

Abstract
Obesity is an important clinical and public health challenge, epitomized by excess adipose tissue accumulation resulting from an imbalance in energy intake and energy expenditure. It is a forerunner for a variety of other diseases such as type-2-diabetes (T2D), cardiovascular diseases, some types of cancer, stroke, hyperlipidaemia and can be fatal leading to premature death. Obesity is highly heritable and arises from the interplay of multiple genes and environmental factors. Recent advancements in Genome-wide association studies (GWAS) have shown important steps towards identifying genetic risks and identification of genetic markers for lifestyle diseases, especially for a metabolic disorder like obesity. According to the 12th Update of Human Obesity Gene Map there are 253 quantity trait loci (QTL) for obesity related phenotypes from 61 genome wide scan studies. Contribution of genetic propensity of individual ethnic and racial variations in obesity is an active area of research. Further, understanding its complexity as to how these variations could influence ones susceptibility to become or remain obese will lead us to a greater understanding of how obesity occurs and hopefully, how to prevent and treat this condition. In this review, various strategies adapted for such an analysis based on the recent advances in genome wide and functional variations in human obesity are discussed.
Keywords: Epigenetics, genetics, GWAS, human obesity, mitochondria, obesity
Introduction
Obesity is considered as a metabolic syndrome resulting from a chronic imbalance of energy intake versus energy expenditure, leading to storage of excessive amounts of triglycerides in adipose tissue1. It is a major risk factor for multiple disorders, such as type 2 diabetes, cancer, fatty liver disease, hormonal disturbances, hypertension, cardiovascular diseases (CVD), increased and morbidity and mortality rates, etc2,3,4,5,6,7. According to the current estimates, by 2015 more than 700 million individuals worldwide will be obese8. Obesity rates are also increasing in children and adolescents all over the world, predisposing them to poor health from an early age8,9.
In clinical practice, it is measured in terms of body mass index (BMI), which gives a surrogate measure of overall obesity and, accordingly the World Health Organization (WHO) classifies a person with a BMI ≥25 kg/m2 as obese and a BMI ≥40 kg/m2 as extremely obese8. It is important to note that sex and age are associated with differences in obesity and body composition. For example, women tend to store more fat subcutaneously rather than in visceral adipose tissue. So at the same BMI, women will tend to carry more body fat than men9. Two general patterns of fat distribution have been observed viz., android type or central obesity (adipose deposition in the abdominal area) common in males and gynoid type (adipose deposition around the hips) common in females10. Android/central obesity is an established independent risk factor for CVD and type 2 diabetes, whereas the gynoid pattern is thought to be protective or inversely correlated11. To account for these differences in fat distribution, waist-to-hip ratio (WHR) is commonly used and BMI and WHR are correlated (r2~0.6)12.
The current lifestyle has driven obesity prevalence to epidemic proportions with a substantial genetic contribution of 40-70 per cent approximately13,14. Since human obesity occurs due to sedentary lifestyle, epigenetics (mitotically and meiotically heritable changes in gene expression without any change in the DNA sequence15) also play an important role in its establishment. There are two important mechanisms involved in epigenetics regulations viz. DNA cytosine methylation and histone modifications16. It has also been observed that microRNAs (miRNAs) extended their role to epigenetic regulation17. Thus, dietary methyl-groups (choline, methionine, genistein and folate) intake during critical periods of developmental stages can alter promoter DNA and histone methylation resulting in the lifelong changes in gene expression and thereby altering the epigenome towards obesity in adulthood18,19.
Genetics
Being a very complex disease, obesity does not appear to be limited to a single gene disorder (Table I) but rather found to occur as symptoms of other disorders (Table II) or as a result of multiple gene disorders (Table III). Hence, depending on the suspected aetiology obesity could be classified into three subgroups: Monogenic, Syndromic and Polygenic or Common obesity. The first single gene defect causing monogenic obesity was first described in 199720, and to date, about 20 such disorders have been reported for an autosomal form of obesity1 (Table I). Interestingly, all these mutations lie in the leptin/melanocortin pathway of the central nervous system (CNS) which are critical in the regulation of whole-body energy homeostasis68. Obesity in these cases appears to be extremely severe due to increased appetite and diminished satiety. The second type, syndromic obesity arises from discrete genetic defects or chromosomal abnormalities at several genes, and can be autosomal or X-linked69 (Table II). The third, polygenic/common obesity is due to the simultaneous presence of DNA variations in multiple genes. Potentially, many such polygenic variants (mostly >100) play a role in body weight regulation70 (Table III). If an individual harbours many such polygenic variants for increased body weight, obesity can ensue and each variant will have a higher frequency than in normal/lean individuals70. A polygenic basis of obesity also implies that a specific set of polygenic variants relevant for obesity can vary from individual to individual.
Table I.
List of genes responsible for monogenic obesity: Autosomal reccessive form of obesity

Table II.
List of syndromic obesity in humans: Autosomal or X-linked
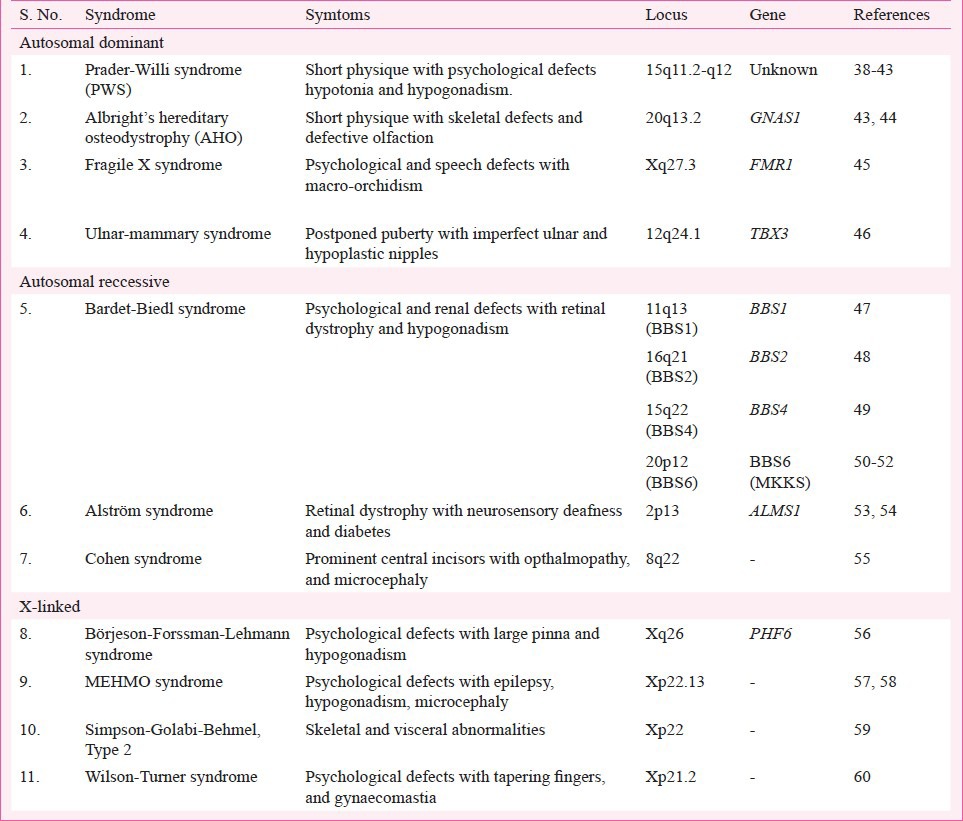
Table III.
List of genetic modifications (SNPs) showing polygenic effects on body weight in terms of BMI in humans
Strategic approaches for detection of obese genes
Human obesity, usually arises from the combined effects of the interactions among multiple genes, environmental factors and behaviour, and this complex aetiology makes the management and prevention of human obesity especially challenging. A genetic basis for obesity exists and has been proven to be a dreadful task. Genetic epidemiologic methods for the gene discovery of complex traits, such as obesity, can be divided into two broad classes: hypothesis-driven (candidate gene or biologic pathway) and hypothesis-free (genome-wide linkage and genome-wide association) approaches.
Candidate gene single nucleotide polymorphism (SNP) analyses: The hypothesis-driven approach (candidate gene or biologic pathway analysis) needs a prior knowledge of the cause(s) for the genetic polymorphisms in a candidate gene or a biologic pathway being studied and their effect(s) on a particular phenotype of interest. This approach has been considered to be an efficient strategy for identifying genetic variants with small or modest effects that underlie susceptibility to common disease, including obesity. The selection of candidate gene(s) should, therefore, consider both the significance of the candidate gene to the pathogenesis of the disease of interest and its functional effects due to a particular polymorphism. So the design of the candidate gene approach is simple; the fundamental requirements are the identification of a gene that is involved in the disease phenotype, a polymorphic marker within that gene and a suitable set of subjects to genotype for that marker. But the identification of the potential candidate gene(s) is the main stumbling block.
There are two main types of candidate genes that are generally considered in such studies: functional and positional. Functional candidates are the genes with products that are involved in the pathogenesis of the disease. Obviously, this is highly dependent on the current state of knowledge about a disease, and in the case of obesity, the discovery of signaling molecules such as leptin and pro-opiomelanocortin (POMC) has provided a great stimulus to the field. Positional candidates are the genes that lie within genomic regions that have been shown to be genetically important in linkage or association studies, or by the detection of chromosomal translocations that disrupt the gene71.
Candidate gene analysis is an indirect test of association to examine the relation between a dense map of SNPs and disease, while candidate SNP analysis is a direct test of association between putative functional variants and disease risk. The advantage of indirect association is that it does not require prior determination of which SNP might be functionally important; however, the disadvantage is that larger numbers of SNPs need to be genotyped72. A combination of functionally important SNPs with a collection of tag SNPs covering the entire candidate gene has been used in many candidate gene association studies73. Genetic variants in multiple candidate genes within the same biologic pathway can be examined, and their interaction can be tested in pathway analysis. But those genetic variants in multiple candidate genes of different biologic pathways and their interactions are very difficult to study through only candidate gene/SNP analysis as in the case of human obesity. Several genes have been analyzed in humans because these were found to be involved in central or peripheral pathways controlling energy intake and expenditure in animal models. Enormous association studies for obesity involving cases and controls or, less regularly, families comprising one or more affected children and both parents have been performed. But only for a small number of genes meta-analyses have been carried out and a list of latest positive results is available at http://obesitygene.pbrc.edu/ which showed positive associations of obesity phenotypes with a total of 113 candidate genes, of which only for 18 genes a minimum of five positive studies had been reported as often the study groups were small74. The first truly validated polygenic effect on body weight detected via a candidate gene analysis was Val103Ile polymorphism in melanocortin-4 receptor (MC4R) gene75.
Generally, these association studies have not given consistent results. Therefore, it is very difficult to get any convincing meta-analysis of variants in candidate genes that are explicitly linked to the genetic risk for obesity76. There is a wide difference in the obesogenic environments from where the subjects are recruited for the study. In many cases, data could not be replicated because of the various limitations in studies with the cohort size in which the association of variant(s) with the disease(s) was first detected. Because the contribution of any given gene to the phenotype of a complex trait is often minimal, a large cohort size is required if statistical significance is to be achieved. Another disadvantage of candidate gene analysis is that it depends on a prior hypothesis about disease mechanisms, therefore, the discovery of new genetic variants or novel genes would be excluded. Thus, the candidate gene approach is more appropriate for single gene disorder and not for the obesity like complex diseases. This type of approach may not give full resolution to the problem, but may help in establishing relationship between disease susceptibility and genetic variation. Hence, the only way forward seems to be investigation of the functional roles of the current candidate genes in model organisms and in vitro cell systems using Genome-Wide Approaches (GWA) which lead to the development of functional assays to test putative activator/inhibitor molecules as potential therapeutics.
Genome-wide approaches
Through genome wide association studies (GWAS) up to 2,000,000 genetic variants can now be analyzed for association with a given phenotype and have been proven extremely successful for various phenotypes77. This approach can be pursued through two ways, viz., Genome-wide linkage scans (GWLS) and Genome-wide association studies (GWAS). The GWLS identify chromosomal regions having gene(s) pertinent for a particular phenotype via linkage data. The regions underlying linkage peaks are narrowed down by fine mapping, so that the candidate gene analyses can be pursued. The first candidate gene for early onset of obesity detected via GWLS was ectonucleotide pyrophosphatase/phophodiesterase 1 (ENPP1) and attempts are ongoing to validate the association78. More than 40 microsatellite-based GWLS have been performed and none of the single candidate genes detected have been validated explicitly which further shows that either the effect sizes of genes influencing adiposity are small or the substantial heterogeneity exists or both. Therefore, to avoid such types of uncertainty GWLS analysis requires large samples size.
The GWAS provide a better devise to identify common variants with low to rational penetrance and are significant as risk factors for the trait of interest. Within a short duration, GWAS have proven to be very successful for the detection of polygenic variants. The advent of high density SNP-chips has made GWAS practicable and has given a new dimension to the study of complex diseases through the increased identification of confirmed genes and thereby revolutionized the molecular genetic analyses of complex disorders. GWAS performed for obesity or BMI (Table III) have established it as an extremely powerful tool to detect genetic variants for the complex phenotype(s). For example, presently known polygenic variants have a mean effect on BMI of approximately 0.03-0.5 kg/m2 which appears reasonable as these effect sizes represent the upper limit of such common variants64,79,80. So far, independent gene variants with small but replicable effects on body weight have been identified unambiguously in 17 gene regions79. FTO gene is the first example of a non-syndromic obesity gene studied thoroughly via GWAS of T2D81. Despite the initial success of the GWAS strategy, the established loci together explain less than 2 per cent of the inter-individual BMI variation and less than 1 per cent of the inter-individual WHR variation12,82. With heritability estimates of 40-70 per cent for BMI and 30-60 per cent for WHR, there must be many more susceptible loci to be discovered32. These types of susceptible loci can only be unraveled if GWAS are expanded to different ethnic groups through large-scale international collaboration and meta-analysis of existing data (Table IV), selection of defined scientific procedures like CT scan, dual-energy X-ray absorptiometry (DEXA) or magnetic resonance imaging (MRI) and consideration of rare and low frequency variants and non-coding RNAs. Though GWAS approach seems to be a strong way of analysis for complex disorders, it has to be extended for different environmental conditions as it is very evident that gene-environment (GXE) interactions also regulate the mechanisms of gene expression90.
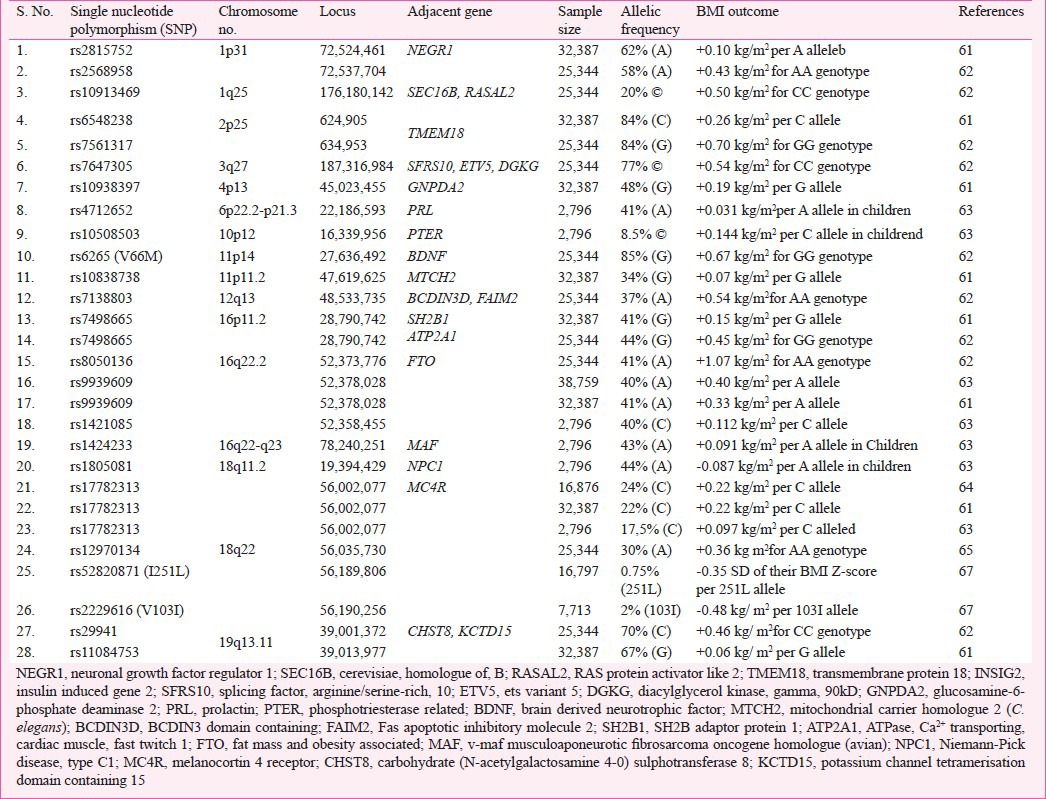
Table IV.
Summary of Genome-wide association studies (GWAS) or meta-analysis for obesity in humans
Epigenetic modifications
Modifications that affect gene expression but do not alter the DNA sequence are termed epigenetic modifications. These include DNA methylation and histone modifications, which are expected to have regulatory roles in the inheritance and vulnerability to obesity, by affecting the expression(s) of associated gene(s). Also, intrauterine atmosphere during specific developmental phases can vary the epigenetics of an individual and may work as a foundation for the obesity and other phenotypes during later stage of life91,92. For example, variations in birth weight are affected by several factors such as maternal genes, maternal in utero and placental factors, maternal BMI, maternal smoking, maternal alcohol consumption, maternal drug use, exercise during pregnancy, paternal genes, birth weight, etc. In the similar manner as during the early post-natal period poor nutrition also affects the mother metabolism to acclimatize to favour the storage of nutrients93. Another study on Pima Indians showed paternally imprinted gene located on chromosome 11 position 80 cm was influencing birth weight94. Similarly, in a study on Mexican Americans quantitative trait loci (QTL) on chromosome 6q was found to be associated with birth weight regulation95. Hence, gene-environment (GXE) interactions play a significant role in the aetiology of obesity may be via modifications in DNA methylation patterns. Thus, apart from variations at DNA sequence level, epigenetic incidents also seem to contribute to the epidemiocity of obesity which are evident from modern day sedentary living style. It has been observed that during the early years of life monozygotic (MZ) twins are epigenetically indistinguishable from each other. But, with increasing age remarkable differences in their overall content and genomic distributions of 5-methylcytosine DNA and histone acetylations become noticeable and similarly environment could have an influence on individual's BMI91. A comparative study of epigenetic metastability of 6,000 unique genomic regions between matched monozygotic (MZ) and dizygotic (DZ) twins demarcated epigenetic differences in both the MZ and DZ twins96. Molecular mechanisms of heritability may not be limited to differences in DNA sequence only, rather epigenetic modifications are also acting as one of the very important governing factors in unraveling the secrets behind the blue prints of DNA sequence.
Mitochondrial contribution to obesity
Being the centre of all the metabolic processes and very susceptible to change, mitochondria play a crucial role in the development of any disorders. Since only mother contributes mitochondria to the next generation, there is a chance of maternal inheritance of diseases affecting physiology of mitochondria in mother. For example, maternal obesity (in utero environment) plays a detrimental role in the development of early embryo(s) during embryogenesis97. It was further elaborated in a study including maternal-diet induced obesity in a murine model through the association of altered mitochondrial activities and redox status of oocytes and zygotes98. These altered mitochondrial properties involved an increase in mitochondrial membrane potential, mitochondrial DNA (mtDNA) content and biogenesis, generation of reactive oxygen species (ROS) and depletion in glutathione level leading to more oxidized - redox state. These effects resulted in oxidative stress which led to the significant developmental impairment at early embryogenesis and might be the explanation for the reduced reproductive status observed in obese women99. Thus, mitochondria were found to be the liable candidates for compromised metabolism in the embryo, and are maternal in origin. Mitochondria also execute various regulatory roles during oocyte maturation, fertilization, initiation and progression of pre-implantation of embryos100,101. As power house of the cell, the central and most vital role of mitochondria is the synthesis of adenosine triphosphate (ATP) by oxidative phosphorylation, a mechanism coupling the oxidation of nutrients and reducing equivalents [NAD(P)H, FADH2] with the phosphorylation of adenosine diphosphate (ADP). Consumption of the energy rich diets results in the excess production of these reducing equivalents [NAD(P)H, FADH2] which ultimately leads to the increased pumping of protons (H+) out of the mitochondrial matrix through mitochondrial electron transport chain (ETC) and results in hyperpolarization of the inner mitochondrial membrane thereby generating excess of proton motive force (PMF) and excess of ATP. Besides being used for energy production, these reducing equivalents [NAD(P)H and FADH2] are known to regulate the intracellular redox status. NADH oxidation in the mitochondria will produce ROS whereas NADPH oxidation (in cytosol and mitochondria) serves to restore the antioxidant protection by reducing per-oxiredoxins, thioredoxin and oxidised glutathione. Thus, mitochondria have dual regulation on the intracellular redox state via regeneration of antioxidants and via ROS production102. Apart from centre of ATP production, mitochondria are also a source of cellular guanosine-5’-triphosphate (GTP) production as well as the site of amino acid synthesis and the reservoir of cellular calcium (Ca2+). Therefore, any change(s) in mitochondrial activity can alter cellular functions. Importance of mitochondria in oocyte quality and embryo development has been highlighted in many studies showing the various impacts of mitochondrial biogenesis defect(s) on mitochondrial mass, failure of oocyte maturation, abnormal embryo development103,104, etc. Both the quality and the quantity of mitochondria are an important prerequisite for successful fertilization and embryo development105. In vitro studies including have emphasized the vulnerability of mitochondria towards various environmental stress either within the oocyte or in the developing embryo and it has been shown that even low-level of acquired mitochondrial injuries may persist into embryonic life106,107. Latent influences of maternal nutritional status during obesity had been indicated in studies showing that periconceptual exposure to high energy substrates such as fatty acids and proteins resulted in perturbed mitochondrial metabolism in oocyte as well as in embryo108,109. Hence, mitochondrial abnormalities in oocytes and in early embryos have been found as a direct environmental consequence of maternal obesity. It is, therefore, noteworthy that alterations in mitochondrial activities (i.e., dysfunctional mitochondria) are not only restricted to the maternal obesity but rather obesity itself results in the development of dysfunctional mitochondria or vice versa. This ultimately leads to various types of obesity associated complications such as development and progression of T2D complications, early mitochondrial adaptations in skeletal muscles to diet-induced obesity, mitochondrial remodelling in adipose tissue associated with obesity, inflammation and mitochondrial fatty acid β-oxidation linking obesity to early tumour promotion in pancreas, etc110,111,112,113,114. It has been observed that mitochondrial genome polymorphisms are also involved in the development of obesity syndrome such as a mtSNP, 8684C>T (T53I) in the mitochondrial ATP synthase subunit 6 gene (ATP6) was detected in five patients of type-2 diabetes and was not detected in any of the young obese adults. Similarly, two mtSNPs, 3497C>T (A64V) in NADH dehydrogenase subunit 1 gene (ND1) and 1119T>C (472U>C) in the 12S rRNA gene, were detected in five young obese adults and were not found in any of the diabetic patients115. Further, several studies have also shown the associations of different mtSNPs with the incidence of obesity in various human ethnic groups during the course of evolution. These include three human mtSNPs viz., ND2, COX2 and ATP8 within genes encoding proteins of electron transport and oxidative phosphorylation in sub-haplogroups of the Pima Indians. These were adaptations toward an energy-efficient metabolism when this population migrated to the desert and adopted a restricted caloric intake. Today these may contribute to obesity116. Polymorphisms of the UCP2 gene (rs660339 and rs659366) were found to be associated with body fat distribution and risk of abdominal obesity in Spanish population117. The UCP2 A55V variant was found to be associated with obesity and related phenotypes in an aboriginal community in Taiwan118. UCP1 variants, g.IVS4-208T>G SNP was associated with obesity in Southern Italy severe obese population119. Two mtSNPs (mt4823 and mt8873) and mtDNA haplogroup X were observed to be significant markers associated with reduced body fat mass120. Linkage and association analyses of the UCP3 gene showed an association with obesity phenotypes in Caucasian families121. A common polymorphism in the promoter of UCP2 gene was found to be associated with obesity and A allele associated with obesity and hyperinsulinaemia in north Indians122. In another study, obesity and hepatosteatosis were observed in human 8-oxoguanine-DNA glycosylase 1 (hOGG1) transgenic (TG) mice with enhanced oxidative damage of mitochondrial DNA due to overexpression of the transgene hOGG1123. Thus, mitochondria might be either a culprit or a victim in obesity and the studies focussing on this organelle may help in better understanding of the cause of diet-induced obesity.
The obesity cauldron – the current focus
Obesity, especially central obesity being heritable arises from the interactions of multiple genes and environmental factors (GXE). Since the sequencing of the human genome, researchers and geneticists are busy in documenting the various interactions among genetic and environmental risks of precursors to chronic illness, including the influence of gene-diet interactions in disorders such as obesity and diabetes. This includes systematic genomic variation(s) in identifying various risks for common diseases along with the knowledge of body compositions in different populations with a consideration of individual's ethnic and racial variations. This might lead to the development of more individualized, more predictive, and more preventive therapeutics against obesity. Thus, if a strategy of GWAS along with epigenetic study of a particular disease like obesity is followed, it will help in finding a solution (Figure). For example, a novel genome wide scan employing a high density SNP array led to the identification of a SNP in the vicinity of the insulin induced gene 2 (INSIG2) and was found to be associated with obesity in both children and adults124,125. In another report, MC4R coding sequence variants were found to be associated with fat mass, body weight and obesity80. The MC4R mutations were also found to be linked with dominantly inherited genetic cause of childhood obesity in Americans of European and African ancestry126.
Fig. 1.
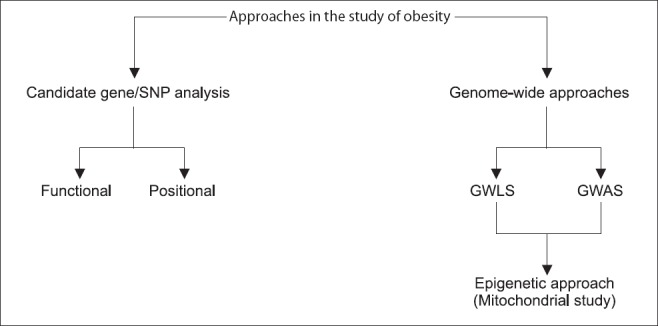
Strategic approach towards analysis of obesity in humans. SNP, single nucleotide polymorphisms; GWLS, genome-wide linkage scans; GWAS, genome-wide association studies.
To detect small gene effects and to narrow down the genomic target region more precisely, the GWAS are expected to be more powerful so that these can analyse a fixed set of genome and try to fix biomarkers. An example is peroxisome proliferater-activated receptor gamma (PPARγ) which is essential for adipocyte differentiation127. Here a zinc finger-containing transcription factor abundantly expressed in adipose tissue Krox 20 is expressed early in adipogenesis and is found to trans-activate the CCAAT/enhancer binding proteins β (C/EBP) β promoter128. Others like Kruphel-like factor (KLF) 6 and KLF 15 have been shown to promote adipogenesis and KLF15 is noticed to upregulate GLUT 4 expression129,130. Recent reports demonstrated that KLF4 functions as an immediate early regulator of adipocyte differentiation. Another one is adiponectin which is an oxidative regulator from adipocytes that modulates insulin sensitivity and thus regulates glucose and lipid metabolism and leads to a reduction in energy homeostasis in obesity131. Hydroxysteroid (11-β) dehydroxgeinase-1 overexpression leads to visceral obesity by regulating glucocorticoid action132. Others in the list are mutation of MC4R, leptin/leptin receptor, prohormone convertase1 and pro-opimelanocartin (POMC)20,21,22,23,24,25,26,30,31.
Genes associated with β-cell dysfunction have also been identified which include hepatocyte nuclear factor 4 and α1 polymorphism in potassium channel Kir6-2 (KCNJ11) and transcription factor 7 like 2 (TCF 7 L 2). Many candidate genes have also been identified such as calpain 10, adiponectin, PPAR γ co-activator 1 (PGC1) and glucose transporter GLUT 2133. Genetic polymorphisms at the perilipin (PLIN) locus that is minor allele at PLIN4 (11482G>A) was associated with higher risk of metabolic syndrome (MS) at baseline, whereas the PLIN6 SNP (14995A>T) was found to be associated with weight loss in obese children and adolescents134. A positive functional relevance of nicotinamide nucleotide transhydrogenase (NNT) in the development of human obesity and visceral fat distribution has been observed in obese patients and correlated with body weight, BMI, percentage body fat, visceral and subcutaneous fat area, waist and hip circumference, and fasting plasma insulin (FPI)135. Association of decreased levels of plasma ghrelin has been observed with obesity in obese Caucasian and Pima Indians136. Interleukin-1 (IL-1) gene polymorphisms may be involved in increased central obesity and the genetic influences are more evident among patients who have a higher level of obesity or inflammatory markers77. Genetic variation in the McKusick-Kaufman gene (MKKS) gene may play a role in the development of obesity and the metabolic syndrome in Greek population138. A study on postmenopausal women suggested that FTO gene was a susceptibility locus for both obesity and type-2-diabetes (T2D). Common genetic variants in the intron 1 of FTO gene may present a natural predisposition to obesity in an ethnicity-specific manner also139. The insulin responsive adiponectin genetic variants of potatin-like phospholipase domain-containing genes 1 (PLPLA1) have moderate effect on obesity and potatin-like phospholipase domain-containing gene 3 (PNPLA3) or adeponectin effects (ADPN) gene shows on insulin sensitivity140. Procolipase (CLPS) is secreted from the exocrine pancreas into the gastrointestinal tract and its genetic variability is associated with the secretary function of insulin in non-diabetic humans141. SNPs in α-2 subunit of neuronal nicotinic acetylcholine receptor gene CHRNA2 rs2043063 SNP might be a risk factor for overweight/obesity in Koreans142. Folate/vitamin B12 plays vital role in the critical stages of foetus development and involves one carbon pool metabolism which may lead to greater insulin resistance, and further to the development of obesity143.
According to twelfth update of Human Obesity Gene Map, 52 genomic regions harbour QTLs supported by two or more studies74. The number of QTLs reported from animal models has reached to 408. The number of human obesity QTLs derived from genome scans continues to grow, and so far 253 QTLs for obesity-related phenotypes from 61 genome-wide scans have been reported. Association studies between the variation(s) of DNA sequence in specific genes and obesity phenotypes have also been increased considerably (426 findings of positive associations with 127 candidate genes). At our institute, two mutant obese rat strains viz., WNIN/Ob (with euglycaemia) and WNIN/GR-Ob (with impaired glucose tolerance) had been developed naturally from a Wistar inbred rat colony144,145. These mutants show hyperinsulinaemia, hypertriglyceridaemia and hypercholesterolaemia, and they also have several obese features such as polydipsia, polyuria and proteinuria. From the preliminary studies it has been found that mutant WNIN/Ob does not exhibit any of the conventional defects either in leptin or leptin receptor locus (unpublished data) but showed the defect on chromosome no. 5, in the upstream region of leptin receptor and the studies are still ongoing to identify and sequence the locus.
Conclusions and the way forward
Numerous analyses have been published discussing about the genetic complications and various types of challenges concerned with the biological pathway of common obesity74,146,147,148,149. However, the major obstruction is the replication of data. The complexity of a trait/disease in an individual's life is a result of accumulation of various interactions of the linked genes to different genetic settings and exposure to diverse environmental factors. Due to scarce knowledge on the extent to which genes finally contribute to complex trait, the significance of subtle environmental factors may not be valued. Thus, at such an early stage of our knowledge to unravel the genetics of obesity both replicated and un-replicated data should be considered equally important. So far, most of the studies on polygenic obesity are SNP based which are located either within or near a candidate gene. Considering the entire candidate gene studies on animal models and in vitro as an initial level studies, their association with the common obese phenotype should be verified through various types of case control and family studies. But, in contrast to monogenic obesity, many genes and chromosomal area contribute to characterize common obese phenotype (polygenic) and have been found linked to an extensive range of biological functions, such as regulation of food intake, energy expenditure, lipid and glucose metabolism, adipose tissue development, etc. Even with this ever increasing gene catalogue at our disposal, unraveling the molecular mechanisms of obesity is still challenging. As not only the number of genes associated with obesity is high, but, the modifications in these gene(s) also show the significant polymorphisms in the elucidation of environmental stimuli. Further, in utero environment and expression of several genes during embryonic development and specifications play an important role in governing the intensity of obesity. For instance, genetically programmed developmental variations in adipocytes and their precursors in different sections of the body play a significant role in the progression of obesity via a complex network of transcription factors like activators, co-activators and repressors150.
With the advancement in the knowledge of the human genome, the development of comprehensive technologies, and new analytical approach it will be feasible to address both the genetic and environmental features of complex traits simultaneously. But the success will eventually lie with international consortiums that pool together expertise and resources to describe and interpret the functional role of the various genetic factors underlying the diverse types of obesity. Undoubtedly, family, twin and adoption studies primarily provide sufficient data for moderate to high heritability of BMI and are a focus of molecular research in finding an explanation at DNA level. Epigenetic research will add a new dimension to this by explaining intra-individual variation in body weight. Thus, with the use of advanced technologies epigenetic profile of the associated obesity genes can be discerned and could also be applied in a genome-wide approach.
References
- 1.O’Rahilly S. Human genetics illuminates the paths to metabolic disease. Nature. 2009;462:307–14. doi: 10.1038/nature08532. [DOI] [PubMed] [Google Scholar]
- 2.Anonymous. Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults-the evidence report. National Institutes of Health. Obes Res. 1998;6(Suppl 2):51S–209S. [PubMed] [Google Scholar]
- 3.Lewis CE, Mc Tique KM, Burke LE, Poirier P, Eckel RH, Howard BV, et al. Mortality, health outcomes, and body mass index in the overweight range: a science advisory from the American Heart Association. Circulation. 2009;119:3263–71. doi: 10.1161/CIRCULATIONAHA.109.192574. [DOI] [PubMed] [Google Scholar]
- 4.Flegal KM, Graubard BI, Williamson DF, Gail MH. Cause specific excess deaths associated with underweight, overweight, and obesity. JAMA. 2007;298:2028–37. doi: 10.1001/jama.298.17.2028. [DOI] [PubMed] [Google Scholar]
- 5.Pischon T, Boeing H, Hoffmann K, Bergmann M, Schulze MB, Overvad K, et al. General and abdominal adiposity and risk of death in Europe. N Engl J Med. 2008;359:2105–20. doi: 10.1056/NEJMoa0801891. [DOI] [PubMed] [Google Scholar]
- 6.Fontaine KR, Redden DT, Wang C, Westfall AO, Allison DB. Years of life lost due to obesity. JAMA. 2003;289:187–93. doi: 10.1001/jama.289.2.187. [DOI] [PubMed] [Google Scholar]
- 7.Teucher B, Rohrmann S, Kaaks R. Obesity: focus on all-cause mortality and cancer. Maturitas. 2010;65:112–6. doi: 10.1016/j.maturitas.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 8.WHO expert consultation, 2004. Appropriate body-mass index for Asian populations and its implications for policy and intervention strategies. Lancet. 2004;363:157–63. doi: 10.1016/S0140-6736(03)15268-3. [DOI] [PubMed] [Google Scholar]
- 9.Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007;132:2087–102. doi: 10.1053/j.gastro.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 10.Bouchard C, Despres JP, Mauriege P. Genetic and nongenetic determinants of regional fat distribution. Endocr Rev. 1993;14:72–93. doi: 10.1210/edrv-14-1-72. [DOI] [PubMed] [Google Scholar]
- 11.Wiklund P, Toss F, Weinehall L, Hallmans G, Franks PW, Nordstorm P, et al. Abdominal and gynoid fat mass are associated with cardiovascular risk factors in men and women. J Clin Endocrinol Metab. 2008;93:4360–6. doi: 10.1210/jc.2008-0804. [DOI] [PubMed] [Google Scholar]
- 12.Lindgren CM, Heid IM, Randall JC, Lamina C, Steinthorsdottir V, Qi L, et al. Genome-wide association scan meta-analysis identifies three Loci influencing adiposity and fat distribution. PLoS Genet. 2009;5:e1000508. doi: 10.1371/journal.pgen.1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stunkard AJ, Foch TT, Hrubec ZA. A twin study of human obesity. JAMA. 1986;256:51–4. [PubMed] [Google Scholar]
- 14.Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27:325–51. doi: 10.1023/a:1025635913927. [DOI] [PubMed] [Google Scholar]
- 15.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–63. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 16.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–7. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 17.Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Körner H, et al. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;15:2591–600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 18.Zeisel SH. Epigenetic mechanisms for nutrition determinants of later health outcomes. Am J Clin Nutr. 2009;89:1488S–93S. doi: 10.3945/ajcn.2009.27113B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dolinoy DC, Weidman JR, Waterland RA, Jirtle RL. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ Health Perspect. 2006;114:567–72. doi: 10.1289/ehp.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–8. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 21.Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet. 1998;18:213–5. doi: 10.1038/ng0398-213. [DOI] [PubMed] [Google Scholar]
- 22.Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 23.Krude H, Biebermann H, Luck W, Horn R, Brabant G, Grüters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19:155–7. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 24.Challis BG, Pritchard LE, Creemers JW, Delplanque J, Keogh JM, Luan J, et al. A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism. Hum Mol Genet. 2002;11:1997–2004. doi: 10.1093/hmg/11.17.1997. [DOI] [PubMed] [Google Scholar]
- 25.Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–6. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 26.Jackson RS, Creemers JW, Farooqi IS, Raffin-Sanson ML, Varro A, Dockray GJ, et al. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest. 2003;112:1550–60. doi: 10.1172/JCI18784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farooqi IS. Monogenic human obesity. Front Horm Res. 2008;36:1–11. doi: 10.1159/000115333. [DOI] [PubMed] [Google Scholar]
- 28.Farooqi IS, O’Rahilly S. Genetics of obesity in humans. Endocr Rev. 2006;27:710–8. doi: 10.1210/er.2006-0040. [DOI] [PubMed] [Google Scholar]
- 29.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–41. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 30.Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111–2. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- 31.Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20:113–4. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- 32.Hinney A, Schmidt A, Nottebom K, Heibü lt O, Becker I, Ziegler A, et al. Several mutations in the melanocortin-4 receptor gene including a nonsense and a frameshift mutation associated with dominantly inherited obesity in humans. J Clin Endocrinol Metab. 1999;84:1483–6. doi: 10.1210/jcem.84.4.5728. [DOI] [PubMed] [Google Scholar]
- 33.Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106:253–62. doi: 10.1172/JCI9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hinney A, Hohmann S, Geller F, Vogel C, Hess C, Wermter AK, et al. Melanocortin-4 receptor gene: case-control study and transmission disequilibrium test confirm that functionally relevant mutations are compatible with a major gene effect for extreme obesity. J Clin Endocrinol Metab. 2003;88:4258–67. doi: 10.1210/jc.2003-030233. [DOI] [PubMed] [Google Scholar]
- 35.Hinney A, Bettecken T, Tarnow P, Brumm H, Reichwald K, Lichtner P, et al. Prevalence, spectrum, and functional characterization of melanocortin-4 receptor gene mutations in a representative population based sample and obese adults from Germany. J Clin Endocrinol Metab. 2006;91:1761–9. doi: 10.1210/jc.2005-2056. [DOI] [PubMed] [Google Scholar]
- 36.Dempfle A, Hinney A, Heinzel-Gutenbrunner M, Raab M, Geller F, Gudermann T, et al. Large quantitative effect of melanocortin-4 receptor gene mutations on body mass index. J Med Genet. 2004;41:795–800. doi: 10.1136/jmg.2004.018614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–95. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 38.MacDonald HR, Wevrick R. The necdin gene is deleted in Prader-Willi syndrome and is imprinted in human and mouse. Hum Mol Genet. 1997;6:1873–8. doi: 10.1093/hmg/6.11.1873. [DOI] [PubMed] [Google Scholar]
- 39.Ozcelik T, Leff S, Robinson W, Donlon T, Lalande M, Sanjines E, et al. Small nuclear ribonucleoprotein polypeptide N (SNRPN), an expressed gene in the Prader-Willi syndrome critical region. Nat Genet. 1992;2:265–9. doi: 10.1038/ng1292-265. [DOI] [PubMed] [Google Scholar]
- 40.Jong MT, Gray TA, Ji Y, Glenn CC, Saitoh S, Driscoll DJ, et al. A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Hum Mol Genet. 1999;8:783–93. doi: 10.1093/hmg/8.5.783. [DOI] [PubMed] [Google Scholar]
- 41.Boccaccio I, Glatt-Deeley H, Watrin F, Roeckel N, Lalande M, Muscatelli F. The human MAGEL2 gene and its mouse homologue are paternally expressed and mapped to the Prader-Willi region. Hum Mol Genet. 1999;8:2497–505. doi: 10.1093/hmg/8.13.2497. [DOI] [PubMed] [Google Scholar]
- 42.De los Santos T, Schweizer J, Rees CA, Francke U. Small evolutionarily conserved RNA, resembling C/D box small nucleolar RNA, is transcribed from PWCR1, a novel imprinted gene in the Prader-Willi deletion region, which is highly expressed in brain. Am J Hum Genet. 2000;67:1067–82. doi: 10.1086/303106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weinstein LS, Yu S, Liu J. Analysis of genomic imprinting of Gs α gene. Methods Enzymol. 2002;344:369–83. doi: 10.1016/s0076-6879(02)44728-3. [DOI] [PubMed] [Google Scholar]
- 44.Weinstein LS, Chen M, Liu J. Gs(α) mutations and imprinting defects in human disease. Ann NY Acad Sci. 2002;968:173–97. doi: 10.1111/j.1749-6632.2002.tb04335.x. [DOI] [PubMed] [Google Scholar]
- 45.Rousseau F, Heitz D, Biancalana V, Blumenfeld S, Kretz C, Boue J, et al. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N Engl J Med. 1991;325:1673–81. doi: 10.1056/NEJM199112123252401. [DOI] [PubMed] [Google Scholar]
- 46.Bamshad M, Lin RC, Law DJ, Watkins WC, Krakowiak PA, Moore ME, et al. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat Genet. 1997;16:311–5. doi: 10.1038/ng0797-311. [DOI] [PubMed] [Google Scholar]
- 47.Mykytyn K ND, Searby CC, Shastri M, Yen HJ, Beck JS, Braun T, et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet. 2002;31:435–8. doi: 10.1038/ng935. [DOI] [PubMed] [Google Scholar]
- 48.Nishimura DY, Searby CC, Carmi R, Elbedour K, Van Maldergem L, Fulton AB, et al. Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome (BBS2) Hum Mol Genet. 2001;10:865–74. doi: 10.1093/hmg/10.8.865. [DOI] [PubMed] [Google Scholar]
- 49.Mykytyn K, Braun T, Carmi R, Haider NB, Searby CC, Shastri M, et al. Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet. 2001;28:188–91. doi: 10.1038/88925. [DOI] [PubMed] [Google Scholar]
- 50.Katsanis N, Beales PL, Woods MO, Lewis RA, Green JS, Parfrey PS, et al. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat Genet. 2000;26:67–70. doi: 10.1038/79201. [DOI] [PubMed] [Google Scholar]
- 51.Katsanis N, Lupski JR, Beales PL. Exploring the molecular basis of Bardet-Biedl syndrome. Hum Mol Genet. 2001;10:2293–9. doi: 10.1093/hmg/10.20.2293. [DOI] [PubMed] [Google Scholar]
- 52.Katsanis N AS, Badano JL, Eichers ER, Lewis RA, Hoskins BE, Scambler PJ, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–9. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- 53.Hearn T, Renforth GL, Spalluto C, Hanley NA, Piper K, Brickwood S, et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alstrom syndrome. Nat Genet. 2002;31:79–83. doi: 10.1038/ng874. [DOI] [PubMed] [Google Scholar]
- 54.Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat Genet. 2002;31:74–8. doi: 10.1038/ng867. [DOI] [PubMed] [Google Scholar]
- 55.Tahvanainen E, Norio R, Karila E, Ranta S, Weissenbach J, Sistonen P, et al. Cohen syndrome gene assigned to the long arm of chromosome 8 by linkage analysis. Nat Genet. 1994;7:201–4. doi: 10.1038/ng0694-201. [DOI] [PubMed] [Google Scholar]
- 56.Lower KM, Turner G, Kerr BA, Mathews KD, Shaw MA, Gedeon AK, et al. Mutations in PHF6 are associated with Borjeson-Forssman-Lehmann syndrome. Nat Genet. 2002;32:661–5. doi: 10.1038/ng1040. [DOI] [PubMed] [Google Scholar]
- 57.DeLozier-Blanchet CD, Haenggeli CA, Bottani A. MEHMO, a novel syndrome: assignment of disease locus to Xp21.1-p22.13. Mental retardation, epileptic seizures, hypogonadism and genitalism, microcephaly, obesity. Eur J Hum Genet. 1999;7:621–2. doi: 10.1038/sj.ejhg.5200364. [DOI] [PubMed] [Google Scholar]
- 58.Leshinsky-Silver E, Zinger A, Bibi CN, Barash V, Sadeh M, Lev D, et al. MEHMO (mental retardation, epileptic seizures, hypogenitalism, microcephaly, obesity): a new X-linked mitochondrial disorder. Eur J Hum Genet. 2002;10:226–30. doi: 10.1038/sj.ejhg.5200791. [DOI] [PubMed] [Google Scholar]
- 59.Brzustowicz LM FS, Khan MB, Weksberg R. Mapping of a new SGBS locus to chromosome Xp22 in a family with a severe form of Simpson-Golabi-Behmel syndrome. Am J Hum Genet. 1999;65:779–83. doi: 10.1086/302527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilson M, Mulley J, Gedeon A, Robinson H, Turner G. New X-linked syndrome of mental retardation, gynecomastia, and obesity is linked to DXS255. Am J Med Genet. 1991;40:406–13. doi: 10.1002/ajmg.1320400405. [DOI] [PubMed] [Google Scholar]
- 61.Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41:18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 63.Meyre D, Delplanque J, Chevre JC, Lecoeur C, Lobbens S, Gallina S, et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet. 2009;41:157–9. doi: 10.1038/ng.301. [DOI] [PubMed] [Google Scholar]
- 64.Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–94. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Loos RJF, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40:768–75. doi: 10.1038/ng.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saunders CL, Chiodini BD, Sham P, Lewis CM, Abkevich V, Adeyemo AA, et al. Metaanalysis of genome-wide linkage studies in BMI and obesity. Obesity (Silver Spring) 2007;15:2263–75. doi: 10.1038/oby.2007.269. [DOI] [PubMed] [Google Scholar]
- 67.Stutzmann F, Vatin V, Cauchi S, Morandi A, Jouret B, Landt O, et al. Non-synonymous polymorphisms in melanocortin-4 receptor protect against obesity: the two facets of a Janus obesity gene. Hum Mol Genet. 2007;16:1837–44. doi: 10.1093/hmg/ddm132. [DOI] [PubMed] [Google Scholar]
- 68.Coll AP, Farooqi IS, Challis BG, Yeo GS, O’Rahilly S. Proopiomelanocortin and energy balance: insights from human and murine genetics. J Clin Endocrinol Metab. 2004;89:2557–62. doi: 10.1210/jc.2004-0428. [DOI] [PubMed] [Google Scholar]
- 69.O’Rahilly S, Farooqi IS. Genetics of obesity. Philos Trans R Soc Lond B Biol Sci. 2006;361:1095–105. doi: 10.1098/rstb.2006.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hinney A, Hebebrand J. Polygenic obesity in humans. Obes Facts. 2008;1:35–42. doi: 10.1159/000113935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bell CG, Walley AJ, Frouguel P. The genetics of human obesity. Nat Rev Genet. 2005;6:221–34. doi: 10.1038/nrg1556. [DOI] [PubMed] [Google Scholar]
- 72.Carlson CS, Eberle MA, Kruglyak L, Nickerson DA. Mapping complex disease loci in whole-genome association studies. Nature. 2004;429:446–52. doi: 10.1038/nature02623. [DOI] [PubMed] [Google Scholar]
- 73.Cowley AW., Jr The genetic dissection of essential hypertension. Nat Rev Genet. 2006;7:829–40. doi: 10.1038/nrg1967. [DOI] [PubMed] [Google Scholar]
- 74.Rankinen T, Zuberi A, Chagnon YC, Weisnagel SJ, Argyropoulos G, Walts B, et al. The human obesity gene map: the 2005 update. Obesity (Silver Spring) 2006;14:529–644. doi: 10.1038/oby.2006.71. [DOI] [PubMed] [Google Scholar]
- 75.Geller F, Reichwald K, Dempfle A, Illig T, Vollmert C, Herpertz S, et al. Melanocortin-4 receptor gene variant I103 is negatively associated with obesity. Am J Hum Genet. 2004;74:572–81. doi: 10.1086/382490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Linlin T, Huaden Y, Quingxiao H, Fei C, Qinwen W, Leiting X, et al. Meta-analyses between 18 candidate genetic markers and overweight/obesity. Diagn Pathol. 2014;9:56. doi: 10.1186/1746-1596-9-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frayling TM. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. 2007;8:657–62. doi: 10.1038/nrg2178. [DOI] [PubMed] [Google Scholar]
- 78.Meyre D, Bouatia-Naji N, Tounian A, Samson C, Lecoeur C, Vatin V, et al. Variants of ENPP1 are associated with childhood and adult obesity and increase the risk of glucose intolerance and type 2 diabetes. Nat Genet. 2005;37:863–7. doi: 10.1038/ng1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scuteri A, Sanna S, Chen W, Uda M, Albai G, Strait J, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115. doi: 10.1371/journal.pgen.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hinney A, Hebebrand J. Three at one swoop! Obesity Facts. 2009;2:3–8. doi: 10.1159/000200020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–41. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rose KM, Newman B, Mayer-Davis EJ, Selby JV. Genetic and behavioral determinants of waist-hip ratio and waist circumference in women twins. Obes Res. 1998;6:383–92. doi: 10.1002/j.1550-8528.1998.tb00369.x. [DOI] [PubMed] [Google Scholar]
- 83.Chambers JC, Elliott P, Zabaneh D, Zhang W, Li Y, Froguel P, et al. Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nat Genet. 2008;40:716–8. doi: 10.1038/ng.156. [DOI] [PubMed] [Google Scholar]
- 84.Heard-Costa NL, Zillikens MC, Monda KL, Johansson A, Harris TB, Fu M, et al. NRXN3 is a novel locus for waist circumference: a genome-wide association study from the CHARGE consortium. PLoS Genet. 2009;5:e1000539. doi: 10.1371/journal.pgen.1000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cotsapas C, Speliotes EK, Hatoum IJ, Greenawalt DM, Dobrin R, Lum PY, et al. Common body mass index-associated variants confer risk of extreme obesity. Hum Mol Genet. 2009;18:3502–7. doi: 10.1093/hmg/ddp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hinney A, Nguyen TT, Scherag A, Friedel S, Brönner G, Müller TD, et al. Genome wide association (GWA) study for early onset extreme obesity supports the role of fat mass and obesity associated gene (FTO) variants. PLoS One. 2007;2:e1361. doi: 10.1371/journal.pone.0001361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scherag A, Dina C, Hinney A, Vatin V, Scherag S, Vogel CIG, et al. Two new Loci for body-weight regulation identified in a joint analysis of genome-wide association studies for early-onset extreme obesity in French and German study groups. PLoS Genet. 2010;6:e1000916. doi: 10.1371/journal.pgen.1000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cho YS, Go MJ, Kim YJ, Heo JY, Oh JH, Ban HJ, et al. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet. 2009;41:527–34. doi: 10.1038/ng.357. [DOI] [PubMed] [Google Scholar]
- 89.Heid IM, Jackson AU, Randall JC, Winkler TW, Qi L, Steinthorsdottir V, et al. Meta-analysis identifies 13 new loci associated with waist–hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010;42:949–60. doi: 10.1038/ng.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;A102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stoger R. Epigenetics and obesity. Pharmacogenomics. 2008;9:1851–60. doi: 10.2217/14622416.9.12.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Waterland RA, Jirtle RL. Early nutrition, epigenetic changes at transposons and imprinted genes and enhanced susceptibility to adult chronic diseases. Nutrition. 2004;20:63–8. doi: 10.1016/j.nut.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 93.Lindsay RS, Kobes S, Knowler WC, Hanson RL. Genome wide linkage analysis assessing parent-of-origin effects in the inheritance of birth weight. Hum Genet. 2002;110:503–9. doi: 10.1007/s00439-002-0718-2. [DOI] [PubMed] [Google Scholar]
- 94.Thompson DB, Ravussin E, Bennett PH, Bogardus C. Structure and sequence variation at the human leptin receptor gene in lean and obese Pima Indians. Hum Mol Genet. 1997;6:675–9. doi: 10.1093/hmg/6.5.675. [DOI] [PubMed] [Google Scholar]
- 95.Arya R, Demerath E, Jenkinson CP, Göring HH, Puppala S, Farook V, et al. A quantitative trait locus (QTL) on chromosome 6q influences birth weight in two independent family studies. Hum Mol Genet. 2006;15:1569–79. doi: 10.1093/hmg/ddl076. [DOI] [PubMed] [Google Scholar]
- 96.Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH, et al. Epigenetic differences arise during the life time of monozygotic twins. Proc Natl Acad Sci USA. 2005;A102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Herbert A, Gerry NB, McQueen MB, Heid IM, Pfeufer A, Illig T, et al. A common genetic varient is associated with adult and childhood obesity. Science. 2006;312:279–83. doi: 10.1126/science.1124779. [DOI] [PubMed] [Google Scholar]
- 98.Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension. 2008;51:383–92. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- 99.Igosheva N, Abramov AY, Poston L, Eckert JJ, Flemming TP, Duchen MR, et al. Maternal diet-induced obesity alters mitochondrial activity and redox status in mouse oocytes and zygotes. PLoS One. 2010;5:e10074. doi: 10.1371/journal.pone.0010074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cummins JM. The role of mitochondria in the establishment of oocyte functional competence. Eur J Obstet Gynecol Reprod Biol. 2004;115(Suppl 1):S23–9. doi: 10.1016/j.ejogrb.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 101.Dumollard R, Duchen M, Carroll J. The role of mitochondrial function in the oocyte and embryo. Curr Top Dev Biol. 2007;77:21–49. doi: 10.1016/S0070-2153(06)77002-8. [DOI] [PubMed] [Google Scholar]
- 102.Dumollard R, Carroll J, Duchen MR, Campbell K, Swann K. Mitochondrial function and redox state in mammalian embryos. Semin Cell Dev Biol. 2009;20:346–53. doi: 10.1016/j.semcdb.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 103.Jansen RP, Burton GJ. Mitochondrial dysfunction in reproduction. Mitochondrion. 2004;4:577–600. doi: 10.1016/j.mito.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 104.Van Blerkom J. Mitochondria as regulatory forces in oocytes, preimplantation embryos and stem cells. Reprod Biomed Online. 2008;16:553–69. doi: 10.1016/s1472-6483(10)60463-4. [DOI] [PubMed] [Google Scholar]
- 105.Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J, et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–72. doi: 10.1038/nature08368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McConnell JM, Petrie L. Mitochondrial DNA turnover occurs during preimplantation development and can be modulated by environmental factors. Reprod Biomed Online. 2004;9:418–24. doi: 10.1016/s1472-6483(10)61277-1. [DOI] [PubMed] [Google Scholar]
- 107.Thouas GA, Trounson AO, Jones GM. Developmental effects of sublethal mitochondrial injury in mouse oocytes. Biol Reprod. 2006;74:969–77. doi: 10.1095/biolreprod.105.048611. [DOI] [PubMed] [Google Scholar]
- 108.Wakefield SL, Lane M, Schulz SJ, Hebart ML, Thompson JG, Mitchell M. Maternal supply of omega-3 polyunsaturated fatty acids alter mechanisms involved in oocyte and early embryo development in the mouse. Am J Physiol Endocrinol Metab. 2008;294:E425–34. doi: 10.1152/ajpendo.00409.2007. [DOI] [PubMed] [Google Scholar]
- 109.Mitchell M, Schulz SL, Armstrong DT, Lane M. Metabolic and mitochondrial dysfunction in early mouse embryos following maternal dietary protein intervention. Biol Reprod. 2009;80:622–30. doi: 10.1095/biolreprod.108.072595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.William IS. Mitochondrial dysfunction in obesity and diabetes. US Endocrinol. 2010;6:20–7. [Google Scholar]
- 111.Vladimir BR, Elizabeth VM, Jing H, Robert EF, Bret HG, David EK. Deficiency of subsarcolemmal mitochondria in obesity and Type 2 Diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 112.Boudina S, Sena S, Sloan C, Tebbi A, Han YH, O’Neill BT, et al. Early mitochondrial adaptations in skeletal muscle to diet-induced obesity are strain dependent and determine oxidative stress and energy expenditure but not insulin sensitivity. Endocrinology. 2012;153:2677–88. doi: 10.1210/en.2011-2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Leanne WF, Sarah N, My C, Mitchell AL, Patricia CC, John L, et al. Mitochondrial remodelling in adipose tissue associated with obesity and treatment with rosiglitazone. J Clin Invest. 2004;114:1281–9. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Khasawneha J, Schulza MD, Walchb A, Rozmanc J, Hrabe de Angelisc M, Klingenspord M, et al. Inflammation and mitochondrial fatty acid β-oxidation link obesity to early tumor promotion. Proc Natl Acad Sci USA. 2009;106:3354–9. doi: 10.1073/pnas.0802864106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li-Jun G, Yoshiharu O, Noriyuki F, Takeshi T, Yasunori F, Miyuki K, et al. Mitochondrial genome polymorphisms associated with type 2 diabetes or obesity. Mitochondrion. 2005;5:15–33. doi: 10.1016/j.mito.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 116.Srikar C, Wesley AB, Mark JR, David AM. The Proceedings of the Biotechnology and Bioinformatics Symposium. Bringham Young University, Provo, Utah; 2006. Oct 20-21, Evolutionary selective pressure on three mitochondrial SNPs is consistent with their influence on metabolic efficiency in Pima Indians. [Google Scholar]
- 117.Martinez-Hervas S, Mansego ML, de Marco G, Martinez F, Alonso MP, Morcillo S, et al. Polymorphisms of the UCP2 gene are associated with body fat distribution and risk of abdominal obesity in Spanish population. Eur J Clin Invest. 2012;42:171–8. doi: 10.1111/j.1365-2362.2011.02570.x. [DOI] [PubMed] [Google Scholar]
- 118.Wang TN, Huang MC, Lin HL, Hsiang CH, Ko AM, Chang WT, et al. UCP2 A55V variant is associated with obesity and related phenotypes in an aboriginal community in Taiwan. Int J Obes. 2007;31:1746–52. doi: 10.1038/sj.ijo.0803648. [DOI] [PubMed] [Google Scholar]
- 119.Labruna G, Pasanisi F, Fortunato G, Nardelli C, Finelli C, Farinaro E, et al. Sequence analysis of the UCP1 gene in a severe obese population from Southern Italy. J Obes 2011. 2011:269043. doi: 10.1155/2011/269043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tie-Lin Y, Yan G, Hui S, Shu-Feng L, Yong-Jun L, Jian L, et al. Genetic association study of common mitochondrial variants on body fat mass. PLoS One. 2011;6:e21595. doi: 10.1371/journal.pone.0021595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu YJ, Liu PY, Long J, Lu Y, Elze L, Recker RR, et al. Linkage and association analyses of the UCP3 gene with obesity phenotypes in Caucasian families. Physiol Genomics. 2005;22:197–203. doi: 10.1152/physiolgenomics.00031.2005. [DOI] [PubMed] [Google Scholar]
- 122.Srivastava N, Prakash J, Lakhan R, Agarwal CG, Pant DC, Mittal B. A common polymorphism in the promoter of UCP2 is associated with obesity and hyperinsulenemia in northern Indians. Mol Cell Biochem. 2010;337:293–8. doi: 10.1007/s11010-009-0311-2. [DOI] [PubMed] [Google Scholar]
- 123.Haihong Z, Chenghui X, Horace JS, Chunlai Z, Masahiro H, Gouri R, et al. Obesity and hepatosteatosis in mice with enhanced oxidative DNA damage processing in mitochondria. Am J Pathol. 2011;178:1715–27. doi: 10.1016/j.ajpath.2010.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Laird NM, Lange C. Family-based designs in the age of large-scale gene association studies. Nat Rev Genet. 2006;7:385–94. doi: 10.1038/nrg1839. [DOI] [PubMed] [Google Scholar]
- 125.Herbert A, Gerry NP, McQueen MB, Heid IM, Pfeufer A, Illig T, et al. A common genetic variant is associated with adult and childhood obesity. Science. 2006;312:279–83. doi: 10.1126/science.1124779. [DOI] [PubMed] [Google Scholar]
- 126.Grant SFA, Brafield JP, Zhang H, Wang K, Kim CE, Annaiah K, et al. Investigation of the locus near MC4R with childhood obesity in Americans of European and African ancestry. Obesity. 2009;17:1461–5. doi: 10.1038/oby.2009.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–56. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 128.Chen Z, Torrens JI, Anand A, Spiegelman BM, Friedman JM. Krox20 stimulates adipogenesis via C/EBPbeta-dependent and-independent mechanisms. Cell Metab. 2005;1:93–106. doi: 10.1016/j.cmet.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 129.Li D, Yea S, Li S, Chen Z, Narla G, Banck M, et al. Krüppel-like factor-6 promotes preadipocyte differentiation through histone deacetylase 3-dependent repression of DLK1. J Biol Chem. 2005;280:26941–52. doi: 10.1074/jbc.M500463200. [DOI] [PubMed] [Google Scholar]
- 130.Mori T, Sakaue H, Iguchi H, Gomi H, Okada Y, Takashima Y, et al. Role of Krüppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem. 2005;1:12867–75. doi: 10.1074/jbc.M410515200. [DOI] [PubMed] [Google Scholar]
- 131.Gu HF, Abulaiti A, Ostenson CG, Humphreys K, Wahlestedt C, Brookes AJ, et al. Single nucleotide polymorphisms in the proximal promoter region of the adiponectin (APM1) gene are associated with type 2 diabetes in Swedish caucasians. Diabetes. 2004;53(Suppl 1):S31–5. doi: 10.2337/diabetes.53.2007.s31. [DOI] [PubMed] [Google Scholar]
- 132.Milagro FI, Campión J, Martínez JA. 11-Beta hydroxysteroid dehydrogenase type 2 expression in white adipose tissue is strongly correlated with adiposity. J Steroid Biochem Mol Biol. 2007;104:81–4. doi: 10.1016/j.jsbmb.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 133.Barroso I. Complex disease: pleiotropic gene effects in obesity and type 2 diabetes. Eur J Hum Genet. 2005;13:1243–4. doi: 10.1038/sj.ejhg.5201514. [DOI] [PubMed] [Google Scholar]
- 134.Deram S, Nicolau CY, Perez-Martinez P, Guazzelli I, Halpern A, Wajchenberg BL, et al. Effects of perilipin (PLIN) gene variation on metabolic syndrome risk and weight-loss in obese children and adolescents. J Clin Endocrinol Metab. 2008;93:4933–40. doi: 10.1210/jc.2008-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.John TH, Matthias K, Joanna K, Gesine F, Michael S, Edward S, et al. Nicotinamide nucleotide transhydrogenase mRNA expression is related to human obesity. Obesity. 2013;21:529–34. doi: 10.1002/oby.20095. [DOI] [PubMed] [Google Scholar]
- 136.Matthias T, Christian W P, Antonio T, Viswanath D, Eric R, Mark LH. Circulating ghrelin levels are decreased in human obesity. Diabetes. 2001;50:707–9. doi: 10.2337/diabetes.50.4.707. [DOI] [PubMed] [Google Scholar]
- 137.Carter KW, Hung J, Powell BL, Wiltshire S, Foo BT, Leow YC, et al. Association of Interleukin-1 gene polymorphisms with central obesity and metabolic syndrome in a coronary heart disease population. Hum Genet. 2008;124:199–206. doi: 10.1007/s00439-008-0540-6. [DOI] [PubMed] [Google Scholar]
- 138.Rouskas K, Paletas K, Kalogeridis A, Sarigianni M, Ioannidou-Papagiannaki E, Tsapas A, et al. Association between BBS6/MKKS gene polymorphisms, obesity and metabolic syndrome in the Greek population. Int J Obes. 2008;32:1618–25. doi: 10.1038/ijo.2008.167. [DOI] [PubMed] [Google Scholar]
- 139.Song Y, You NC, Hsu YH, Howard BV, Langer RD, Manson JE, et al. FTO polymorphisms are associated with obesity but not diabetes risk in postmenopausal women. Obesity. 2008;16:2472–80. doi: 10.1038/oby.2008.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Johansson LE, Johansson LM, Danielsson P, Norgren S, Johansson S, Marcus C, et al. Genetic variance in the adiponutrin gene family and childhood obesity. PLoS One. 2009;4:e5327. doi: 10.1371/journal.pone.0005327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Weyrich P, Albet S, Lammers R, Machicao F, Fritsche A, Stefan N, et al. Genetic variability of procolipase associates with altered insulin secretion in non-diabetic caucasians. Exp Clin Endocrinol Diabetes. 2009;117:83–7. doi: 10.1055/s-2008-1078733. [DOI] [PubMed] [Google Scholar]
- 142.Kim J. Association of CHRNA2 polymorphisms with overweight/obesity and clinical characteristics in a Korean population. Clin Chem Lab Med. 2008;46:1085–9. doi: 10.1515/CCLM.2008.230. [DOI] [PubMed] [Google Scholar]
- 143.Yajnik C. Nutritional control of fetal growth. Nutr Rev. 2006;64:S50–1. doi: 10.1301/nr.2006.may.s50-s51. discussion, S72-91. [DOI] [PubMed] [Google Scholar]
- 144.Giridharan NV, Harishankar N, Satyavani M. WNIN/Ob, A new rat model for the studies of obesity. Scand J Lab Anim Sci. 1996;3:131–8. [Google Scholar]
- 145.Giridharan NV, Lakshmi CN, Raghuramulu N. Identification of impaired glucose tolerant animals from a Wistar inbred rat colony. Lab Ani Sci. 1997;47:428–31. [PubMed] [Google Scholar]
- 146.Clement K. Genetics of human obesity. Proc Nutr Soc. 2005;64:133–42. doi: 10.1079/pns2005416. [DOI] [PubMed] [Google Scholar]
- 147.Roche HM, Phillips C, Gibney MJ. The metabolic syndrome: The crossroads of diet and genetics. Proc Nutr Soc. 2005;64:371–7. doi: 10.1079/pns2005445. [DOI] [PubMed] [Google Scholar]
- 148.Swarbrick MM, Vaisse C. Emerging trends in the search for genetic variants predisposing to human obesity. Curr Opin Clin Nutr Metab Care. 2003;6:369–75. doi: 10.1097/01.mco.0000078997.96795.03. [DOI] [PubMed] [Google Scholar]
- 149.Hebebrand J, Friedel S, Schaüble N, Geller F, Hinney A. Perspectives: molecular genetic research in human obesity. Obes Rev. 2003;4:139–46. doi: 10.1046/j.1467-789x.2003.00106.x. [DOI] [PubMed] [Google Scholar]
- 150.Mina D, Michael GR. Fetal programming of adipose tissue: effects of IUGR and maternal obesity/high fat diet. Semin Reprod Med. 2011;29:237–45. doi: 10.1055/s-0031-1275517. [DOI] [PMC free article] [PubMed] [Google Scholar]