

Abstract
An overproduction of reactive oxygen species (ROS) mediated by NADPH oxidase 2 (NOX2) has been related to airway inflammation typical of influenza infection. Virus-induced oxidative stress may also control viral replication, but the mechanisms underlying ROS production, as well as their role in activating intracellular pathways and specific steps of viral life cycle under redox control have to be fully elucidated. In this study, we demonstrate that influenza A virus infection of lung epithelial cells causes a significant ROS increase that depends mainly on NOX4, which is upregulated at both mRNA and protein levels, while the expression of NOX2, the primary source of ROS in inflammatory cells, is downregulated. Inhibition of NOX4 activity through chemical inhibitors or RNA silencing blocks the ROS increase, prevents MAPK phosphorylation, and inhibits viral ribonucleoprotein (vRNP) nuclear export and viral release. Overall these data, obtained in cell lines and primary culture, describe a so far unrecognized role for NOX4-derived ROS in activating redox-regulated intracellular pathways during influenza virus infection and highlight their relevance in controlling specific steps of viral replication in epithelial cells. Pharmacological modulation of NOX4-mediated ROS production may open the way for new therapeutic approaches to fighting influenza by targeting cell and not the virus.
Introduction
An alteration of the intracellular redox balance, which occurs during several viral infections, is associated with the progression of virus-induced diseases (Beck et al., 2000; reviewed in Nencioni et al., 2011). This redox imbalance can be determined by several factors, including an overproduction of reactive oxygen species (ROS) (Petherans et al., 1987; Elbim et al., 2001; Machida et al., 2006; Imai et al., 2008; Hu et al., 2011) and/or a decrease of reduced glutathione (GSH), the main intracellular non-enzymatic antioxidant (Cai et al., 2003; Nencioni et al., 2003). We previously reported that, during viral infections, the decrease of intracellular GSH varies in intensity, duration and mechanism of induction depending on the type of virus and the infected host cell (Garaci et al., 1992; 1997,; Palamara et al., 1995; 1996,; Ciriolo et al., 1997). Regarding influenza virus infection, we found that such GSH decrease was pivotal for viral replication (Nencioni et al., 2003) by allowing the folding and maturation of viral haemagglutinin (HA) (Sgarbanti et al., 2011). We also demonstrated that administration of GSH-C4 (a hydrophobic glutathione derivative that increases intracellular GSH) strongly impaired the replication of several influenza virus strains (Sgarbanti et al., 2011), confirming the important role of low GSH levels in the life cycle of this virus.
Since GSH exerts an efficient buffering role against ROS (Bindoli et al., 2008; Mieyal et al., 2008), a deficiency of GSH puts the cell at risk for oxidative damage from ROS and related reactive species. The main source of deliberate cellular ROS production is the family of NADPH (nicotinamide adenine dinucleotide phosphate) oxidases (NOXs), which consists of seven members – NOX1- to -5 and the two dual oxidases, Duox1 and Duox2 – expressed in most cell types (Bedard and Krause, 2007). These enzymes, which generate superoxide anion or hydrogen peroxide as primary products (Katsuyama et al., 2011), function in physiological and pathological processes as well as in the defence against infection. In phagocytic cells, NOX2 is the main isoform responsible for the respiratory burst that results in the release of superoxide in the phagocytic vacuole and promotes bacterial killing. At the same time, NOX2 production of ROS aggravates airway inflammation during influenza A virus infection: as shown in mice lacking NOX2, in the absence of ROS production, the severity of symptoms was mitigated (Snelgrove et al., 2006; Vlahos et al., 2011). NOXs are also involved in viral infections in non-phagocytic cells. For example, in airway epithelial cells, NOX2 was the specific source of ROS responsible for NF-κB activation and cytokine production during respiratory syncytial virus and Sendai virus infections (Fink et al., 2008). In hepatocytes, hepatitis C virus stimulated NOX4 expression and ROS generation, implicating this enzyme in inflammatory liver disease (Boudreau et al., 2009). In non-infected endothelial cells, NOX4 has been shown to stimulate intracellular signalling cascades such as that of mitogen-activated protein kinase (MAPK) pathways including p38 (Jaulmes et al., 2009) and ERK (Wu et al., 2010).
These cascades may also play a role in influenza virus replication. In particular, activation of p38 and ERK MAPK is required for the nuclear export of viral ribonucleoprotein (vRNP) complexes (Pleschka et al., 2001; Marjuki et al., 2006; Nencioni et al., 2009). The mechanisms through which influenza and other viruses activates MAPK have been deeply investigated. Marjuki et al. (2006) showed that activation of the ERK cascade is related to membrane accumulation of viral HA. In Rhinovirus infection, the activation of p38 MAPK depends on the accumulation of double-strand RNA (Gern et al., 2003). To date, however, the potential role of virus-induced ROS production on MAPK activation, the steps of viral life cycle under redox control, as well as the mechanisms through which influenza virus induces ROS production remain to be fully elucidated. Therefore, in the present study, we investigated, in lung epithelial cell lines and primary airway epithelial cells, the mechanisms underlying influenza virus-induced redox imbalance and its role in virus life cycle. Here we demonstrate that: (1) influenza virus induces oxidative stress through a moderate but significant increase of the production of ROS and a depletion of GSH; (2) influenza virus infection transiently upregulates NOX4 mRNA and protein expression; (3) NOX4-derived ROS activate p38 and ERK1-2 MAPK that, in turn, are involved in nuclear export of vRNP; and (4) NOX4 activity is required for viral replication.
Results
Influenza virus alters GSH redox homeostasis and increases ROS in lung epithelial cells
Since GSH plays a fundamental role in buffering ROS, we investigated whether the GSH decrease induced by influenza virus was associated with changes in intracellular ROS levels. Confluent monolayers of human mucoepidermoid pulmonary carcinoma cells (NCI-H292) were infected with 1 MOI (multiplicity of infection) of influenza virus (A/Puerto Rico/8/34 H1N1) (PR8) to allow single-cycle replication. After viral adsorption, at different time points, the content of intracellular GSH was evaluated by high-performance liquid chromatography (HPLC), while ROS production was measured in a cytofluorimetric assay using the ROS-sensitive probe dichlorodihydrofluorescein diacetate (DCF-DA). As expected (Sgarbanti et al., 2011), viral infection was associated with a decrease in GSH levels over time (Fig. 1A). GSH content in infected cells was significantly different from that of mock-infected control cells at 6 h after infection (37% lower, P < 0.01, Student's t-test). ROS production underwent a transient but significant increase in infected cells, starting at 3 h and disappearing by 6 h (P < 0.05) (Fig. 1B). Since the GSH drop caused by infection was temporally downstream of the ROS increase, we hypothesized that the GSH decrement was, at least in part, due to ROS scavenging activity. To test this possibility, mock-infected and infected cells were treated with the antioxidant Trolox, a cell-permeable water-soluble analogue of vitamin E, and ROS and GSH levels were quantified when the greatest differences between infected and control cells were observed (Fig. 1B and A respectively). Treatment with Trolox prevented the ROS increase (Fig. 1C) and blocked the GSH decrease due to infection (Fig. 1D), implying that depletion of GSH resulted from its consumption while buffering against ROS. The fact that Trolox addition also reduced ROS accumulation in control cells confirmed its ability to scavenge free radicals (Fig. 1C).
Fig 1.

Influenza A virus infection depletes GSH and increases ROS production.A. Intracellular GSH content in mock-infected (Ctr) and PR8 virus-infected (I) NCI-H292 cells over time; data are from three experiments each done in duplicate (n = 6).B. Time-course of intracellular ROS levels in PR8 virus-infected NCI-H292 cells; data are from three separate experiments each done in triplicate (n = 9). DCF+ cells, cells positive for the ROS-sensitive probe DCF-DA.C. ROS levels in mock-infected and infected NCI-H292 cells, cultured in the absence or presence of 200 μM Trolox (Tx) for 3 h after the viral challenge. Treatment with tert-butyl hydroperoxide (tb) was used as positive control. Data are from three separate experiments each done in triplicate (n = 9).D. GSH content in mock-infected and infected NCI-H292 cells, cultured without or with 200 μM Trolox for 6 h after the viral challenge; data are from three separate experiments each done in duplicate (n = 6). All values are expressed as mean and SD. *P versus control, §P versus infected, Student's t-test.
NADPH oxidases are involved in influenza virus-induced ROS production
Given the increasing evidence that NOXs are implicated in the severity of influenza and other viral infections, we investigated whether these enzymes were responsible for the ROS overproduction during influenza infection of epithelial cells. To this aim, NCI-H292 cells were infected as described above and treated with diphenyleneiodonium (DPI), a broad-spectrum inhibitor of flavoenzymes including NOXs (Wind et al., 2010), just after viral challenge. DPI treatment reduced ROS levels in control cells and, importantly, inhibited any increase in ROS content in infected cells (Fig. 2A). In parallel, DPI treatment raised GSH levels in control cells (possibly because, in cells producing less ROS, there was lower consumption of GSH) and prevented the intracellular GSH depletion observed during infection (Fig. 2B). These results support the hypothesis that the GSH depletion during infection is due to its ROS buffering function and suggest that NOXs are involved in the observed virus-induced redox imbalance.
Fig 2.
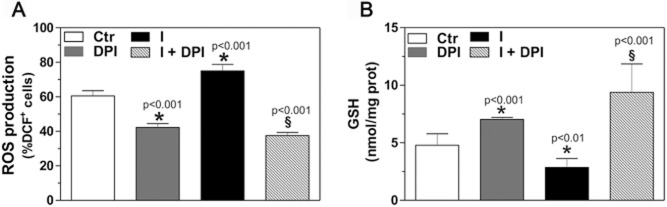
NADPH oxidases are involved in influenza virus-induced oxidative stress.A. Intracellular ROS levels in mock-infected (Ctr) and infected (I) NCI-H292 cells, cultured in the absence or presence of 10 μM DPI for 3 h after the viral challenge; data are from three separate experiments each performed in triplicate (n = 9). DCF+ cells, cells positive for the ROS-sensitive probe dichlorodihydrofluorescein diacetate.B. Intracellular GSH content in mock-infected (Ctr) and infected (I) NCI-H292 cells, cultured without or with DPI for 6 h after the viral challenge; data are from three separate experiments each performed in duplicate (n = 6). All values are expressed as mean and SD. *P versus control, §P versus infected, Student's t-test.
NCI-H292 cells express all seven NOX isoforms (Kim et al., 2011 and data not shown). To determine which isoform is involved in virus-induced ROS overproduction, we measured the expression of these enzymes during viral replication. To this aim, the cells were infected with PR8, cultured for up to 6 h, and assayed for NOX mRNA using RT-qPCR. In these experimental conditions, influenza virus only modulated the transcription of NOX2 and NOX4 mRNA (Fig. 3A), whereas the other NOX mRNAs were not significantly affected by infection (data not shown). Interestingly, the virus had differential effects on these two isoforms, reducing NOX2 mRNA while increasing NOX4 mRNA transiently with a peak at 3 h. Similar results were obtained at the protein level: NOX2 protein decreased (Fig. 3B) while that of NOX4 increased (Fig. 3C). The trend of NOX4 protein increase mirrored that of the ROS elevation (Fig. 1B), suggesting that NOX4 is the isoform that plays the leading role in virus-induced redox imbalance.
Fig 3.

NOX4 expression is increased during influenza virus infection.A. Real-time qPCR assay of NOX isoform levels in PR8 virus-infected NCI-H292 cells, normalized to levels in mock-infected cells. NOX2 and NOX4 levels changed over time. Values are mean and SD of three independent experiments (**P < 0.01; ***P < 0.001, Student's t-test).B. Western blot of uninfected (Ctr) and PR8 virus-infected NCI-H292 cells up to 6 h, using antibodies against NOX2 (left panels) and NOX4 (right panels). Actin was used as loading control for densitometric analysis of three independent experiments (bottom).
NOX4-derived ROS activate p38 and ERK1-2 MAPK to regulate the influenza virus life cycle
Given that NOX4-derived ROS are known to activate p38 and ERK1-2 MAPK in non-phagocytic cells, we investigated whether this was true even in the context of influenza virus infection by analysing the phosphorylation state of these kinases. As expected, influenza virus infection activated the kinases, seen as an increase in the phosphorylated forms (Fig. 4A and B). Treatment with DPI strongly inhibited the phosphorylation of p38 MAPK (Fig. 4A, upper panel) and of ERK1 (Fig. 4A, bottom panel). Since NOX4 is the only NOX isoform whose expression is upregulated in our experimental model, these data suggest that also during influenza virus infection NOX4-derived ROS activate MAPK. Because these kinase cascades are needed for vRNP trafficking, we evaluated the effect of DPI on the intracellular localization of influenza A nucleoprotein (NP) over time (Fig. 4C). Without DPI (top row of images), viral nucleoprotein progressively migrated from the nucleus to the cytoplasm; instead, the presence of DPI (bottom row) impaired NP export, leaving it confined within nuclei even at 6 h. Altogether, these data suggest that NOX4-related ROS production is pivotal for p38 and ERK MAPK activation, which, in turn, stimulate nuclear export of NP.
Fig 4.

NOX-produced ROS activate p38 MAPK and ERK1-2, permitting nuclear export of viral ribonucleoprotein.A. Mock-infected (Ctr) and PR8 virus-infected (I) NCI-H292 cells, treated or not with DPI, were collected at 6 h and subjected to Western blotting with antibodies against p38 MAPK and its phosphorylated form (P-p38) (top), and ERK1-2 and their phosphorylated forms (bottom).B. Densitometric analysis of p38 (top) and ERK1 (bottom) protein levels. Values are ratios of the phosphorylated isoform to the total, normalized to the ratio obtained in control cells. Data are representative of two independent experiments (*P < 0.05; **P < 0.001, Student's t-test).C. Immunofluorescence photomicrographs of NCI-H292 cells infected with PR8 virus (I) and then cultured in the absence (top) or presence (bottom) of DPI for up to 6 h. Cells were labelled with monoclonal anti-influenza A nucleoprotein (NP) antibody followed by an Alexa Fluor 568-conjugated secondary antibody (red); nuclei were stained with DAPI (blue). Images are representative of two experiments.
We next examined if NOX4 is needed for viral replication. First, we assayed the release of viral particles from cells infected with PR8 virus strain over 8 h, in the absence or presence of DPI at different concentrations (5, 10, 20 μM) through the haemagglutination assay. We found that viral titre was reduced in a dose-dependent manner in DPI-treated cells by about 90% at 10 and 20 μM (P < 0.01, Student's t-test) (Fig. 5A). Because the degree of inhibition at 10 and 20 μM was superimposable, we decided to utilize the concentration 10 μM in the next experiments. These findings were confirmed also in plaque assay, where the number of plaque forming units (PFU) per millilitre of culture medium from DPI-treated cells was reduced by about 1.5 log (P < 0.001, Student's t-test) (Fig. 5B), and in an assay of the 50% tissue culture infectious dose (TCID50) which showed a 90% reduction due to DPI (data not shown). Interestingly, the ability of DPI in inhibiting viral titre was also observed in NCI-H292 cells infected with A/parrot/Ulster/73 and A/NWS/33 influenza viruses (Fig. S1A>). Furthermore, A549 human lung carcinoma cells were infected with human and avian viruses (10 MOI) and treated or not with DPI for 8 h; the haemagglutination assay demonstrated that DPI inhibited replication of the viruses, by about 60% (Fig. S1B>). Since DPI is a general inhibitor of flavoenzymes, we also used VAS2870, a broad-spectrum NOX inhibitor (Wingler et al., 2012) that does not possess intrinsic antioxidant activity and does not inhibit other flavoproteins (Altenhofer et al., 2012). The haemagglutination assay showed a dose-dependent reduction in viral titre, reaching 85% at 30 μM (P < 0.0001, Student's t-test) (Fig. 5C). Similar results were obtained with the plaque assay (1.3 log inhibition versus untreated cells, P < 0.01; Fig. 5D). To further investigate the role of NOX4 in controlling viral replication, we verified the effect of DPI and VAS2870 on the replication of H3N2 and pandemic H1N1 2009, two currently circulating influenza A viruses. The TCID50 assay showed that both the inhibitors strongly reduced the replication of the viruses up to 48 h after the viral challenge (P < 0.001; P < 0.01) (Fig. 5E and F). These results demonstrated that the effect of DPI and VAS2870 was not only due to a delay on virus replication and that was independent of the strain used. To confirm the role of NOX4 in regulating the influenza virus life cycle, we used short hairpin RNA (shRNA) to specifically knockdown NOX4 gene expression. NCI-H292 cells were transfected with a pool of NOX4 shRNA expression plasmids and 24 h later were collected for analysis: Western blotting revealed that the level of NOX4 protein was reduced by about 60% compared to that measured in cells transfected with non-targeting shRNA (Fig. 6A). In this condition, transfected cells were infected with PR8 for 24 h. A significant reduction in viral particle release was observed in both haemagglutination (Fig. 6B) and plaque assays (Fig. 6C). Importantly, ROS production in cells silenced for NOX4 and infected with influenza virus was significantly lower compared to infected cells transfected with non-targeting shRNA (Fig. 6D). Similar results were obtained when the experiment was repeated in human embryonic kidney (HEK-293) cells (Fig. S2). Altogether, these data indicate that NOX4 plays an essential role in virus-induced ROS production and in supporting influenza virus replication in different cell lines.
Fig 5.

NOX4 supports influenza virus replication in NCI-H292 cells.A. Cells were infected with PR8 virus and cultured in the absence or presence of DPI for 8 h before the conditioned medium was assayed. Haemagglutination assay (**P < 0.01).B. Plaque assay for PR8-infected cells treated or not with DPI 10 μM. PFU, plaque-forming units (**P < 0.001).C and D. PR8-infected cells were cultured in the absence or presence of VAS2870 for 8 h. (C) Haemagglutination assay (***P < 0.0001). (D) Plaque assay (*P < 0.01).E. MDCK cells were infected with H3N2 virus and cultured in the absence or presence of DPI or VAS2870 for 24 and 48 h. TCID50 assay (**P < 0.001; *P < 0.01).F. MDCK cells were infected with pandemic 2009 H1N1 virus and cultured in the absence or presence of DPI or VAS2870 for 24 and 48 h. TCID50 assay (*P < 0.01).
Fig 6.

A. NCI-H292 cells were transfected with plasmids encoding shRNAs specific to NOX4 or with a control plasmid encoding a non-targeting shRNA. The Western blot was sequentially probed with anti-NOX4 antibody and anti-tubulin antibody. Densitometry values are ratios of intensities of the NOX4 band to the tubulin band, normalized to the ratio obtained from non-targeting cells. The blot is shown from one representative experiment out of the two performed.B and C. Viral load in medium of NCI-H292 cells transfected with NOX4-targeting or non-targeting shRNA expression plasmids before infection with PR8. (B) Haemagglutination assay (**P < 0.001). (C) Plaque assay (**P < 0.01).D. Intracellular ROS levels in NCI-H292 cells transfected with NOX4-targeting or non-targeting shRNA expression plasmids before infection with PR8. Data are from three separate experiments (n = 3). Data are expressed as ratio of the infected cells to the non-infected ones (**P < 0.01).
NOX4 supports influenza virus replication in primary lung epithelial cells
To determine if the results obtained in cell lines could also be observed in primary cultures, we repeated the experiments in primary airway epithelial cells from BALB/c mice using mouse-adapted PR8 virus. Viral infection increased the level of NOX4 protein while it decreased NOX2 (Fig. 7A), as already observed in NCI-H292 cells. Viral infection also increased the phosphorylation state of p38 and of ERK1-2 MAPK, but the addition of DPI inhibited the activation of the two kinases (Fig. 7B). In the presence of DPI, the localization of NP was mainly nuclear (Fig. 7C). Western blotting analysis demonstrated that DPI treatment strongly inhibited the expression of HA, NP and matrix 1 protein to about 20–40% of that of infected, untreated cells (Fig. 7D). Finally, the viral titre of the culture medium of infected cells treated with DPI was significantly lower than that from untreated cells (Fig. 7E). Altogether, these results demonstrate that NOX4 activity is also important for influenza virus propagation in primary airway epithelial cells.
Fig 7.

NOX4 is required for influenza virus replication in primary airway epithelial cells from BALB/c mice.A. Western blot of uninfected (Ctr) and PR8-infected (I) cells, using antibodies against NOX4 (upper panel) and NOX2 (bottom panels). Tubulin was used as loading control. Data are representative of three independent experiments.B. Mock-infected (Ctr) and PR8-infected (I) cells, treated or not with DPI, were collected at 8 h and subjected to Western blotting with antibodies against p38 MAPK and its phosphorylated form (P-p38) (top), and ERK1-2 and their phosphorylated forms (bottom). Data are representative of two independent experiments.C. Immunofluorescence photomicrographs of cells infected with PR8 and then cultured in the absence (left) or presence (right) of DPI for up to 8 h. Cells were labelled with monoclonal anti-influenza A nucleoprotein antibody followed by an Alexa Fluor 568-conjugated secondary antibody (red).D. Mock-infected (Ctr) and PR8-infected (I) cells, treated or not with DPI, were collected at 8 h and subjected to Western blotting with goat polyclonal anti-influenza A virus antibody. Actin was used as loading control.E. Viral load in medium above PR8-infected cells, cultured in the absence or presence of DPI for 24 h. Haemagglutination assay (***P < 0.001).
Discussion
This study has shown that influenza A virus infection of lung epithelial cells transiently increases intracellular ROS as an essential step in the progression of the virus life cycle. This process results in the activation of the p38 and ERK1-2 MAPK pathways that, in turn, promote the nuclear export of vRNP, a key event for viral assembly and release. In human pulmonary carcinoma cell lines and in murine primary airway epithelial cells, the NADPH oxidase NOX4 was the prime actor in the virus-induced oxidative stress and, as a consequence, in the promotion of viral replication: NOX4 expression transiently increased during infection, while inhibition or knockdown of NOX4 reduced infected cells' ability to produce viral particles.
The ROS increase observed upon influenza A virus infection in our experimental system was attributed to an upregulation of NOX4 at the mRNA and protein levels, accompanied by the downregulation of NOX2 (and no change in other NOXs). The activity of both NOX4 and NOX2 is regulated, at least in part, at the transcriptional level. Therefore, it is possible to speculate that influenza virus exerts a positive and a negative regulatory effect on NOX4 and NOX2 promoters, respectively, through the binding of some viral proteins to DNA, or through the activation of specific intracellular signalling pathways and their downstream transcription factors (Boudreau et al., 2009; Katsuyama et al., 2011). A number of DNA-binding proteins were shown to interact with the NOX2 promoter region, among which the transcriptional repressors CDP and HoxA10 (Skalnik et al., 1991; Lindsey et al., 2005). Therefore, influenza virus could increase DNA-binding activity of these repressors. As concern NOX4 several factors have been identified that regulate NOX4 transcription among which NF-κB, the JAK/STAT pathway, the TGF-β1, IRE-1 (Katsuyama et al., 2011). Given that NOX4 is mainly localized in nuclei and endoplasmic reticulum (ER), two cellular compartments under redox control and exploited by influenza virus for two important steps of its own replication, we hypothesized that influenza virus increases the transcriptional level of NOX4 to favouring its replication cycle.
On the other hand, since NOX2 regulates the antiviral response of host cells in other viral infections (Soucy-Faulkner et al., 2010), it is possible that in lung epithelial cells influenza virus downregulates the NOX2 isoform to evade the host innate responses, in order to complete its life cycle. Indeed, it is known that influenza virus uses another mechanism to contrast the innate immune response, namely the blocking of type I IFN-mediated antiviral responses by the viral non-structural (NS1) protein (Garcia-Sastre et al., 1998; Geiss et al., 2002; Imai et al., 2010).
Recent research has shown that the ROS overproduction that follows infection by HCV in liver (Boudreau et al., 2009) and encephalomyocarditis virus in brain (Ano et al., 2010) is due to the activation of NOX4 and NOX2 respectively. The latter enzyme is involved in NF-κB activation during infection with respiratory syncytial and Sendai viruses, leading to an excessive inflammatory response (Fink et al., 2008). Moreover, NOX2 activity is responsible for airway inflammation following infection with low and high pathogenicity influenza A virus strains (Snelgrove et al., 2006; Vlahos et al., 2011).
An excessive ROS production due to an overactive innate immune response was responsible for much of the acute lung injury caused by highly pathogenic (H5N1) influenza virus (Imai et al., 2008). Accordingly ROS generation may act as a stress signal for the formation of the inflammasome complex, which is crucial for the production of pro-inflammatory cytokines (Rubartelli et al., 2011; Kaushik et al., 2012). A mild ROS flux which is in line with the low ROS increase observed in our results, is able to induce signalling cascades including the MAPK cascade (Filomeni et al., 2003; Torres and Forman, 2003; Vigilanza et al., 2008). These pathways are also important in stimulating the overproduction of cytokines and chemokines during acute influenza (Lee et al., 2005; Borgeling et al., 2013). Interestingly, activation of MAPK cascades is also necessary for progression of the virus life cycle, as shown here and earlier (Pleschka et al., 2001; Marjuki et al., 2006; Nencioni et al., 2009; Marchant et al., 2010).
Our results highlight a novel signalling pathway induced by influenza virus infection that brings about MAPK activation involving NOX4 as a central regulator. Interestingly, p38 and ERK1-2 MAPK are involved in the regulation of influenza virus replication by controlling nucleocytoplasmic traffic of vRNP through different mechanisms (Pleschka et al., 2001; Marjuki et al., 2006; Nencioni et al., 2009). In particular, we previously demonstrated the involvement of p38 MAPK in nucleoprotein phosphorylation, a key event for the export of vRNP complexes from the nucleus to the cytoplasm (Nencioni et al., 2009). Indeed, inhibition of p38 MAPK impaired nucleoprotein phosphorylation, vRNP nucleocytoplasmic translocation and, consequently, viral replication. As concerns the ERK1-2 pathway, Pleschka et al. (2001) showed that ERK inhibition caused the nuclear retention of vRNP complexes, by impairing the function of the viral nuclear export protein (NEP/NS2), and reduced viral production. In line with these findings, DPI treatment in this study blocked the nuclear export of NP, confirming the role of p38 MAPK and ERK1-2 in vRNP trafficking. Importantly, the same results were also observed in primary lung cells from mice, suggesting that our results may also be valid in vivo.
The use of DPI in our experimental model impaired viral replication more than that achieved with specific MAPK inhibitors as reported by others (Pleschka et al., 2001; Nencioni et al., 2009), probably due to inhibition of redox-sensitive cellular factors controlling various steps of virus life cycle, including nucleocytoplasmic NP export and viral protein synthesis. Also, we cannot rule out that DPI could inhibit other intracellular signalling molecules involved in vRNP traffic, such as protein kinase C (Bui et al., 2000; Palamara et al., 2005) and redox-sensitive folding factors responsible for viral glycoprotein maturation (Sgarbanti et al., 2011). The latter is largely mediated by the redox-sensitive host-cell protein disulfide isomerase (PDI), whose redox state determines its ability to form disulfide bonds (oxidation) or reduce them. Oxidation of this isomerase is one of the rate-limiting steps in oxidative protein folding (Chakravarthi and Bulleid, 2004). Sgarbanti et al. (2011) demonstrated that treatment of influenza virus infected cells with a GSH derivative (GSH-C4) increased the reduced form of the isomerase and inhibited the disulfide bond formation needed for HA maturation, by restoring intracellular GSH content. Here we showed a strong reduction of HA expression after DPI treatment. Thus, since NOX4 localizes in endoplasmic reticulum of various cell types (Martyn et al., 2006) we can speculate that in our model, NOX4-derived ROS can favour the oxidation of PDI and, consequently, the maturation process of HA. Further studies are in progress to deepen this concern.
Overall, this study shows that redox-sensitive intracellular pathways play an important role in viral replication and suggests that modulation of NOX4 activity could represent a new pharmacological approach for controlling the replication of different influenza viruses. The global spread of the 2009 pandemic H1N1 influenza virus and the emergence of viral strains resistant to antiviral agents underlined the limits of currently used strategies for controlling influenza (reviewed in De Clercq, 2012). To date, vaccination represents the best anti-influenza strategy (both for prophylaxis and control), but vaccines cannot be produced quickly enough to be ready at the start of a pandemic. Therefore, the discovery of molecules affecting host-cell factors essential for viral replication may help design therapies with greater effectiveness towards different virus types and lower probability that the infectious agents develop resistance. Interestingly, it has recently been demonstrated that modulation of redox state with GSH or its derivatives altered the Th1/Th2 balance in favour of a Th1-type immune response, which is important for the antiviral response (Fraternale et al., 2011). Therefore, restoration of the redox state in infected cells may be an effective way to control viral infection, by inhibiting viral replication, moderating the inflammatory response, and improving the host cells' immune responses.
Experimental procedures
Cell lines and primary cultures
NCI-H292 human mucoepidermoid pulmonary carcinoma cells and MDCK Madine Darby canine kidney cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 0.3 mg ml−1 glutamine, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. A549 human carcinoma cells and HEK-293 human embryonic kidney cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 0.3 mg ml−1 glutamine, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin.
Harlan BALB/c mice (6- to 8-week-old females) were housed and studied under Institutional Animal Care and Use Committee–approved protocols. Primary alveolar epithelial cells (AEC) were isolated as described previously (Corti et al., 1996) with some modifications. Briefly, mice were anesthetized by an overdose of isofluorane, heparinized and exsanguinated by cutting the inferior vena cava. Lungs were perfused with sterile phosphate-buffered saline (PBS) via the right ventricle until the effluent was visually free of blood. A small incision was made into the exposed trachea to insert a shortened cannula and 1.5 ml sterile dispase solution (BD Biosciences) was administered into the lung. After 2 minutes, the lungs were excised and placed in culture tubes each containing 2 ml dispase solution and gently agitated at room temperature for 45 minutes. Lungs were then minced and suspended in 2 ml dispase solution containing 2 μg ml−1 collagenase and 0.01% DNase. Digested lungs were resuspended in DMEM containing 10% FBS, and sequentially filtered through 70-, 40-, 20-μm nylon mesh filters (BD Falcon). The resulting cell suspension was centrifuged. After resuspension in DMEM, cells underwent negative selection by using biotinylated anti-CD16/32, anti-CD45 and anti-CD31 antibodies (Miltenyi Biotec) followed by binding to streptavidin-coated magnetic particles (Miltenyi Biotec), according to the manufacturer's instructions. The non-bound cell fraction consisting of AECs had > 90% purity, as assessed by immunofluorescence staining for cytokeratin. The cells were seeded in 24-well culture plates at a density of 3 × 105 cells per well in DMEM supplemented with 10% FBS and antibiotics, and subjected to virus infection on day 3 when they were about 90% confluent.
Influenza virus production, cell infection and viral titre assays
Influenza virus A/Puerto Rico/8/34 H1N1 (PR8), Influenza A/parrot/Ulster/73 (H7N1) (Ulster 73), a subtype of low pathogenicity obtained from the Istituto Zooprofilattico delle Tre Venezie, Padua, Italy, and A/NWS/33 (H1N1) (NWS 33) (ATCC VR-219), a neurotropic strain of human influenza virus derived from the WS strain by intracerebral inoculation of mice, were grown in the allantoic cavities of 10-day-old embryonated chicken eggs. Influenza pandemic 2009/H1N1 isolated from clinical samples at ‘Sapienza’ University of Rome was kindly provided by Prof. Antonelli, while Influenza A H3N2/Florence/07 isolated from clinical samples at University of Florence was kindly provided by Prof. Azzi and Dr. Giannecchini.
NCI-H292 and A549 cells were plated, grown for 24 h, and then challenged with influenza virus diluted in serum-free medium at different multiplicity of infection (MOI). Mock infection was performed with the same dilution of allantoic fluid from uninfected eggs. Cells were incubated in presence of the virus for 1 h at 37°C. After the viral challenge, mock-infected and virus-infected cells were washed with PBS and then cultured with fresh medium containing 2% FBS and experimental reagents until use. Primary AECs were challenged with mouse-adapted PR8 diluted in serum-free medium at 0.05 MOI for 2 h, after which the inoculum was removed and the cells were washed with PBS and then cultured with fresh medium containing 2% FBS and experimental reagents.
Viral titre of conditioned culture medium was assayed using the haemagglutination assay (Mahy, 1991), the plaque assay and the 50% tissue culture infective dose (TCID50) assay. The plaque assay was done using confluent monolayers of MDCK cells in 24-well plates according to the standard procedure (Gaush and Smith, 1968). For the TCID50 assay, confluent monolayers of MDCK cells in 96-well plates were inoculated with 10-fold dilutions of the samples (8 wells per dilution), and incubated for 3 days; wells showing positive cytopathic effects were counted, and the TCID50 titre was interpolated using the Reed-Muench method (Reed and Muench, 1938).
Cell treatments
For treatment with the antioxidant 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox; Sigma), the compound was dissolved in ethanol and then applied to cells at a final concentration of 200 μM in RPMI containing 2% FBS. Trolox is a vitamin E analogue acting as a direct radical scavenger (Iuliano et al., 1999). The enzyme inhibitor diphenyleneiodonium chloride (DPI; Sigma) was dissolved in dimethylsulfoxide (DMSO) and applied to the cells at a final concentration of 10 μM. No toxic effects were observed. The enzyme inhibitor VAS2870 (Calbiochem) was dissolved in DMSO and used at a final concentration of 10, 20 or 30 μM.
The highest DMSO concentration present in the culture medium was 0.05%. Control cells were treated with an equivalent concentration of vehicle.
Glutathione and ROS assays
Intracellular GSH was assayed upon formation of S-carboxymethyl derivatives of free thiol with iodoacetic acid, followed by the conversion of free amino groups to 2,4-dinitrophenyl derivatives by reaction with 1-fluoro-2,4-dinitrobenzene as previously described (Baldelli et al., 2008). Data are expressed as nmol GSH/mg protein. Protein concentration was assayed by the Lowry method (Lowry et al., 1951).
ROS were detected by cytofluorimetric analysis as previously reported (Aquilano et al., 2006). Briefly, cells were incubated for 1 h at 37°C with 50 μM dichlorodihydrofluorescein diacetate (DCF-DA) in RPMI. Incubation with 0.1 mM tert-butyl hydroperoxide for 30 min was used as positive control. Then, cells were scraped, washed and resuspended in PBS. The fluorescence intensity of 10 000 cells from each sample was measured using a FACScalibur instrument (Becton Dickinson, San Jose, USA). Data were analyzed using WinMDI 2.8 software.
shRNA expression plasmid transfection
The following shRNA plasmids specific for NOX4 were obtained from Sigma-Aldrich: NM_016931.2-412S1C1; NM_016931.2-1015S1C1; NM_016931.2-619S1C1; NM_016931.2-559 S1C1; and NM_016931.2-1457S1C1. NCI-H292 and HEK-293 cells were transfected using the jetPEI transfection reagent (Polyplus-Transfection, Illkirch, France) according to the manufacturer's protocol. Briefly, a transfection mixture containing jetPEI, NaCl and plasmid DNA was incubated at room temperature for 20 minutes and then added drop-wise to the culture medium of cells at 60% confluence (24 h after plating). NCI-H292 cells were exposed to the transfection mixture for 24 h, as this time was sufficient to significantly decrease NOX4 expression, while HEK-293 cells were exposed for about 72 h. Then, cells were washed with PBS and infected with PR8 at 0.1 MOI or mock-infected at 37°C for 1 h. After the viral challenge, cells were washed with PBS and cultured with fresh medium containing 2% FBS (up to 24 h).
Western blotting
Cells were detached, washed with cold PBS and centrifuged at 700 g for 10 min. The pellet was lysed in cold lysis buffer (10 mM Tris pH 7.4, 150 mM NaCl and 0.25% NP-40) containing protease and phosphatase inhibitors, for 30 min on ice. Lysates were centrifuged at 10 000 g for 30 min at 4°C to remove debris. Total protein in the supernatant was quantified using the Bradford method (Bio-Rad). Lysates were diluted in SDS sample buffer containing 10% β-mercaptoethanol, separated by SDS-PAGE, and blotted onto nitrocellulose membranes. The membranes were blocked with 10% non-fat dry milk in Tris-buffered saline containing 0.01% Tween-100 for 1 h at room temperature (RT). Primary antibodies, used at final concentration of 1 μg ml−1, included mouse monoclonal anti-gp91-phox IgG1 (anti-NOX2; Santa Cruz Biotechnology), rabbit polyclonal anti-NOX4 IgG (Santa Cruz Biotechnology), mouse monoclonal anti-actin (Sigma Aldrich), mouse monoclonal anti-tubulin (Sigma Aldrich), rabbit polyclonal anti-p38 IgG (Santa Cruz Biotechnology), rabbit polyclonal anti-phospho-p38 IgG (Santa Cruz Biotechnology), rabbit polyclonal anti-phospho-p44/42 MAPK (ERK1-2) IgG (Cell Signaling Technology), rabbit polyclonal anti-p44/42 MAPK (ERK1-2) IgG (Cell Signaling Technology), and goat polyclonal anti-influenza A virus IgG (Chemicon). Bound antibodies were revealed using horseradish peroxidase-conjugated secondary antibodies (Jackson) followed by enhanced chemiluminescence (GE Healthcare Life Sciences). Densitometry was done using Quantity One 1-D Analysis software (Bio-Rad).
Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature and washed with PBS. Cells were permeabilized with 0.1% Triton X-100 in PBS at room temperature for 5 min. After blocking with 3% non-fat dry milk for 30 min, cells were incubated with mouse monoclonal anti-influenza A nucleoprotein (AbD Serotec); bound antibodies were revealed with goat anti-mouse IgG conjugated with Alexa Fluor 568 (Molecular Probes). After washing, nuclei were stained with 1 μg ml−1 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes) in PBS for 15 min at room temperature. Fluorescent images were acquired on an Olympus IX70 microscope equipped with Nanomover and softWoRx DeltaVision image acquisition software (Applied Precision, WA, USA) and a U-PLAN-APO 60× objective. Images were captured under constant exposure time, gain and offset. To improve the contrast and resolution of digital images captured in the microscope, they were elaborated with deconvolution software.
Statistical analyses
Differences between samples groups were tested for significance using Student's t-test or two-way anova. A value of P < 0.05 was considered to indicate significance.
Acknowledgments
This work was partially supported by the Italian Ministry of Instruction, Universities and Research (Projects PON, FIRB Internazionale and PRIN 2010-2011), the Institute Pasteur Cenci-Bolognetti Foundation (grant 2012), and Ateneo grant 2012. The authors thank Philippe J. Sansonetti for helpful discussion; Prof. Antonelli (Rome), Prof. Azzi and Dr. Giannecchini (Florence) for kindly provided viruses isolated from clinical samples; Dr. Paolo Coluccio for technical assistance with the ex vivo experiments; Dr. Palma Mattioli (Centro di Microscopie Avanzate, Department of Biology, Tor Vergata University of Rome) for the acquisition and analysis of immunofluorescent microscope images, and Valerie Matarese for scientific editing of the manuscript.
Supporting Information
Fig. S1. NOX4 supports the replication of different influenza virus strains in both NCI-H292 and A549 cells. A. Cells were infected with PR8, NWS or ULSTER virus, and cultured in the absence or presence of DPI for 8 h before the conditioned medium was assayed. Haemagglutination assay (***P < 0.0001; *P < 0.01).
Fig. S2. NOX4 supports influenza virus replication in HEK-293 cells.
A. HEK-293 cells were transfected with plasmids encoding shRNAs specific to NOX4 or with a control plasmid encoding a non-targeting shRNA. The Western blot was sequentially probed with anti-NOX4 antibody and anti-tubulin antibody. Data are representative of two independent experiments.
B and C. Viral load in medium of HEK-293 cells transfected with non-targeting or NOX4-specific shRNA expression plasmids before infection with PR8. (B) Haemagglutination assay (***P < 0.001). (C) Plaque assay (**P < 0.01).
References
- Altenhofer S, Kleikers PW, Radermacher KA, Scheurer P, Rob Hermans JJ, Schiffers P, et al. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci. 2012;69:2327–2343. doi: 10.1007/s00018-012-1010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ano Y, Sakudo A, Kimata T. Uraki R. Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neurosci Lett. 2010;469:39–43. doi: 10.1016/j.neulet.2009.11.040. [DOI] [PubMed] [Google Scholar]
- Aquilano K, Vigilanza P, Rotilio G. Ciriolo MR. Mitochondrial damage due to SOD1 deficiency in SH-SY5Y neuroblastoma cells: a rationale for the redundancy of SOD1. FASEB J. 2006;20:1683–1685. doi: 10.1096/fj.05-5225fje. [DOI] [PubMed] [Google Scholar]
- Baldelli S, Aquilano K, Rotilio G. Ciriolo MR. Glutathione and copper, zinc superoxide dismutase are modulated by overexpression of neuronal nitric oxide synthase. Int J Biochem Cell Biol. 2008;40:2660–2670. doi: 10.1016/j.biocel.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Beck MA, Handy J. Levander OA. The role of oxidative stress in viral infections. Ann N Y Acad Sci. 2000;917:906–912. doi: 10.1111/j.1749-6632.2000.tb05456.x. [DOI] [PubMed] [Google Scholar]
- Bedard K. Krause KZ. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bindoli A, Fukuto JM. Forman HJ. Thiol chemistry in peroxidase catalysis and redox signaling. Antiox Redox Signal. 2008;10:1549–1564. doi: 10.1089/ars.2008.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgeling Y, Schmolke M, Viemann D, Nordhoff C, Roth J. Ludwig S. Inhibition of p38 Mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal H5N1 infection. J Biol Chem. 2013;289:13–27. doi: 10.1074/jbc.M113.469239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau HE, Emerson SU, Korzeniowska A, Jendrysik MA. Leto TL. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor β-dependent manner: a new contributor to HCV-induced oxidative stress. J Virol. 2009;83:12934–12946. doi: 10.1128/JVI.01059-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui M, Willis EG, Helenius A. Whittaker GR. Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J Virol. 2000;74:1781–1786. doi: 10.1128/jvi.74.4.1781-1786.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Chen Y, Seth S, Furukawa S, Compans RW. Jones DP. Inhibition of influenza infection by glutathione. Free Radic Biol Med. 2003;34:928–936. doi: 10.1016/s0891-5849(03)00023-6. [DOI] [PubMed] [Google Scholar]
- Chakravarthi S. Bulleid NJ. Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J Biol Chem. 2004;279:39872–39879. doi: 10.1074/jbc.M406912200. [DOI] [PubMed] [Google Scholar]
- Ciriolo MR, Palamara AT, Incerpi S, Lafavia E, Buè MC, De Vito P, et al. Loss of GSH, oxidative stress, and decrease of intracellular pH as sequential steps in viral infection. J Biol Chem. 1997;272:2700–2708. doi: 10.1074/jbc.272.5.2700. [DOI] [PubMed] [Google Scholar]
- Corti M, Brody AR. Harrison JH. Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol. 1996;14:309–315. doi: 10.1165/ajrcmb.14.4.8600933. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Human viral diseases: what is next for antiviral drug discovery? Curr Opin Virol. 2012;2:572–579. doi: 10.1016/j.coviro.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Elbim C, Pillet S, Prevost MH, Preira A, Girard PM, Rogine N, et al. The role of phagocytes in HIV-related oxidative stress. J Clin Virol. 2001;20:99–109. doi: 10.1016/s1386-6532(00)00133-5. [DOI] [PubMed] [Google Scholar]
- Filomeni G, Aquilano K, Rotilio G. Ciriolo MR. Reactive oxygen species-dependent c-Jun NH2-terminal kinase/c-Jun signaling cascade mediates neuroblastoma cell death induced by diallyl disulfide. Cancer Res. 2003;63:5940–5949. [PubMed] [Google Scholar]
- Fink K, Duval A, Martel A, Soucy-Faulkner A. Grandvaux N. Dual role of NOX2 in respiratory syncitial virus-induced activation of NF-κB in airway epithelial cells. J Immunol. 2008;180:6911–6922. doi: 10.4049/jimmunol.180.10.6911. [DOI] [PubMed] [Google Scholar]
- Fraternale A, Paoletti MF, Dominici S, Buondelmonte C, Caputo A, Castaldello A, et al. Modulation of Th1/Th2 immune responses to HIV-1 Tat by new pro-GSH molecules. Vaccine. 2011;29:6823–6829. doi: 10.1016/j.vaccine.2011.07.101. [DOI] [PubMed] [Google Scholar]
- Garaci E, Palamara AT, Di Francesco P, Favalli C, Ciriolo MR. Rotilio G. Glutathione inhibits replication and expression of viral proteins in cultured cells infected with Sendai virus. Biochem Biophys Res Commun. 1992;188:1090–1096. doi: 10.1016/0006-291x(92)91343-o. [DOI] [PubMed] [Google Scholar]
- Garaci E, Palamara AT, Ciriolo MR, D'Agostini C, Abdel-Latif MS, Aquaro S, et al. Intracellular GSH content and HIV replication in human macrophages. J Leukoc Biol. 1997;62:54–59. doi: 10.1002/jlb.62.1.54. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- Gaush CR. Smith TF. Replication and plaque assay of influenza virus in an established line of canine kidney cells. Appl Microbiol. 1968;16:588–594. doi: 10.1128/am.16.4.588-594.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss GK, Salvatore M, Tumpey TM, Carter VS, Wang X, Basler CF, et al. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc Natl Acad Sci USA. 2002;99:10736–10741. doi: 10.1073/pnas.112338099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gern JE, French DA, Grindle KA, Brockman-Schneider RA, Konno S-I. Busse WW. Double-stranded RNA induces the synthesis of specific chemokines by bronchial epithelial cells. Am J Respir Cell Mol Biol. 2003;28:731–737. doi: 10.1165/rcmb.2002-0055OC. [DOI] [PubMed] [Google Scholar]
- Hu S, Sheng WS, Schachtele SJ. Lokensgard JR. Reactive oxygen species drive herpes simplex virus (HSV)-1-induced proinflammatory cytokine production by murine microglia. J Neuroinflamm. 2011;8:123. doi: 10.1186/1742-2094-8-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H, Shinya K, Takano R, Kiso M, Muramoto Y, Sakabe S, et al. The HA and NS1 genes of human H5N1 influenza A virus contribute to high virulence in ferrets. PLoS Pathog. 2010;6:e1001106. doi: 10.1371/journal.ppat.1001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iuliano L, Pedersen JZ, Camastra C, Bello V, Ceccarelli S. Violi F. Protection of low density lipoprotein oxidation by the antioxidant agent IRFI005, a new synthetic hydrophilic vitamin E analogue. Free Radic Biol Med. 1999;26:858–868. doi: 10.1016/s0891-5849(98)00271-8. [DOI] [PubMed] [Google Scholar]
- Jaulmes A, Sansilvestri-Morel P, Rolland-Valognes G, Bernhardt F, Gaertner R, Lockhart BP, et al. Nox4 mediates the expression of plasminogen activator inhibitor-1 via p38 MAPK pathway in cultured human endothelial cells. Thromb Res. 2009;124:439–446. doi: 10.1016/j.thromres.2009.05.018. [DOI] [PubMed] [Google Scholar]
- Katsuyama M, Matsuno K. Yabe-Nishimura C. Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J Clin Biochem Nutr. 2011;50:9–22. doi: 10.3164/jcbn.11-06SR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik DK, Gupta M, Kumawat KL. Basu A. NLRP3 inflammasome: key mediator of neuroinflammation in murine Japanese encephalitis. PLoS ONE. 2012;2:e3227. doi: 10.1371/journal.pone.0032270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim CH, Ryu JH, Joo JH, Lee SN, Kim MJ, et al. Crosstalk between platelet-derived growth factor-induced Nox4 activation and MUC8 gene overexpression in human airway epithelial cells. Free Radic Biol Med. 2011;50:1039–1052. doi: 10.1016/j.freeradbiomed.2011.01.014. [DOI] [PubMed] [Google Scholar]
- Lee DC, Cheung CY, Law AH, Mok CK, Peiris M. Lau AS. p38 mitogen-activated protein kinase-dependent hyperproduction of tumor necrosis factor alpha expression in response to avian influenza virus H5N1. J Virol. 2005;79:10147–10154. doi: 10.1128/JVI.79.16.10147-10154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey S, Zhu C, Lu YF. Eklund EA. HoxA10 represses transcription of the gene encoding p67phox in phagocytic cells. J Immunol. 2005;175:5269–5279. doi: 10.4049/jimmunol.175.8.5269. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL. Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Machida K, Cheng KT, Lai CK, Jeng KS, Sung VM. Lai MM. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J Virol. 2006;80:7199–7207. doi: 10.1128/JVI.00321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahy BWJ. Virology: A Practical Approach. Oxford: IRL Press; 1991. pp. 119–150. [Google Scholar]
- Marchant D, Singhera GK, Utokaparch S, Hackett TL, Boyd JH, Luo Z, et al. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J Virol. 2010;84:11359–11373. doi: 10.1128/JVI.00804-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjuki H, Alam MI, Ehrhardt C, Wagner R, Planz O, Klenk HD, et al. Membrane accumulation of influenza A virus hemagglutinin triggers nuclear export of the viral genome via protein kinase Cα-mediated activation of ERK signaling. J Biol Chem. 2006;281:16707–16715. doi: 10.1074/jbc.M510233200. [DOI] [PubMed] [Google Scholar]
- Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC. Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA. Shelton MD. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antiox Redox Signal. 2008;10:1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nencioni L, Iuvara A, Aquilano K, Ciriolo MR, Cozzolino F, Rotilio G, et al. Influenza A virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. FASEB J. 2003;17:758–760. doi: 10.1096/fj.02-0508fje. [DOI] [PubMed] [Google Scholar]
- Nencioni L, De Chiara G, Sgarbanti R, Amatore D, Aquilano K, Marcocci ME, et al. Bcl-2 expression and p38MAPK activity in cells infected with influenza A virus. Impact on virally induced apoptosis and viral replication. J Biol Chem. 2009;284:16004–16015. doi: 10.1074/jbc.M900146200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nencioni L, Sgarbanti R, Amatore D, Checconi P, Celestino I, Limongi D, et al. Intracellular redox signaling as therapeutic target for novel antiviral strategy. Curr Pharm Des. 2011;17:3898–3904. doi: 10.2174/138161211798357728. [DOI] [PubMed] [Google Scholar]
- Palamara AT, Perno CF, Ciriolo MR, Dini L, Balestra E, D'Agostini C, et al. Evidence for antiviral activity of glutathione: in vitro inhibition of herpes simplex virus type 1 replication. Antiviral Res. 1995;27:237–253. doi: 10.1016/0166-3542(95)00008-a. [DOI] [PubMed] [Google Scholar]
- Palamara AT, Garaci E, Rotilio G, Ciriolo MR, Casabianca A, Fraternale A, et al. Inhibition of murine AIDS by reduced glutathione. AIDS Res Hum Retrovir. 1996;12:1373–1381. doi: 10.1089/aid.1996.12.1373. [DOI] [PubMed] [Google Scholar]
- Palamara AT, Nencioni L, Aquilano K, De Chiara G, Hernandez L, Cozzolino F, et al. Inhibition of influenza A virus replication by resveratrol. J Infect Dis. 2005;191:1719–1729. doi: 10.1086/429694. [DOI] [PubMed] [Google Scholar]
- Petherans E, Grob M, Burge T. Zanoni R. Virus-induced formation of reactive oxygen intermediates in phagocytic cells. Free Radic Res Commun. 1987;3:39–46. doi: 10.3109/10715768709069768. [DOI] [PubMed] [Google Scholar]
- Pleschka S, Wolff T, Ehrhardt C, Hobom G, Planz O, Rapp UR. Ludwig S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat Cell Biol. 2001;3:301–305. doi: 10.1038/35060098. [DOI] [PubMed] [Google Scholar]
- Reed LJ. Muench H. A simple method of estimating fifty percent endpoints. Am J Hygiene. 1938;27:493–497. [Google Scholar]
- Rubartelli A, Gattorno M, Netea MG. Dinarello CA. Interplay between redox status and inflammasome activation. Trends Immunol. 2011;12:559–566. doi: 10.1016/j.it.2011.08.005. [DOI] [PubMed] [Google Scholar]
- Sgarbanti R, Nencioni L, Amatore D, Coluccio P, Fraternale A, Sale P, et al. Redox regulation of the influenza hemagglutinin maturation process: a new cell-mediated strategy for anti-influenza therapy. Antiox Redox Signal. 2011;15:593–606. doi: 10.1089/ars.2010.3512. [DOI] [PubMed] [Google Scholar]
- Skalnik DG, Strauss EC. Orkin SH. CCAAT dysplacement protein as a repressor of the myelomonocytic-specific gp91-phox gene promoter. J Biol Chem. 1991;266:16736–16744. [PubMed] [Google Scholar]
- Snelgrove RJ, Edwards L, Rae AJ. Hussell T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur J Immunol. 2006;36:1364–1373. doi: 10.1002/eji.200635977. [DOI] [PubMed] [Google Scholar]
- Soucy-Faulkner A, Mukawera E, Fink K, Martel A, Jouan L, Nzengue Y, et al. Requirement of NOX2 and reactive oxygen species for efficient RIG-I-mediated antiviral response through regulation of MAVS expression. PLoS Pathog. 2010;6:e1000930. doi: 10.1371/journal.ppat.1000930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres M. Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17:287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- Vigilanza P, Aquilano K, Rotilio G. Ciriolo MR. Transient cytoskeletal alterations after SOD1 depletion in neuroblastoma cells. Cell Mol Life Sci. 2008;65:991–1004. doi: 10.1007/s00018-008-7526-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos R, Stambas J, Bozinovski S, Broughton BRS, Drummond GR. Selemidis S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog. 2011;7:e1001271. doi: 10.1371/journal.ppat.1001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wind S, Beuerlein K, Eucker T, Müller H, Scheurer P, Armitage ME, et al. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol. 2010;161:885–898. doi: 10.1111/j.1476-5381.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingler K, Altenhoefer SA, Kleikers PW, Radermacher KA, Kleinschnitz C. Schmidt HH. VAS2870 is a pan-NADPH oxidase inhibitor. Cell Mol Life Sci. 2012;69:159–160. doi: 10.1007/s00018-012-1107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RF, Ma Z, Liu Z. Terada LS. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local ras activation. Mol Cell Bio. 2010;30:3553–3568. doi: 10.1128/MCB.01445-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. NOX4 supports the replication of different influenza virus strains in both NCI-H292 and A549 cells. A. Cells were infected with PR8, NWS or ULSTER virus, and cultured in the absence or presence of DPI for 8 h before the conditioned medium was assayed. Haemagglutination assay (***P < 0.0001; *P < 0.01).
Fig. S2. NOX4 supports influenza virus replication in HEK-293 cells.
A. HEK-293 cells were transfected with plasmids encoding shRNAs specific to NOX4 or with a control plasmid encoding a non-targeting shRNA. The Western blot was sequentially probed with anti-NOX4 antibody and anti-tubulin antibody. Data are representative of two independent experiments.
B and C. Viral load in medium of HEK-293 cells transfected with non-targeting or NOX4-specific shRNA expression plasmids before infection with PR8. (B) Haemagglutination assay (***P < 0.001). (C) Plaque assay (**P < 0.01).