolic disease is required [70].
Antisynthetase syndrome is characterised by myositis and interstitial lung disease in the presence of anti-tRNA-synthetase antibodies (most commonly Jo-1). A recent French study calculated a right heart catheter-based prevalence of pulmonary hypertension of 7.9% [71]. The majority of patients had severe pre-capillary pulmonary hypertension in the presence of significant coexisting ILD while 3-year survival was 58%. PAH appears to be less common in other forms of CTD, with an estimated prevalence of 2% in mixed connective tissue disease [72], while studies estimating the prevalence of PAH in other forms of CTD (including Sjogren's syndrome and rheumatoid arthritis) have used echocardiography to calculate systolic pulmonary arterial pressures with significant risk of over-estimation [73,74].
Conclusion
Survival in systemic sclerosis-PAH remains poor. This may be as a result of the underlying pulmonary arterial vasculopathy and the poor ability of the right ventricle to cope with increased afterload, together with the multisystem nature of the condition including the involvement of myocardium and lungs. Although early detection of systemic sclerosis-PAH in patients is required, further work to demonstrate a definite impact on the natural history of the condition is needed. More extensive investigation of the underlying molecular and genetic pathogenesis is warranted, while specific trials of currently available and novel PAH-specific therapies in systemic sclerosis-PAH are urgently required.
Abbreviations
- 6MWD
six minute walk distance
- CTD
connective tissue disease
- CTEPH
chronic thromboembolic pulmonary hypertension
- DLCO
lung gas transfer
- Ea
arterial elastance
- Ees
end-systolic elastance
- FC
Functional Class
- FVC
forced vital capacity
- ILD
interstitial lung disease
- mPAP
mean pulmonary arterial pressure
- PAH
pulmonary arterial hypertension
- NT-proBNP
N-terminal pro-brain natriuretic peptide
- PVOD
pulmonary veno-occlusive disease
- PVR
pulmonary vascular resistance
- RCT
randomized controlled trial
- SLE
systemic lupus erythematosus
- TGF-β
transforming growth factor beta
- WHO
World Health Organisation
Disclosures
Robin Condliffe and Luke S. Howard have received honoraria for lecturing and advisory boards from Actelion, Bayer, Glaxo Smith Kline, Pfizer and United Therapeutics.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/m/7/6
References
- 1.Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, Langleben D, Manes A, Satoh T, Torres F, Wilkins MR, Badesch DB. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D42–50. doi: 10.1016/j.jacc.2013.10.032. [DOI] [PubMed] [Google Scholar]
- 2.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718215559
- 3.Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, Howard LS, Pepke-Zaba J, Sheares KK, Corris PA, Fisher AJ, Lordan JL, Gaine S, Coghlan JG, Wort SJ, Gatzoulis MA, Peacock AJ. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: Results from the pulmonary hypertension registry of the united kingdom and ireland. Am J Respir Crit Care Med. 2012;186:790–6. doi: 10.1164/rccm.201203-0383OC. [DOI] [PubMed] [Google Scholar]
- 4.Allcock RJ, Forrest I, Corris PA, Crook PR, Griffiths ID. A study of the prevalence of systemic sclerosis in northeast england. Rheumatology (Oxford) 2004;43:596–602. doi: 10.1093/rheumatology/keh124. [DOI] [PubMed] [Google Scholar]
- 5.Mayes MD, Lacey JV, Jr., Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, Schottenfeld D. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large us population. Arthritis Rheum. 2003;48:2246–55. doi: 10.1002/art.11073. [DOI] [PubMed] [Google Scholar]
- 6.Hachulla E, Gressin V, Guillevin L, Carpentier P, Diot E, Sibilia J, Kahan A, Cabane J, Frances C, Launay D, Mouthon L, Allanore Y, Tiev KP, Clerson P, de Groote P, Humbert M. Early detection of pulmonary arterial hypertension in systemic sclerosis: A french nationwide prospective multicenter study. Arthritis Rheum. 2005;52:3792–800. doi: 10.1002/art.21433. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/8301
- 7.Avouac J, Airo P, Meune C, Beretta L, Dieude P, Caramaschi P, Tiev K, Cappelli S, Diot E, Vacca A, Cracowski JL, Sibilia J, Kahan A, Matucci-Cerinic M, Allanore Y. Prevalence of pulmonary hypertension in systemic sclerosis in european caucasians and metaanalysis of 5 studies. J Rheumatol. 2010;37:2290–8. doi: 10.3899/jrheum.100245. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/724376583
- 8.Hachulla E, de Groote P, Gressin V, Sibilia J, Diot E, Carpentier P, Mouthon L, Hatron PY, Jego P, Allanore Y, Tiev KP, Agard C, Cosnes A, Cirstea D, Constans J, Farge D, Viallard JF, Harle JR, Patat F, Imbert B, Kahan A, Cabane J, Clerson P, Guillevin L, Humbert M. The three-year incidence of pulmonary arterial hypertension associated with systemic sclerosis in a multicenter nationwide longitudinal study in france. Arthritis Rheum. 2009;60:1831–9. doi: 10.1002/art.24525. [DOI] [PubMed] [Google Scholar]
- 9.Overbeek MJ, Mouchaers KT, Niessen HM, Hadi AM, Kupreishvili K, Boonstra A, Voskuyl AE, Belien JA, Smit EF, Dijkmans BC, Vonk-Noordegraaf A, Grunberg K. Characteristics of interstitial fibrosis and inflammatory cell infiltration in right ventricles of systemic sclerosis-associated pulmonary arterial hypertension. Int J Rheumatol. 2010 doi: 10.1155/2010/604615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorfmuller P, Humbert M, Perros F, Sanchez O, Simonneau G, Muller KM, Capron F. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol. 2007;38:893–902. doi: 10.1016/j.humpath.2006.11.022. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1072025
- 11.Gore B, Izikki M, Mercier O, Dewachter L, Fadel E, Humbert M, Dartevelle P, Simonneau G, Naeije R, Lebrin F, Eddahibi S. Key role of the endothelial tgf-beta/alk1/endoglin signaling pathway in humans and rodents pulmonary hypertension. PLoS One. 2014;9:e100310. doi: 10.1371/journal.pone.0100310. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718463002
- 12.Wipff J, Kahan A, Hachulla E, Sibilia J, Cabane J, Meyer O, Mouthon L, Guillevin L, Junien C, Boileau C, Allanore Y. Association between an endoglin gene polymorphism and systemic sclerosis-related pulmonary arterial hypertension. Rheumatology (Oxford) 2007;46:622–5. doi: 10.1093/rheumatology/kel378. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1063852
- 13.Koenig M, Dieude M, Senecal JL. Predictive value of antinuclear autoantibodies: The lessons of the systemic sclerosis autoantibodies. Autoimmun Rev. 2008;7:588–93. doi: 10.1016/j.autrev.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 14.Denton CP, Bickerstaff MC, Shiwen X, Carulli MT, Haskard DO, Dubois RM, Black CM. Serial circulating adhesion molecule levels reflect disease severity in systemic sclerosis. Br J Rheumatol. 1995;34:1048–54. doi: 10.1093/rheumatology/34.11.1048. [DOI] [PubMed] [Google Scholar]
- 15.Choi JJ, Min DJ, Cho ML, Min SY, Kim SJ, Lee SS, Park KS, Seo YI, Kim WU, Park SH, Cho CS. Elevated vascular endothelial growth factor in systemic sclerosis. J Rheumatol. 2003;30:1529–33. [PubMed] [Google Scholar]
- 16.Tamby MC, Humbert M, Guilpain P, Servettaz A, Dupin N, Christner JJ, Simonneau G, Fermanian J, Weill B, Guillevin L, Mouthon L. Antibodies to fibroblasts in idiopathic and scleroderma-associated pulmonary hypertension. Eur Respir J. 2006;28:799–807. doi: 10.1183/09031936.06.00152705. [DOI] [PubMed] [Google Scholar]
- 17.Becker MO, Kill A, Kutsche M, Guenther J, Rose A, Tabeling C, Witzenrath M, Kuhl AA, Heidecke H, Ghofrani HA, Tiede H, Schermuly RT, Nickel N, Hoeper MM, Lukitsch I, Gollasch M, Kuebler WM, Bock S, Burmester GR, Dragun D, Riemekasten G. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am J Respir Crit Care Med. 2014;190:808–17. doi: 10.1164/rccm.201403-0442OC. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718553025
- 18.Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3:187–96. doi: 10.1586/ers.09.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steen V, Medsger TA., Jr Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Arthritis Rheum. 2003;48:516–22. doi: 10.1002/art.10775. [DOI] [PubMed] [Google Scholar]
- 20.Chung L, Liu J, Parsons L, Hassoun PM, McGoon M, Badesch DB, Miller DP, Nicolls MR, Zamanian RT. Characterization of connective tissue disease-associated pulmonary arterial hypertension from reveal: Identifying systemic sclerosis as a unique phenotype. Chest. 2010;138:1383–94. doi: 10.1378/chest.10-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campo A, Mathai SC, Le Pavec J, Zaiman AL, Hummers LK, Boyce D, Housten T, Champion HC, Lechtzin N, Wigley FM, Girgis RE, Hassoun PM. Hemodynamic predictors of survival in scleroderma-related pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182:252–60. doi: 10.1164/rccm.200912-1820OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hachulla E, Launay D, Mouthon L, Sitbon O, Berezne A, Guillevin L, Hatron PY, Simonneau G, Clerson P, Humbert M. Is pulmonary arterial hypertension really a late complication of systemic sclerosis? Chest. 2009;136:1211–9. doi: 10.1378/chest.08-3042. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/725274767
- 23.Nihtyanova SI, Tang EC, Coghlan JG, Wells AU, Black CM, Denton CP. Improved survival in systemic sclerosis is associated with better ascertainment of internal organ disease: A retrospective cohort study. QJM. 2010;103:109–15. doi: 10.1093/qjmed/hcp174. [DOI] [PubMed] [Google Scholar]
- 24.Chang B, Schachna L, White B, Wigley FM, Wise RA. Natural history of mild-moderate pulmonary hypertension and the risk factors for severe pulmonary hypertension in scleroderma. J Rheumatol. 2006;33:269–74. [PubMed] [Google Scholar]
- 25.Hurdman J, Condliffe R, Elliot CA, Davies C, Hill C, Wild JM, Capener D, Sephton P, Hamilton N, Armstrong IJ, Billings C, Lawrie A, Sabroe I, Akil M, O'Toole L, Kiely DG. Aspire registry: Assessing the spectrum of pulmonary hypertension identified at a referral centre. Eur Respir J. 2012;39:945–55. doi: 10.1183/09031936.00078411. [DOI] [PubMed] [Google Scholar]
- 26.Condliffe R, Kiely DG, Peacock AJ, Corris PA, Gibbs JS, Vrapi F, Das C, Elliot CA, Johnson M, DeSoyza J, Torpy C, Goldsmith K, Hodgkins D, Hughes RJ, Pepke-Zaba J, Coghlan JG. Connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med. 2009;179:151–7. doi: 10.1164/rccm.200806-953OC. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1156982
- 27.Launay D, Sitbon O, Hachulla E, Mouthon L, Gressin V, Rottat L, Clerson P, Cordier JF, Simonneau G, Humbert M. Survival in systemic sclerosis-associated pulmonary arterial hypertension in the modern management era. Ann Rheum Dis. 2013;72:1940–6. doi: 10.1136/annrheumdis-2012-202489. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717965310
- 28.Chung L, Domsic RT, Lingala B, Alkassab F, Bolster M, Csuka ME, Derk C, Fischer A, Frech T, Furst DE, Gomberg-Maitland M, Hinchcliff M, Hsu V, Hummers LK, Khanna D, Medsger TA, Jr., Molitor JA, Preston IR, Schiopu E, Shapiro L, Silver R, Simms R, Varga J, Gordon JK, Steen VD. Survival and predictors of mortality in systemic sclerosis-associated pulmonary arterial hypertension: Outcomes from the pulmonary hypertension assessment and recognition of outcomes in scleroderma registry. Arthritis Care Res (Hoboken) 2014;66:489–95. doi: 10.1002/acr.22121. [DOI] [PubMed] [Google Scholar]
- 29.Lefevre G, Dauchet L, Hachulla E, Montani D, Sobanski V, Lambert M, Hatron PY, Humbert M, Launay D. Survival and prognostic factors in systemic sclerosis-associated pulmonary hypertension: A systematic review and meta-analysis. Arthritis Rheum. 2013;65:2412–23. doi: 10.1002/art.38029. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718017762
- 30.Koh ET, Lee P, Gladman DD, Abu-Shakra M. Pulmonary hypertension in systemic sclerosis: An analysis of 17 patients. Br JRheumatol. 1996;35:989–93. doi: 10.1093/rheumatology/35.10.989. [DOI] [PubMed] [Google Scholar]
- 31.Rubenfire M, Huffman MD, Krishnan S, Seibold JR, Schiopu E, McLaughlin VV. Survival in systemic sclerosis with pulmonary arterial hypertension has not improved in the modern era. Chest. 2013;144:1282–90. doi: 10.1378/chest.12-0653. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718018237
- 32.Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS, Gomez-Sanchez MA, Jondeau G, Klepetko W, Opitz C, Peacock A, Rubin L, Zellweger M, Simonneau G. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34:1219–63. doi: 10.1183/09031936.00139009. [DOI] [PubMed] [Google Scholar]
- 33.Humbert M, Yaici A, de Groote P, Montani D, Sitbon O, Launay D, Gressin V, Guillevin L, Clerson P, Simonneau G, Hachulla E. Screening for pulmonary arterial hypertension in patients with systemic sclerosis: Clinical characteristics at diagnosis and long-term survival. Arthritis Rheum. 2011;63:3522–30. doi: 10.1002/art.30541. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/724435177
- 34.Galie N, Rubin L, Hoeper M, Jansa P, Al-Hiti H, Meyer G, Chiossi E, Kusic-Pajic A, Simonneau G. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (early study): A double-blind, randomised controlled trial. Lancet. 2008;371:2093–100. doi: 10.1016/S0140-6736(08)60919-8. [DOI] [PubMed] [Google Scholar]
- 35.Mukerjee D, St George D, Knight C, Davar J, Wells AU, Du Bois RM, Black CM, Coghlan JG. Echocardiography and pulmonary function as screening tests for pulmonary arterial hypertension in systemic sclerosis. Rheumatology (Oxford) 2004;43:461–6. doi: 10.1093/rheumatology/keh067. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/720742074
- 36.Coghlan JG, Denton CP, Grunig E, Bonderman D, Distler O, Khanna D, Muller-Ladner U, Pope JE, Vonk MC, Doelberg M, Chadha-Boreham H, Heinzl H, Rosenberg DM, McLaughlin VV, Seibold JR, DETECT study group Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: The detect study. Ann Rheum Dis. 2013;73:1340–9. doi: 10.1136/annrheumdis-2013-203301. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718011359
- 37.Valerio CJ, Schreiber BE, Handler CE, Denton CP, Coghlan JG. Borderline mean pulmonary artery pressure in patients with systemic sclerosis: Transpulmonary gradient predicts risk of developing pulmonary hypertension. Arthritis Rheum. 2013;65:1074–84. doi: 10.1002/art.37838. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/717997200
- 38.Bae S, Saggar R, Bolster MB, Chung L, Csuka ME, Derk C, Domsic R, Fischer A, Frech T, Goldberg A, Hinchcliff M, Hsu V, Hummers L, Schiopu E, Mayes MD, McLaughlin V, Molitor J, Naz N, Furst DE, Maranian P, Steen V, Khanna D. Baseline characteristics and follow-up in patients with normal haemodynamics versus borderline mean pulmonary arterial pressure in systemic sclerosis: Results from the pharos registry. Ann Rheum Dis. 2012;71:1335–42. doi: 10.1136/annrheumdis-2011-200546. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/717957242
- 39.Dumitrescu D, Oudiz RJ, Karpouzas G, Hovanesyan A, Jayasinghe A, Hansen JE, Rosenkranz S, Wasserman K. Developing pulmonary vasculopathy in systemic sclerosis, detected with non-invasive cardiopulmonary exercise testing. PLoS One. 2010;5:e14293. doi: 10.1371/journal.pone.0014293. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/724181260
- 40.Saggar R, Khanna D, Shapiro S, Furst DE, Maranian P, Clements P, Abtin F, Dua S, Belperio J, Saggar R. Brief report: Effect of ambrisentan treatment on exercise-induced pulmonary hypertension in systemic sclerosis: A prospective single-center, open-label pilot study. Arthritis Rheum. 2012;64:4072–7. doi: 10.1002/art.34614. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/724434795
- 41.Kovacs G, Maier R, Aberer E, Brodmann M, Scheidl S, Hesse C, Troester N, Salmhofer W, Stauber R, Fuerst FC, Thonhofer R, Ofner-Kopeinig P, Gruenig E, Olschewski H. Assessment of pulmonary arterial pressure during exercise in collagen vascular disease: Echocardiography vs right-sided heart catheterization. Chest. 2010;138:270–8. doi: 10.1378/chest.09-2099. [DOI] [PubMed] [Google Scholar]
- 42.Galie N, Corris PA, Frost A, Girgis RE, Granton J, Jing ZC, Klepetko W, McGoon MD, McLaughlin VV, Preston IR, Rubin LJ, Sandoval J, Seeger W, Keogh A. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D60–72. doi: 10.1016/j.jacc.2013.10.031. [DOI] [PubMed] [Google Scholar]
- 43.Badesch DB, Tapson VF, McGoon MD, Brundage BH, Rubin LJ, Wigley FM, Rich S, Barst RJ, Barrett PS, Kral KM, Jobsis MM, Loyd JE, Murali S, Frost A, Girgis R, Bourge RC, Ralph DD, Elliott CG, Hill NS, Langleben D, Schilz RJ, McLaughlin VV, Robbins IM, Groves BM, Shapiro S, Medsger TA., Jr Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med. 2000;132:425–34. doi: 10.7326/0003-4819-132-6-200003210-00002. [DOI] [PubMed] [Google Scholar]
- 44.Oudiz RJ, Schilz RJ, Barst RJ, Galie N, Rich S, Rubin LJ, Simonneau G. Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. Chest. 2004;126:420–7. doi: 10.1378/chest.126.2.420. [DOI] [PubMed] [Google Scholar]
- 45.Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, Landzberg M, Simonneau G. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718722409
- 46.Badesch DB, Hill NS, Burgess G, Rubin LJ, Barst RJ, Galie N, Simonneau G. Sildenafil for pulmonary arterial hypertension associated with connective tissue disease. J Rheumatol. 2007;34:2417–22. [PubMed] [Google Scholar]
- 47.Goh NS, Desai SR, Veeraraghavan S, Hansell DM, Copley SJ, Maher TM, Corte TJ, Sander CR, Ratoff J, Devaraj A, Bozovic G, Denton CP, Black CM, du Bois RM, Wells AU. Interstitial lung disease in systemic sclerosis: A simple staging system. Am J Respir Crit Care Med. 2008;177:1248–54. doi: 10.1164/rccm.200706-877OC. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1108827
- 48.Clements PJ, Tan M, McLaughlin VV, Oudiz RJ, Tapson VF, Channick RN, Rubin LJ, Langer A, Pulmonary Arterial Hypertension Quality Enhancement Research Initiative I The pulmonary arterial hypertension quality enhancement research initiative: Comparison of patients with idiopathic pah to patients with systemic sclerosis-associated pah. Ann Rheum Dis. 2012;71:249–52. doi: 10.1136/annrheumdis-2011-200265. [DOI] [PubMed] [Google Scholar]
- 49.Kawut SM, Taichman DB, Archer-Chicko CL, Palevsky HI, Kimmel SE. Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest. 2003;123:344–50. doi: 10.1378/chest.123.2.344. [DOI] [PubMed] [Google Scholar]
- 50.Fisher MR, Mathai SC, Champion HC, Girgis RE, Housten-Harris T, Hummers L, Krishnan JA, Wigley F, Hassoun PM. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum. 2006;54:3043–50. doi: 10.1002/art.22069. [DOI] [PubMed] [Google Scholar]
- 51.Overbeek MJ, Vonk MC, Boonstra A, Voskuyl AE, Vonk-Noordegraaf A, Smit EF, Dijkmans BA, Postmus PE, Mooi WJ, Heijdra Y, Grunberg K. Pulmonary arterial hypertension in limited cutaneous systemic sclerosis: A distinctive vasculopathy. Eur Respir J. 2009;34:371–9. doi: 10.1183/09031936.00106008. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1163330
- 52.Gunther S, Jais X, Maitre S, Berezne A, Dorfmuller P, Seferian A, Savale L, Mercier O, Fadel E, Sitbon O, Mouthon L, Simonneau G, Humbert M, Montani D. Computed tomography findings of pulmonary venoocclusive disease in scleroderma patients presenting with precapillary pulmonary hypertension. Arthritis Rheum. 2012;64:2995–3005. doi: 10.1002/art.34501. [DOI] [PubMed] [Google Scholar]
- 53.Mathai SC, Bueso M, Hummers LK, Boyce D, Lechtzin N, Le Pavec J, Campo A, Champion HC, Housten T, Forfia PR, Zaiman AL, Wigley FM, Girgis RE, Hassoun PM. Disproportionate elevation of n-terminal pro-brain natriuretic peptide in scleroderma-related pulmonary hypertension. Eur Respir J. 2010;35:95–104. doi: 10.1183/09031936.00074309. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/722035706
- 54.Overbeek MJ, Groepenhoff H, Voskuyl AE, Smit EF, Peeters JW, Vonk-Noordegraaf A, Spreeuwenberg MD, Dijkmans BC, Boonstra A. Membrane diffusion- and capillary blood volume measurements are not useful as screening tools for pulmonary arterial hypertension in systemic sclerosis: A case control study. Respir Res. 2008;9:68. doi: 10.1186/1465-9921-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tedford RJ, Mudd JO, Girgis RE, Mathai SC, Zaiman AL, Housten-Harris T, Boyce D, Kelemen BW, Bacher AC, Shah AA, Hummers LK, Wigley FM, Russell SD, Saggar R, Saggar R, Maughan WL, Hassoun PM, Kass DA. Right ventricular dysfunction in systemic sclerosis-associated pulmonary arterial hypertension. Circ Heart Fail. 2013;6:953–63. doi: 10.1161/CIRCHEARTFAILURE.112.000008. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718022833
- 56.Follansbee WP, Miller TR, Curtiss EI, Orie JE, Bernstein RL, Kiernan JM, Medsger TA., Jr A controlled clinicopathologic study of myocardial fibrosis in systemic sclerosis (scleroderma) J Rheumatol. 1990;17:656–62. [PubMed] [Google Scholar]
- 57.Hachulla AL, Launay D, Gaxotte V, de Groote P, Lamblin N, Devos P, Hatron PY, Beregi JP, Hachulla E. Cardiac magnetic resonance imaging in systemic sclerosis: A cross-sectional observational study of 52 patients. Ann Rheum Dis. 2009;68:1878–84. doi: 10.1136/ard.2008.095836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fox BD, Shimony A, Langleben D, Hirsch A, Rudski L, Schlesinger R, Eisenberg MJ, Joyal D, Hudson M, Boutet K, Serban A, Masetto A, Baron M. High prevalence of occult left heart disease in scleroderma-pulmonary hypertension. Eur Respir J. 2013;42:1083–91. doi: 10.1183/09031936.00091212. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718128697
- 59.Robbins IM, Hemnes AR, Pugh ME, Brittain EL, Zhao DX, Piana RN, Fong PP, Newman JH. High prevalence of occult pulmonary venous hypertension revealed by fluid challenge in pulmonary hypertension. Circ Heart Fail. 2014;7:116–22. doi: 10.1161/CIRCHEARTFAILURE.113.000468. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718196846
- 60.Steen V. Predictors of end stage lung disease in systemic sclerosis. Ann Rheum Dis. 2003;62:97–9. doi: 10.1136/ard.62.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mathai SC, Hummers LK, Champion HC, Wigley FM, Zaiman A, Hassoun PM, Girgis RE. Survival in pulmonary hypertension associated with the scleroderma spectrum of diseases: Impact of interstitial lung disease. Arthritis Rheum. 2009;60:569–77. doi: 10.1002/art.24267. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/1147497
- 62.Rhodes CJ, Howard LS, Busbridge M, Ashby D, Kondili E, Gibbs JS, Wharton J, Wilkins MR. Iron deficiency and raised hepcidin in idiopathic pulmonary arterial hypertension: Clinical prevalence, outcomes, and mechanistic insights. J Am Coll Cardiol. 2011;58:300–9. doi: 10.1016/j.jacc.2011.02.057. [DOI] [PubMed] [Google Scholar]
- 63.Ruiter G, Lanser IJ, de Man FS, van der Laarse WJ, Wharton J, Wilkins MR, Howard LS, Vonk-Noordegraaf A, Voskuyl AE. Iron deficiency in systemic sclerosis patients with and without pulmonary hypertension. Rheumatology (Oxford) 2014;53:285–92. doi: 10.1093/rheumatology/ket331. [DOI] [PubMed] [Google Scholar]
- 64.Cefle A, Inanc M, Sayarlioglu M, Kamali S, Gul A, Ocal L, Aral O, Konice M. Pulmonary hypertension in systemic lupus erythematosus: Relationship with antiphospholipid antibodies and severe disease outcome. Rheumatol Int. 2009;31:183–9. doi: 10.1007/s00296-009-1255-2. [DOI] [PubMed] [Google Scholar]
- 65.Winslow TM, Ossipov MA, Fazio GP, Simonson JS, Redberg RF, Schiller NB. Five-year follow-up study of the prevalence and progression of pulmonary hypertension in systemic lupus erythematosus. Am Heart J. 1995;129:510–5. doi: 10.1016/0002-8703(95)90278-3. [DOI] [PubMed] [Google Scholar]
- 66.Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: A comparison of worldwide disease burden. Lupus. 2006;15:308–18. doi: 10.1191/0961203306lu2305xx. [DOI] [PubMed] [Google Scholar]
- 67.Ruiz-Irastorza G, Garmendia M, Villar I, Egurbide MV, Aguirre C. Pulmonary hypertension in systemic lupus erythematosus: Prevalence, predictors and diagnostic strategy. Autoimmun Rev. 2013;12:410–5. doi: 10.1016/j.autrev.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 68.Schreiber BE, Connolly MJ, Coghlan JG. Pulmonary hypertension in systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2013;27:425–34. doi: 10.1016/j.berh.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 69.Jais X, Launay D, Yaici A, Le Pavec J, Tcherakian C, Sitbon O, Simonneau G, Humbert M. Immunosuppressive therapy in lupus- and mixed connective tissue disease-associated pulmonary arterial hypertension: A retrospective analysis of twenty-three cases. Arthritis Rheum. 2008;58:521–31. doi: 10.1002/art.23303. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718231323
- 70.Dhala A. Pulmonary arterial hypertension in systemic lupus erythematosus: Current status and future direction. Clin Dev Immunol. 2012;2012:854941. doi: 10.1155/2012/854941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hervier B, Meyer A, Dieval C, Uzunhan Y, Devilliers H, Launay D, Canuet M, Tetu L, Agard C, Sibilia J, Hamidou M, Amoura Z, Nunes H, Benveniste O, Grenier P, Montani D, Hachulla E. Pulmonary hypertension in antisynthetase syndrome: Prevalence, aetiology and survival. Eur Respir J. 2013;42:1271–82. doi: 10.1183/09031936.00156312. [DOI] [PubMed] [Google Scholar]
- 72.Gunnarsson R, Andreassen AK, Molberg O, Lexberg AS, Time K, Dhainaut AS, Bertelsen LT, Palm O, Irgens K, Becker-Merok A, Nordeide JL, Johnsen V, Pedersen S, Proven A, Garabet LS, Garen T, Aalokken TM, Gilboe IM, Gran JT. Prevalence of pulmonary hypertension in an unselected, mixed connective tissue disease cohort: Results of a nationwide, norwegian cross-sectional multicentre study and review of current literature. Rheumatology (Oxford) 2013;52:1208–13. doi: 10.1093/rheumatology/kes430. [DOI] [PubMed] [Google Scholar]
- 73.Kobak S, Kalkan S, Kirilmaz B, Orman M, Ercan E. Pulmonary arterial hypertension in patients with primary sjogren's syndrome. Autoimmune diseases. 2014;2014:710401. doi: 10.1155/2014/710401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Keser G, Capar I, Aksu K, Inal V, Danaoglu Z, Savas R, Oksel F, Tunc E, Kabasakal Y, Kitapcioglu G, Doganavsargil E. Pulmonary hypertension in rheumatoid arthritis. Scand J Rheumatol. 2004;33:244–5. doi: 10.1080/03009740410005809. [DOI] [PubMed] [Google Scholar]
- 75.Le Pavec J, Girgis RE, Lechtzin N, Mathai SC, Launay D, Hummers LK, Zaiman A, Sitbon O, Simonneau G, Humbert M, Hassoun PM. Systemic sclerosis-related pulmonary hypertension associated with interstitial lung disease: Impact of pulmonary arterial hypertension therapies. Arthritis Rheum. 2011;63:2456–64. doi: 10.1002/art.30423. [DOI] [PubMed] [Google Scholar]
- 76.Le Pavec J, Humbert M, Mouthon L, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;181:1285–93. doi: 10.1164/rccm.200909-1331PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kiely DG, Elliot CA, Sabroe I, Condliffe R. Pulmonary hypertension: Diagnosis and management. BMJ. 2013;346:f2028. doi: 10.1136/bmj.f2028. [DOI] [PubMed] [Google Scholar]