

Abstract
Purpose:
To assess the functional consequences of silencing of tuberin, an inhibitor of the mTOR signaling pathway, in a preclinical model of retinitis pigmentosa (RP) in order to test the hypothesis that insufficient induction of the protein kinase B (PKB)-regulated tuberin/mTOR self-survival pathway initiates apoptosis.
Methods:
In an unbiased genome-scale approach, kinase peptide substrate arrays were used to analyze self-survival pathways at the onset of photoreceptor degeneration. The mutant Pde6bH620Q/Pde6bH620Q at P14 and P18 photoreceptor outer segment (OS) lysates were labeled with P-ATP and hybridized to an array of 1,164 different synthetic peptide substrates. At this stage, OS of Pde6bH620Q/Pde6bH620Q rods are morphologically normal. In vitro kinase assays and immunohistochemistry were used to validate phosphorylation. Short hairpin RNA (shRNA) gene silencing was used to validate tuberin’s role in regulating survival.
Results:
At the onset of degeneration, 162 peptides were differentially phosphorylated. Protein kinases A, G, C (AGC kinases), and B exhibited increased activity in both peptide array and in vitro kinase assays. Immunohistochemical data confirmed altered phosphorylation patterns for phosphoinositide-dependent kinase-1 (PDK1), ribosomal protein S6 (RPS6), and tuberin. Tuberin gene silencing rescued photoreceptors from degeneration.
Conclusions:
Phosphorylation of tuberin and RPS6 is due to the upregulated activity of PKB. PKB/tuberin cell growth/survival signaling is activated before the onset of degeneration. Substrates of the AGC kinases in the PKB/tuberin pathway are phosphorylated to promote cell survival. Knockdown of tuberin, the inhibitor of the mTOR pathway, increased photoreceptor survival and function in a preclinical model of RP.
INTRODUCTION
GENE-BASED AND MECHANISM-BASED THERAPIES FOR RETINITIS PIGMENTOSA
Retinitis pigmentosa (RP) is a devastating form of inherited retinal degenerative disease, affecting 1.5 million people worldwide.1 One in 10 in the US population carries a recessive RP allele.2,3 A major barrier to treatment of many inherited retinal diseases is the limited set of feasible therapeutic tools. Long-term successful gene therapies are mostly observed in nonphotoreceptor diseases,4 such as retinoschisis or RPE65-related early-onset retinal dystrophy. Mouse photoreceptor diseases successfully treated with gene therapy are thus far limited to those with minimal photoreceptor death.5 Current therapies used in humans and mouse models are primarily based on gene addition at a stage in which the retina has not yet significantly degenerated, or in which photoreceptors are not the primary target of gene delivery. Moreover, many autosomal dominant diseases, which cannot be treated by gene addition, are excluded from treatment even though they require the most clinical attention.
Achievements in preclinical gene therapy prompted the first-ever gene therapy trial for patients with RPE65 early-onset retinal dystrophy.6–8 Unfortunately, analyses of RPE65 monopartite vector transduction revealed a failure to slow photoreceptor degeneration.9 This was possibly due to insufficient activation of the mammalian target of rapamycin (mTOR)-mediated self-survival pathway, thus reaching a “point of no return” for treatment10 in midstage disease. Preclinical models that help define the earliest steps for intervention are essential.
PDE6 is a rod-specific enzyme that plays a critical role in phototransduction and specifically in the preservation of rods.2,11–22 Pde6brd1/Pde6brd1 and other knockout mouse models have been used to study retinal degeneration pathways for nearly 80 years.23,24 It was found that mutations in PDE6 (Figure 1) are responsible for approximately 72,000 cases of RP each year worldwide (roughly 8% of cases).2,11–13 Within PDE6-affected patients, about 70% of human RP-associated PDE6 alleles contain missense mutations in the catalytic domain in the β subunit of PDE (PDE6β). In general, mutations in PDE6α and PDE6β are linked to lowered activity in rods.
FIGURE 1.
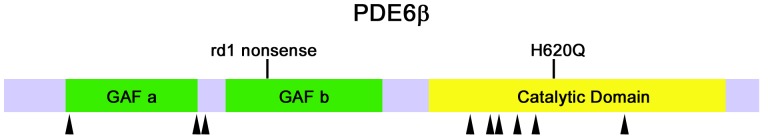
PDE6B alleles. Most PDE6β missense mutations in retinitis pigmentosa occur in the catalytic domain. PDE6β contains two GAF domains (green) and a catalytic domain (yellow). Black arrowheads indicate the position of missense mutations in retinitis pigmentosa. Mouse Pde6brd1 nonsense and Pde6bH620Q missense mutations are indicated on top.
Because of the role of PDE6 in photoreceptor G-protein signaling, defects in PDE6 contribute to RP pathogenesis (Figure 2). Phototransduction begins when a photon is first absorbed into the photoreceptor. First, rhodopsin becomes photoexcited and stimulates the G-protein transducin (GNAT1). GNAT1 then activates PDE6, its effector. The PDE6αβγ complex can be broken down into four parts: two catalytic subunits (PDE6α and PDE6β) and two inhibitory subunits (PDE6γ2).25–41
FIGURE 2.

Low levels of PDE6 result in high concentrations of cGMP. PDE6 is a heterotetrameric enzyme consisting of a catalytic α-subunit, a catalytic β-subunit, and two inhibitory γ-subunits in the rod photoreceptors. PDE6 is highly important in the phototransduction cascade, as it is the primary regulator of cytoplasmic cGMP concentration in the photoreceptor cells. In the dark, PDE6 is in an inactive form, with the γ inhibitory subunits bound (PDE6αβγ), and cGMP levels within the photoreceptor cell outer segment are high (several micromolars). The main molecular components of phototransduction are known; however, the links between abnormally high cGMP and intracellular Ca2+ and cell death are unclear.
PDE6 MOUSE MODELS
One of the most extensively studied RP models thus far has been the Pde6brd1/Pde6brd1 mouse, developed in 1924.23,24 The Pde6brd1/Pde6brd1 mouse has a null allele caused by a nonsense mutation N-terminal to the catalytic domain (Figure 1). Since photoreceptors are nonfunctional, it is considerably more complicated to measure rescue following experimental intervention. The Pde6brd1 mutation is a null allele due to N-terminal truncation, whereas most RP-associated PDE6B alleles are missense mutations in the catalytic domain, and there is no expression of the gene product.
The Pde6brd1/Pde6brd1 mice do not express RNA, protein, or detectable activity.42,43 Pde6brd1/Pde6brd1 mutants degenerate quickly; they exhibit truncated and disorganized outer segments at postnatal day 7 (P7) and complete rod degeneration by P21.42,43 Although cones do not express PDE6B, they eventually degenerate as well, approximately 3 weeks later than rods. In this context, cone degeneration is nonautonomous.44
As expected for a PDE6 loss-of-function phenotype, Pde6brd1/Pde6brd1 mice show abnormal cGMP regulation, leading to interruptions in the visual cycle. It has been shown that cGMP increases despite normal guanylate cyclase activity.45 Since high cGMP levels result in the opening of CNG channels, it follows that high intracellular Na+/Ca2+ concentrations should accompany high cGMP levels in Pde6brd1/Pde6brd1 mice. Prior expression microarray analyses support that Ca2+ levels do indeed increase, since transcripts for Ca2+-binding proteins (eg, calmodulin, calbindin, calpastatin, calpain) are elevated in Pde6brd1/Pde6brd1 mice.46 These changes in transcription are likely downstream of changes in intracellular signaling caused by elevated cGMP/Ca2+ levels.
Studies prior to those on the mouse model also showed that there was a correlation between a rise in cGMP/Ca2+ levels and photoreceptor apoptosis, making it a prime model for studying cellular pathways that lead to RP. However, there are most likely other abnormally regulated signaling pathways that lead to degenerative changes in these mutants.47,48
Despite the strengths of the Pde6brd1/Pde6brd1 mutant, there are distinct disadvantages to using it to identify signaling pathways that lead to RP. Degeneration occurs quickly; it is almost simultaneous to photoreceptor growth and differentiation. Farber and Lolley49 have found that compared to age-matched controls, cGMP levels in Pde6brd1/Pde6brd1 mutants dramatically increase between P10 and P14, and then quickly decrease between P16 and P21. In addition, due to the unique and profoundly abnormal outer segment (OS) development of the Pde6brd1/Pde6brd1 strain, functional and histological changes in the eye over time are difficult to measure. In the Pde6brd1/Pde6brd1 mice, a severe degeneration occurs quickly, complicating any understanding of what sequence of events causes photoreceptors to die at the cellular level.
PDE6BH620Q/PDE6BH620Q MOUSE AS AN IMPROVED PRECLINICAL MODEL FOR RP
Our laboratory studied an alternative model to overcome the limitations of the Pde6brd1/Pde6brd1 mutant. Mice with a single point mutation, Pde6bH620Q/Pde6bH620Q, show RP-like features and dramatic elevation of retinal cGMP.14 Degeneration in Pde6bH620Q/Pde6bH620Q occurs over a significantly longer period (6 weeks) than in the related Pde6brd1/Pde6brd1 model. For the first 3 weeks after birth, Pde6bH620Q/Pde6bH620Q mutants display relatively normal OS histology and quantifiable rod physiology, providing a window of opportunity for observing photoreceptor death. Pde6bH620Q/Pde6bH620Q photoreceptors are functional with activity that is measurable by electroretinography. Overt photoreceptor degeneration begins after week 2 (ie, there is a 2-week window between differentiation and degeneration). The Pde6bH620Q/Pde6bH620Q allele is a missense mutation in the PDE6β catalytic domain—as in most RP-associated PDE6β alleles.50 Moreover, Pde6bH620Q/Pde6bH620Q is a hypomorphic loss-of-function allele, as shown by compound heterozygous Pde6brd1/Pde6bH620Q mice that have a phenotype intermediate between Pde6brd1/Pde6brd1 and Pde6bH620Q/Pde6bH620Q homozygotes.50
Despite the advantageously slower degeneration of the Pde6bH620Q/Pde6bH620Q mouse, there remain key similarities between Pde6brd1/Pde6brd1 and Pde6bH620Q/Pde6bH620Q mutants that allow us to explore the underlying theme of this study, ie, the identification of downstream signaling pathways that link Ca2+ levels to photoreceptor death. Appropriately, in both mutants, elevation of cGMP and Ca2+ levels precede any detectable photoreceptor death. Overall, the Pde6bH620Q/Pde6bH620Q mouse provides a larger temporal window to study early mechanisms of photoreceptor degeneration.
Although studies of RP using knockout mice have contributed to a rich body of knowledge about the disease, including potential gene therapy-based treatments, newer proteomics techniques provide another avenue for studying disease. Peptide kinase arrays, for example, allow a more inclusive view of the disease that can reveal activation of its molecular pathways.
SURVEY OF THE OUTER SEGMENT KINOME
An unbiased proteome-wide survey of all disturbed states of the kinome can potentially identify novel candidate pathways that initiate the rod apoptotic program. For a systems biology approach to explain the survival pathways in RP, it is necessary to consider the vast array of interacting signal transduction networks that are functionally linked with photoreceptor degeneration. Kinases can serve as readouts of degeneration and have provided some of the most successful drug targets in medicine. For the current study, we used peptide substrate arrays to provide relatively unbiased genome-scale data about kinase activities in the outer segments of PDE6 mutants.
CA2+-REGULATED KINASES IN RP MOUSE MODELS
Given the established links between high cGMP,42,49 intracellular Ca2+ levels,51 and cell death in RP, changes of activity in cGMP-and/or Ca2+-effectors would be expected to appear in mouse models of the disease. Presumably, high levels of cGMP/Ca2+ lead to abnormal signaling in rod and cones, both within and between photoreceptor cells. Kinases stimulated by cGMP/Ca2+ may exhibit abnormally high activity in Pde6b mutant retinas, and therefore an increase in the phosphorylation of effectors of cell growth and survival. Despite these intriguing possibilities, the role of kinases in Pde6 mice has not been examined in detail.
Thus, our first hypothesis was that elevated cGMP and/or Ca2+ second messengers generate complex changes in intracellular signaling, including the stimulation of both pro-growth/survival and pro-apoptotic pathways. Our aim was to measure kinase activities and provide a basis for identifying the pathways and substrates that mediate the pathogenic response.
Our studies had shown that in Pde6 mice, as cGMP and Ca2+ become elevated, there is an accompanying increase in AGC kinases (protein kinase A, G, C, and B) (Figure 3). At P11, protein kinase C (PKC) transcription and activity have been found in Pde6brd1/Pde6brd1 retinas—at precisely the time when degeneration is under way.52 Similarly, Ca2+/calmodulin kinase and PKB have been shown to be activated in Pde6brd1/Pde6brd1 retinas.53–55 These studies support a role for cGMP- and Ca2+-regulated kinases as participants or indicators of photoreceptor health. Therefore, we predicted that in dying PDE6 photoreceptors, we would be able to identify kinase activity that was higher than normal. We expected to see increased phosphorylation of kinase substrates (Figure 4).
FIGURE 3.
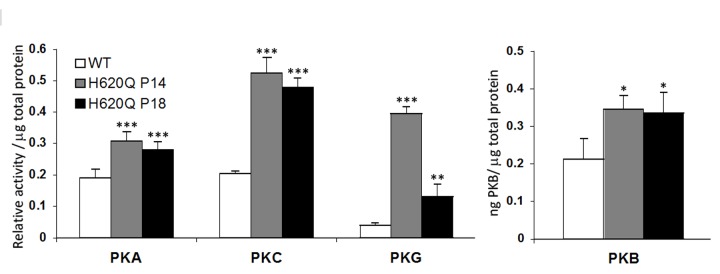
AGC kinases are activated before the onset of degeneration. Pde6bH620Q/Pde6bH620Q retinal lysates exhibit increased PKG, PKA, PKC, and PKB activity compared to wild-type (WT) lysates at P14. WT and H620Q P14 and P18 retinal lysates were tested in different in vitro enzyme immunometric kinase assays for PKA, PKC, PKG, and PKB activity. Differences between WT and H620Q P14 or P18, respectively, are calculated with the Student t test P values at P14 (WT vs H620Q): PKA, P<.001, n=3; PKC, P<.001, n=3; PKG, P<.001, n=3; and PKB, P<.05, n=3. Levels of significance: *significant, P<.05; **highly significant, P< .01; ***very highly significant, P<.001.
FIGURE 4.

Kinase substrate array and kinases in the mTOR survival pathway. Representative kinase substrates (oligopeptides) were immobilized on arrays and hybridized with outer segment (OS) extract labeled with 33P-ATP to detect kinase activities in P14 (left panel) and P18 Pde6bH620Q/Pde6bH620Q OS (right panel). Lysates were analyzed using 1,164 different synthetic peptide substrates and 12 controls; substrates were spotted twice.57,58 Dots represent phosphorylated peptide substrates (based on incorporation of 33P). Circled dots represent preferentially phosphorylated substrates (left panel, P14; right panel, P18).
This would indicate a nonspecific downstream effect associated with elevated cGMP and Ca2+, but it would not necessarily indicate a connection to cell death or survival. Accordingly, our second hypothesis was that some types of AGC kinase activity are directly tied to photoreceptor death and/or survival. We proposed to test this hypothesis by manipulating the expression of downstream effectors of cGMP and Ca2+, and then measure the changes in mouse photoreceptors, quantifying these in terms of both visual function and photoreceptor survival. The peptide array studies detailed below allowed us to identify tuberin, a substrate of the AGC kinase PKB, as a focus to test our hypothesis that activation of mTOR signaling parallels Ca2+ increases and occurs prior to significant histological evidence of degeneration (Figure 5).
FIGURE 5.
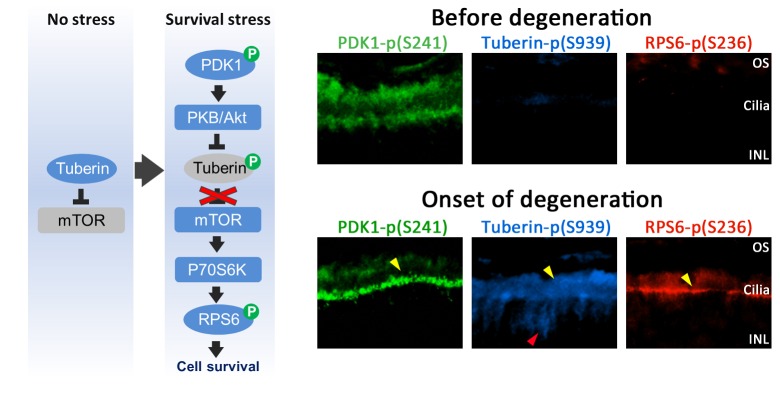
Increased activity of the mTOR survival pathway at the onset of degeneration. Left, Schematized version of the mTOR survival pathway when stress is absent and during the stress response when the pathway is activated. Right, Phosphosubstrate immunostaining in Pde6bH620Q/Pde6bH620Q mutant P14 (before degeneration) and P18 (onset of degeneration) retinas: PDK1 p(S241), left column; tuberin p(S939), middle column; RPS6 p(S236), right column. Yellow and red arrowheads indicate regions of increased staining using anti-PDK1 pS241, anti-tuberin pS939, and anti-RPS6 pS236 antibodies. OS, photoreceptor outer segments; INL, inner nuclear layer.
Tuberin, which was active in high levels in our RP mouse model, is a negative regulator of growth factor–induced signaling in the mTOR pathway. This substrate of PKB is a ∼200 kD cytoplasmic protein. In many healthy organs and cell lines, including photoreceptors, tuberin is present but is expressed at low levels. The role it plays within the mTOR pathway is that of a GTPase-activating protein for other proteins such as Rheb.56 Tuberin’s activity on Rheb leads to inactivation of mTOR, blocking growth factor signals, which in turn inhibits cell growth and survival.
The mTOR pathway controls cell growth and proliferation, protein synthesis, and autophagy. Typically, growth factors or insulin (among other factors) activate PKB. It was recently found that PKB has a role in cellular metabolism through the regulatory proteins hamartin and tuberin (TSC1 and TSC2). PKB inhibits these tumor suppressors found in the pathway. This complex acts as a GTPase-activating protein for the GTP-binding protein Rheb, found in the mTOR1 protein complex.
The activated mTOR1 complex phosphorylates target proteins 4E-BP1 (eukaryotic initiation factor 4E binding protein-1), p70S6K (ribosomal p70S6 kinase), and GRB10. These serve to enhance mRNA and protein production. Phosphorylation of S6K activates the kinase to phosphorylate multiple target proteins, including S6. The S6K protein, found in both the cytoplasm and the nucleus, enhances transcription and translation through ribosome biogenesis.
Preliminary studies found that activation of certain mTOR signaling proteins parallels pathologic increases in Ca2+ levels in RP-mutant mouse retinas and occurs prior to detectable histologic or microscopic evidence of degeneration. Moreover, exogenous inhibition of mTOR produces a similar pattern of photoreceptor degeneration in wild-type (WT) mice to that seen in RP models. Based on these findings, we tested the hypothesis that additionally augmenting the mTOR survival pathway will retard retinal degeneration.
METHODS
All experiments were prospectively approved by Columbia University’s Institutional Animal Care and Use Committee (IACUC) under protocol AAAB-4306. Mice were used in accordance with the Statement for the Use of Animals in Ophthalmic and Vision Research of the Association for Research in Vision and Ophthalmology, as well as the Policy for the Use of Animals in Neuroscience Research established by the Society for Neuroscience.
PEPTIDE ARRAY ANALYSIS OF KINASE ACTIVITIES
Kinase profiling was performed using the PepScan (PepChip Kinase Peptide Microarrays, Lelystad, The Netherlands). Peptide substrates with increased phosphorylation in mutant lysates at P14 were compared to controls. Kinase substrates (oligopeptides) were immobilized on arrays and P-ATP to detect kinase activities. Lysates were analyzed using 1,164 different synthetic peptide substrates and 12 controls; substrates were spotted twice.57,58 Substrates with PepScan Pde6bH620Q/DBA2J ratios of 1.3 and higher were identified. The ratio of 1.3 was defined by the PepScan algorithm57,58 to be significant for the detection of peptide substrate with increased phosphorylation.
POLYACRYLAMIDE GEL ELECTROPHORESIS, SDS-PAGE, AND IMMUNOBLOT ANALYSIS
The following reagents were used for immunoblots as previously described17,26,29,51,59–65: polyclonal antibodies to tuberin (Cell Signaling Technology, Danvers, Massachusetts), phospho-tuberin (phosphorylated Ser939, Cell Signaling), ribosomal protein S6 (RPS6) (Bethyl Laboratories, Montgomery, Texas), phospho-RPS6 (Bethyl), and monoclonal anti-α-tubulin (Sigma-Aldrich, St Louis, Missouri).
LENTIVIRAL shRNA TRANSDUCTION
Mission lentiviral transduction particles, Sigma-Aldrich, Clone ID TRCN0000042723-7, are referred to in this manuscript as shRNA_tuberin.20,21
TRIPARTITE AAV8 VECTOR
AAV serotype 2/8 capsids22,66 containing a point mutation in surface-exposed tyrosine residues, AAV2/8 (Y733F) were used for packaging our tripartite construct. Vector plasmids were constructed by inserting 1.1 kb of the murine rhodopsin promoter region [Ensembl, rhodopsin, chromosome 6: (115, 930, 881–115, 931, 988); Ensembl, rhodopsin, ATG = 1: (−1125, −17)] and a full-length murine Pde6α cDNA fragment, tuberin_shRNA, and Cnga1_shRNA into the pZac2.1 plasmid to generate the tripartite construct. AAV vectors were packaged, and purified at the Penn Vector Core (University of Pennsylvania, Philadelphia) to become AAV2/8(Y733F). The AAV2/8(Y733F), mRhodopsin promoter: Pde6α, tuberin_shRNA, and Cnga1_shRNA SV40 vector will be referred to as tripartite vector in this report. Targeting sequences for Tuberin_shRNA was cgagtgtatgagagcctcatt, and CNGA1_shRNA was gctgttaaggatctctcgaat.
TRANSDUCTION INTO SUBRETINAL SPACE
Lenti (0.7μL, 1.00e8 genome copy (GC)/mL) (Figure 6) or AAV8 (0.7μL, 1.26e13 genome copy (GC)/mL) were transduced into the subretinal space of the right eye of Pde6 mutants at postnatal day 5 (P5). Saline was injected into the subretinal space of the left eye of Pde6 mutant mice at P5 for control experiments. A 100-μm-diameter pulled capillary needle was passed through the sclera and the injection was delivered. Virus particles were injected at the 6 o’clock position of the eye, ∼1.5 mm from the limbus, to produce a subretinal bleb in the midperiphery of the retina. The left eyes of all mice were kept as a matched control for experimental analyses.
FIGURE 6.

Monotherapy with shRNA_tuberin can decrease tuberin levels in a dose-dependent manner. Top, Schematic representation of the shRNA lentiviral vector. This self-inactivating (SIN) vector consists of a 5′ long terminal repeat (LTR), a packaging signal ψ, a tRNA primer binding site, a lentiviral reverse response element (RRE), the shRNA driven by U6 promoter, and a 3′LTR. This pLKO.1 viral vector also contains a central polypurine tract/DNA flap (cPPT) and a Woodchuck hepatitis virus posttranscriptional regulatory element (WPRE). The Simian virus 40 (SV40) polyadenylation signal is located at the 3′ end of the cDNA. Arrows indicate the direction of transcription. AmpR, ampicillin resistance gene; ori, origin of replication. Bottom, Immunoblot showing that phosphotuberin levels are inversely proportional to the amount of tuberin shRNA. Phosphorylated residue detected by the phosphotuberin antibody (phospho-Ser939).
Pde6bH620Q/Pde6bH620Q (C3H background) mice were obtained.42,49,50,67 DBA, which are closely related to C3H, was used as controls for viral toxicity because C3H did not have photoreceptors due to the Pde6rd1 allele. At P5, one subretinal injection (6 o’clock) of 1.2 μL of lentivirus particles [∼3.2×107 transducing unit (TU) per mL or AAV8 (1.26e13 genome copy (GC)/mL)] was given to the right eyes.68 Left eyes were not injected and were used as a control.
ELECTRORETINOGRAMS
Electroretinograms (ERGs) were conducted as described.18,19,63,65,69–73 Espion ERG Diagnosys equipment (Diagnosys LLC, Littleton, Massachusetts) was used on dark-adapted mice. Adult C57BL/6 mice were used as controls. Mice were anesthetized with 0.1 mL mix (1 mL ketamine 100 mg/mL and 0.5 mL xylazine 20 mg/mL in 8.5 mL PBS) per 10 gm body weight. Corneas were anesthetized with a drop of 0.5% proparacaine hydrochloride (Alcon Laboratories, Inc, Fort Worth, Texas), and pupils were dilated with topical 2.5% phenylephrine hydrochloride and 1% tropicamide. Gonioscopic prism solution (Alcon Labs) was applied, and Burian-Allen bipolar mouse contact lens electrodes (Hansen Lab, Coralville, Iowa) were placed on each cornea. Reference electrodes were placed subcutaneously in the anterior scalp between the eyes, and ground electrodes were inserted into the tail. Electrode impedance was balanced for each eye pair measured. Both eyes were recorded simultaneously. For rod and mixed-rod cone responses, pulses of 0.00130 cd/m2 and 3 cd/m2 (White-6500K) were used. Each result represents the average of 40 trials. For cone responses, mice were light-adapted in the Ganzfeld dome for at least 10 minutes. A background of 30 cd/m2 (White-6500K) was present throughout the trials to suppress rod function. ERGs were recorded using white flashes of light up to 4.35 cd/m2 (xenon).
HISTOLOGY
Following ERGs, mice were sacrificed and the eyes were rapidly removed and placed in a fixing solution (2% paraformaldehyde, 2.5% glutaraldehyde, 0.01 M sodium phosphate buffer). After fixing, dehydrated eyes were embedded in paraffin, sectioned, and stained with hematoxylin-eosin as described.15–22,51,74
RESULTS
Pde6bH620Q/Pde6bH620Q mutants underwent biochemical, microarray, and immunohistochemical testing at P11, P14, P18, and P21, after retinal degeneration was already under way. Initial biochemical assays detected elevated kinase A, G, C (AGC kinase), and B activity in P14 and P18 retinas (Figure 3). The increased phosphorylation of PKB was consistent with activation of the PKB/tuberin survival pathway (Figures 4 and 5), which has been linked to growth and survival in cells of several organs.75–77
PEPTIDE ARRAY ANALYSES
We conducted an unbiased proteome-wide search for differences in kinase activity on postnatal day 14 (P 14) (before onset of degeneration) compared with P18 (at the onset of degeneration) in Pde6bH620Q/Pde6bH620Q retinal outer segments using PepScan (PepChip Kinase Peptide Microarrays). At this stage, OS of Pde6bH620Q/Pde6bH620Q rods are morphologically normal. Substrates exhibiting PepScan P18/P14 ratios of 1.3 and higher were defined by the PepScan algorithm54,55 as significant for detection of relevant substrates. The results of this peptide assay (Figure 4) display a marked increase in AGC kinase activity (Figure 3).
KINASE PEPTIDE SUBSTRATE ARRAY (KINOME) ANALYSIS OF P14 AND P18 IN PDE6-DEFICIENT RETINAS
Peptide microarray and immunohistochemical analyses (Figure 4) identified phosphorylation differences between P14 and P18 mutant OS. At this stage, OS of Pde6bH620Q/Pde6bH620Q rods are morphologically normal. We found that the mTOR pathway was the most physiologically relevant of 40 potential Ca2+-dependent substrates in mediating rod photoreceptor survival responses to initial Ca2+ increases. Activity of mTOR was upregulated in Pde6bH620Q/Pde6bH620Q mice after development of mature OS (P14), but preceding degeneration (P18).
Peptide array results indicated increased phosphorylation of tuberin by PKB. These results were consistent with activation of the PDK1/PKB/mTOR pathway. Investigators from our laboratory and elsewhere have previously observed increased PKB/Akt activity in Pde6b mutants (Figure 5). PKB is thus an important candidate for further testing. To confirm these in vitro assays, we examined whether tuberin was preferentially phosphorylated with immunohistochemical assays.
IMMUNOHISTOCHEMICAL DETECTION OF KINASE SUBSTRATE PHOSPHORYLATION
We found altered phosphorylation patterns for several kinases involved in mTOR signaling from purified OS from P18 compared with P14 photoreceptors, including phosphoinositide-dependent kinase-1 (PDK1), tuberin, and RPS6 (Figure 5). These altered phosphorylation patterns were observed at the onset of degeneration, when rod OS were morphologically normal, as determined by electron microscopy. Similarly, mTOR activation contributed to cone survival.78
Phosphotuberin (Ser939), phospho-RPS6 (Ser236), and phospho-PDK1 (Ser241) antibodies were used to analyze P14 mutant and DBA/2 control sections (Figure 5).79 Preferential expression of phosphotuberin was apparent in mutant outer and inner segments of photoreceptors (Figure 5). This high tuberin expression points to increased PKB activity. We also observed localized expression of phospho-RPS6 in the IS (Figure 5). These results formed the basis of the second part of our study—testing a specific link between kinase expression and cell survival in photoreceptors.
TUBERIN IN THE mTOR SELF-SURVIVAL PATHWAY
A phosphospecific antibody assay showed activation of mTOR pathway kinases in photoreceptor OS at the onset of degeneration. Increased PKB phosphorylation is consistent with mTOR activation (Figure 5). Tuberin had been inactivated by phosphorylation at Ser939 (phospho-Ser939). Phosphotuberin (Ser939), phospho-RPS6 (Ser236), and phospho-PDK1 (Ser241) antibodies were used to analyze P14 and P18 sections (Figure 5). Preferential expression of phosphotuberin was apparent at the mutant outer/inner segment junction, marked by yellow arrows (Figure 5), and in photoreceptors, indicated by a red arrow (Figure 5). We also observed localized expression of phospho-RPS6 in IS, as indicated by yellow arrows (Figure 5).
Based on our observation of higher phosphotuberin levels in Pde6bH620Q/Pde6bH620Q eyes, we devised a strategy to activate the mTOR pathway by knocking down expression of tuberin using lentiviral vectors (Figure 6). Transduced eyes showed rescued photoreceptor a-wave ERG function, suggesting that retinal degeneration was antagonized by tuberin silencing and by induced activation of mTOR.
TRANSIENT INDUCTION OF THE mTOR PATHWAY
To knock down tuberin in cells, we used an shRNA approach. Lentiviral vectors were chosen to transduce cells with shRNA expression cassettes. In order to identify a virus that effectively reduced tuberin function, five lentiviral constructs were tested in NIH 3T3 cells. Each construct contained a different shRNA targeted to a specific sequence within Tuberin. Lentiviral vector with a cgagtgtatgagagcctcatt targeting sequence was found to be the most effective. Phosphotuberin levels were initially used to evaluate tuberin knockdown. The residue detected by the phosphotuberin antibody (Ser939) is phosphorylated when tuberin is inactive.
TUBERIN KNOCKDOWN IMPROVED RETINAL FUNCTION AND SURVIVAL
To determine whether tuberin knockdown had effects on a degenerating retina, tuberin_shRNA lentiviral vector was injected into the right eyes of 17 Pde6bH620Q/Pde6bH620Q mice. Two bolus injections of the tuberin_shRNA virus were injected between the retinal pigment epithelial and photoreceptor layers at postnatal day 5. It was expected that the virus would infect the photoreceptors, leading to shRNA expression and decreased tuberin levels, and that this tuberin_shRNA knockdown would result in increased survival of the photoreceptors.
At week 8, transduced eyes were examined for retinal detachment, ERGs were recorded for each eye (Figure 7), mice were sacrificed, and the eyes were fixed, sectioned, and stained with hematoxylin-eosin. Nine of 17 virus-injected eyes showed improved ERG responses compared to control eyes. All eyes with improved ERG had corresponding increase in photoreceptor survival (Figure 7). Six additional eyes showed retinal detachment and no ERG rescue. Treated retinas showed a 26% average increase in outer nuclear layer thickness compared to control eyes, suggesting that tuberin knockdown slows or prevents photoreceptor loss (Figure 7).
FIGURE 7.

Monotherapy with shRNA_tuberin improved photoreceptor survival and electroretinogram (ERG) function in 6-week-old mouse Pde6bH620Q/ Pde6bH620Q retinas. Upper left, Survival of photoreceptors. Retinal sections were stained to assess changes in the outer nuclear layer (ONL), which contains photoreceptor nuclei. Treated retinas showed a 26% average increase in ONL thickness compared to control eyes, suggesting that tuberin knockdown slows or prevents photoreceptor loss. Vertical yellow bars represent the ONL thickness. GC, ganglion cells; INL, inner nuclear layer. Upper right, ONL enhancement in mice (n=4) 8 weeks post tuberin shRNA transduction as indicated by the solid bar. Lower panel, Representative maximum ERG responses (in μV) of shRNA-transduced eyes (blue) compared to saline-injected control eyes (red). ERGs were performed on both eyes (injected and control) simultaneously.
Based on these results, we also tested the hypothesis that a tripartite vector delivery could prolong the survival effects of gene therapy by augmenting the mTOR survival pathway, limiting excessive Ca2+ influx, and delivering the wild-type gene to retinal cells (Figure 8). Increased photoreceptor survival was observed in eyes transduced with tripartite AAV8 transduction compared to control eyes (Figure 8).
FIGURE 8.

Improved photoreceptor survival of PDE mutant photoreceptors after tripartite AAV8 transduction. Top, Schematic representation of the AAV2/8(Y733F)-Rho::Pde6α tripartite vector. pZac2.1 plasmid vector displaying the Pde6α complementary DNA (cDNA) fragment driven by 1.1 kb of the murine rhodopsin promoter. The Simian virus 40 (SV40) polyadenylation signal is located at the 3’ end of the cDNA. The H1 promoter drives expression of the tuberin_shRNA. The U6 promoter drives expression of the Cnga1_shRNA. Arrows indicate the direction of transcription. 5′- and 3′-ITR, inverted terminal repeats of AAV; AmpR, ampicillin resistance gene; F1 ori, origin of replication. Left, Hematoxylin-eosin stained retinal section of a 1-month-old C57BL/6J control. Middle, Pde6aD670G/Pde6aD670G eye transduced by Rho::Pde6a-shRNA_tuberin-shRNA_Cnga1 at 1 month. Right, Untreated Pde6aD670G/Pde6aD670 at 1 month. Vertical yellow bars represent the thickness of the outer nuclear layer. Images were obtained at 40× magnification at the same location of the retina, 0.3 mm distal from the optic nerve head.
DISCUSSION
The first human gene therapy trials to treat a form of RP reported enhanced visual function for at least 3 years in 33% to 50% of patients.6–8 This was profoundly encouraging given that these phase 1 safety trials enrolled only patients with end-stage disease (no visual function). However, follow-up studies revealed that monotherapy ultimately failed to halt or slow photoreceptor degeneration as evaluated by optical coherence tomography imaging studies.9 Failure to halt progressive photoreceptor degeneration in these landmark gene therapy trials was hypothesized to be due to a retinal “point of no return.”10 Several hypotheses provide explanations for this retinal “point of no return,” including the following: (1) changes in Ca2+ homeostasis may disrupt the mitochondrial membrane potential and induce apoptosis22,51,66,80 and (2) insufficient activation of the mTOR-mediated self-survival pathway for rod photoreceptor cells may cause initiation of apoptosis.
Mutations in genes encoding subunits of the rod-specific enzyme, cGMP PDE6, are responsible for approximately 72,000 cases of RP worldwide each year,2,11–13 making therapeutic models highly relevant to human pathophysiology.
In PDE6 patients, α, β, and γ subunit malfunctions can all be observed,26,28,29,62,81 and elevated Ca2+ levels can trigger self-survival pathways. In the absence of PDE6 activity, (1) rod OS formation is severely affected, (2) cGMP accumulates to cytotoxic levels triggering PKG, (3) increased intracellular Na+ induces changes in osmotic pressure, activating c-Jun NH2-terminal protein kinase (JNK), and (4) high intracellular Ca2+ triggers self-survival pathways and PKC/PKA.17 PKB, PKC, PKG, PKA, and RPS6 are members of the cAMP-dependent, cGMP-dependent protein kinase C (AGC) family. PKB phosphorylates tuberin and RPS6, which are gatekeepers for mTOR signaling in regulation of protein synthesis; RPS6 and PKB are both regulated by 3-PDK1 phosphorylation. In addition, inhibition of tuberin, a substrate of PKB, appears to result in activation of the mTOR pathway. Immunohistochemistry showed activation of several factors associated with this pathway in photoreceptor cells.
Phosphorylation of tuberin is known to facilitate transmission of PKB signals through two mechanisms. First, high-energy signals lead to the inhibition of tuberin, allowing mTOR to promote cell division through its actions on ribosomal protein S6 kinase (S6K) and eukaryotic translation initiation factor 4E binding protein (4EBP).82–84 Second, activation of mTOR leads to inhibition of apoptosis through the apoptosis factor BCL2.85
In peptide array analyses, we found that the mTOR pathway promotes rod survival through the inhibition of tuberin at the onset of degeneration. To test the hypothesis that insufficient activation of the mTOR-mediated self-survival pathway for rod photoreceptor cells can initiate apoptosis, we tested the consequences of knocking down tuberin. Ablating tuberin preserved retinal function by allowing induction of the mTOR survival pathway.75,86 We have shown that the silencing of tuberin can rescue both quantity of photoreceptors and photoreceptor function (Figure 7). Based on these results, we also showed that a tripartite vector delivery could prolong the survival effects of gene therapy by augmenting the mTOR survival pathway and delivering the wild-type gene to retinal cells.
FUTURE DIRECTIONS
Stimulating the mTOR survival pathway may reset the rheostat for the “point of no return” and slow retinal degeneration in RP. A tripartite AAV8 vector could simultaneously overexpress wild-type version of the defective gene and knockdown tuberin to delay the “point of no return” in a preclinical arRP model.
Acknowledgments
Funding/Support: The Bernard & Shirlee Brown Glaucoma Laboratory and Barbara & Donald Jonas Stem Cell Laboratory are supported by NIH core grants 5P30CA013696 and 5P30EY019007 and unrestricted funds from Research to Prevent Blindness, New York, New York. Dr Tsang is a member of the RD-CURE Consortium and is supported by the Tistou and Charlotte Kerstan Foundation. This work is also supported by NIH R01EY018213 (Dr Tsang), K08EY020530 (Dr Mahajan), the Research to Prevent Blindness Physician-Scientist Award, the Nancy and Kobi Karp Foundation, the Schneeweiss Stem Cell Fund, New York State (N09G-302), the Foundation Fighting Blindness New York Regional Research Center Grant (C-NY05-0705-0312), the Joel Hoffman Fund, the Gale and Richard Siegel Stem Cell Fund, Charles Culpeper Scholarship, the Irma T. Hirschl Charitable Trust, the Professor Gertrude Rothschild Stem Cell Foundation, and the Justin Manus Fund and Gebroe Family Foundation.
Financial disclosures: None.
Author Contributions: Design and conduct of study (S.H.T.); Collection and management of data:(W.H.W., C.W.H., Y.T.T., J.Y., J.T., K.J.W.); Analysis and interpretation of data: (V.B.M., S.H.T., Y.T.T., L.C., R.J.D.); Preparation, review, approval of manuscript: (S.H.T., L.C., Y.T.T., W.H.W., C.W.H., J.Y., J.T., K.J.W., R.J.D., V.B.M.).
Other Acknowledgments: We thank members of the Bernard and Shirlee Brown Glaucoma Laboratory for their support, especially Diane K. Fiander and Kerstin Janisch. We also thank Deniz Erol for her hard work on the initial submission of this manuscript and Allegra Pincus for her help revising the final manuscript.
REFERENCES
- 1.Berson EL. Retinitis pigmentosa: The Friedenwald lecture. Invest Ophthal Vis Sci. 1993;34(5):1659–1676. [PubMed] [Google Scholar]
- 2.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 3.Sohocki MM, Daiger SP, Bowne SJ, et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001;17(1):42–51. doi: 10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dinculescu A, Glushakova L, Min SH, Hauswirth WW. Adeno-associated virus-vectored gene therapy for retinal disease. Hum Gene Ther. 2005;16(6):649–663. doi: 10.1089/hum.2005.16.649. [DOI] [PubMed] [Google Scholar]
- 5.Schlichtenbrede FC, da Cruz L, Stephens C, et al. Long-term evaluation of retinal function in Prph2Rd2/Rd2 mice following AAV-mediated gene replacement therapy. J Gene Med. 2003;5(9):757–764. doi: 10.1002/jgm.401. [DOI] [PubMed] [Google Scholar]
- 6.Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 7.Jacobson SG, Cideciyan AV, Aleman TS, et al. Photoreceptor layer topography in children with Leber congenital amaurosis caused by RPE65 mutations. Invest Ophthalmol Vis Sci. 2008;49(10):4573–4577. doi: 10.1167/iovs.08-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cideciyan AV, Jacobson SG, Beltran WA, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci U S A. 2013;110(6):E517–525. doi: 10.1073/pnas.1218933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cepko CL, Vandenberghe LH. Retinal gene therapy coming of age. Hum Gene Ther. 2013;24(3):242–244. doi: 10.1089/hum.2013.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McLaughlin ME, Ehrhart TL, Berson EL, Dryja TP. Mutation spectrum of the gene encoding the b subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc Nat Acad Sci U S A. 1995;92:3249–3253. doi: 10.1073/pnas.92.8.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bird AC. Retinal photoreceptor dystrophies: the LI. Edward Jackson Memorial Lecture. Am J Ophthalmol. 1995;119:543–562. doi: 10.1016/s0002-9394(14)70212-0. [DOI] [PubMed] [Google Scholar]
- 13.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125(2):151–158. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis JW, Nakanishi H, Kumar VS, et al. Circulating tumor cells in peripheral blood samples from patients with increased serum prostate specific antigen: initial results in early prostate cancer. J Urol. 2008;179(6):2187–2191. doi: 10.1016/j.juro.2008.01.102. discussion 2191. [DOI] [PubMed] [Google Scholar]
- 15.Tsang SH, Tsui I, Chou CL, et al. A novel mutation and phenotypes in phosphodiesterase 6 deficiency. Am J Ophthalmol. 2008;146(5):780–788. doi: 10.1016/j.ajo.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woodruff ML, Janisch KM, Peshenko IV, Dizhoor AM, Tsang SH, Fain GL. Modulation of phosphodiesterase6 turnoff during background illumination in mouse rod photoreceptors. J Neurosci. 2008;28(9):2064–2074. doi: 10.1523/JNEUROSCI.2973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janisch KM, Kasanuki JM, Naumann MC, et al. Light-dependent phosphorylation of the gamma subunit of cGMP-phophodiesterase (PDE6gamma) at residue threonine 22 in intact photoreceptor neurons. Biochem Biophys Res Commun. 2009;390(4):1149–1153. doi: 10.1016/j.bbrc.2009.10.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Naumann MC, Tsai YT, et al. Vigabatrin-induced retinal toxicity is partially mediated by signaling in rod and cone photoreceptors. PLoS One. 2012;7(8):e43889. doi: 10.1371/journal.pone.0043889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sancho-Pelluz J, Tosi J, Hsu CW, et al. Mice with a D190N mutation in the gene encoding rhodopsin: a model for human autosomal-dominant retinitis pigmentosa. Mol Med. 2012;18(1):549–555. doi: 10.2119/molmed.2011.00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tosi J, Davis RJ, Wang NK, Naumann M, Lin CS, Tsang SH. shRNA knockdown of guanylate cyclase 2e or cyclic nucleotide gated channel alpha 1 increases photoreceptor survival in a cGMP phosphodiesterase mouse model of retinitis pigmentosa. J Cell Mol Med. 2011;15(8):1778–1787. doi: 10.1111/j.1582-4934.2010.01201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tosi J, Sancho-Pelluz J, Davis RJ, et al. Lentivirus-mediated expression of cDNA and shRNA slows degeneration in retinitis pigmentosa. Exp Biol Med (Maywood) 2011;236(10):1211–1217. doi: 10.1258/ebm.2011.011053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wert KJ, Davis RJ, Sancho-Pelluz J, Nishina PM, Tsang SH. Gene therapy provides long-term visual function in a pre-clinical model of retinitis pigmentosa. Hum Mol Genet. 2013;22(3):558–567. doi: 10.1093/hmg/dds466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keeler CE. The inheritance of a retinal abnormality in white mice. Proc Natl Acad Sci U S A. 1924;10:329–333. doi: 10.1073/pnas.10.7.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keeler C. Retinal degeneration in the mouse is rodless retina. J Hered. 1966;57:47–50. doi: 10.1093/oxfordjournals.jhered.a107462. [DOI] [PubMed] [Google Scholar]
- 25.Tsang SH, Gouras P. Molecular physiology and pathology of the retina. In: Tasman W, Jaeger EA, editors. Duane’s Clinical Ophthalmology. Vol. 3. Philadelphia: Lippincott; 1996. chap 2. [Google Scholar]
- 26.Tsang SH, Burns ME, Calvert PD, et al. Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science. 1998;282(5386):117–121. doi: 10.1126/science.282.5386.117. [DOI] [PubMed] [Google Scholar]
- 27.Tsang SH, Woodruff ML, Janisch KM, Cilluffo MC, Farber DB, Fain GL. Removal of phosphorylation sites of gamma subunit of phosphodiesterase 6 alters rod light response. J Physiol. 2007;579(Pt 2):303–312. doi: 10.1113/jphysiol.2006.121772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsang S, Salchow D, Jiang M, et al. Toward repair of retinal degenerations with differentiated and embryonic stem (ES) cells. Am J Hum Genet. 2001;69:A2836. [Google Scholar]
- 29.Tsang SH, Yamashita CK, Lee WH, et al. The positive role of the carboxyl terminus of the gamma subunit of retinal cGMP-phosphodiesterase in maintaining phosphodiesterase activity in vivo. Vision Res. 2002;42(4):439–445. doi: 10.1016/s0042-6989(01)00213-9. [DOI] [PubMed] [Google Scholar]
- 30.Fain GL, Matthews HR, Cornwall MC, Koutalos Y. Adaptation in vertebrate photoreceptors. Physiol Rev. 2001;81(1):117–151. doi: 10.1152/physrev.2001.81.1.117. [DOI] [PubMed] [Google Scholar]
- 31.Stryer L. Visual excitation and recovery. J Biol Chem. 1991;266:10711–10714. [PubMed] [Google Scholar]
- 32.Yarfitz S, Hurley JB. Transduction mechanisms of vertebrate and invertebrate photoreceptors. J Biol Chem. 1994 May 20;269:14329–14332. [PubMed] [Google Scholar]
- 33.Burns ME, Baylor DA. Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annu Rev Neurosci. 2001;24:779–805. doi: 10.1146/annurev.neuro.24.1.779. [DOI] [PubMed] [Google Scholar]
- 34.Burns ME, Arshavsky VY. Beyond counting photons: trials and trends in vertebrate visual transduction. Neuron. 2005;48(3):387–401. doi: 10.1016/j.neuron.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 35.Fung BK-K, Hurley JB, Stryer L. Flow of information in the light-triggered cyclic nucleotide cascade of vision. Proc Natl Acad Sci U S A. 1981;78:152–156. doi: 10.1073/pnas.78.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arshavsky VY, Lamb TD, Pugh EN., Jr G proteins and phototransduction. Annu Rev Physiol. 2002;64:153–187. doi: 10.1146/annurev.physiol.64.082701.102229. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X, Cote RH. cGMP signaling in vertebrate retinal photoreceptor cells. Front Biosci. 2005;10:1191–1204. doi: 10.2741/1612. [DOI] [PubMed] [Google Scholar]
- 38.Baehr W, Devlin MJ, Applebury ML. Isolation of bovine ROS phosphiesterase. J Biol Chem. 1979;254:11669–11677. [PubMed] [Google Scholar]
- 39.Fung BKK, Young JH, Yamane HK, Griswold-Prenner I. Subunit stoichiometry of retinal rod cGMP phosphodiesterase. Biochemistry. 1990;29:2657–2664. doi: 10.1021/bi00463a006. [DOI] [PubMed] [Google Scholar]
- 40.Guo LW, Grant JE, Hajipour AR, et al. Asymmetric interaction between rod cyclic GMP phosphodiesterase gamma subunits and alpha/beta subunits. J Biol Chem. 2005;280(13):12585–12592. doi: 10.1074/jbc.M410380200. [DOI] [PubMed] [Google Scholar]
- 41.Guo LW, Muradov H, Hajipour AR, Sievert MK, Artemyev NO, Ruoho AE. The inhibitory gamma subunit of the rod cGMP phosphodiesterase binds the catalytic subunits in an extended linear structure. J Biol Chem. 2006;281(22):15412–15422. doi: 10.1074/jbc.M600595200. [DOI] [PubMed] [Google Scholar]
- 42.Farber DB, Lolley RN. Enzyme basis for cyclic GMP accumulation in degenerative photoreceptor cells of mouse retina. J Cyclic Nucleotide Res. 1976;2:139–148. [PubMed] [Google Scholar]
- 43.Farber DB. From mice to men: the cyclic GMP phosphodiesterase gene in vision and disease. The Proctor Lecture. Invest Opthalmol Vis Sci. 1995 Feb;36:263–275. [PubMed] [Google Scholar]
- 44.Cepko CL. Effect of gene expression on cone survival in retinitis pigmentosa. Retina. 2005;25(8 Suppl):S21–S24. doi: 10.1097/00006982-200512001-00008. [DOI] [PubMed] [Google Scholar]
- 45.Farber DB, Flannery JG, Bowes-Rickman C. The rd mouse story: seventy years of research on an animal model of inherited retinal degeneration. Prog Retinal Res. 1994;13:31–64. [Google Scholar]
- 46.Rohrer B, Pinto FR, Hulse KE, Lohr HR, Zhang L, Almeida JS. Multidestructive pathways triggered in photoreceptor cell death of the rd mouse as determined through gene expression profiling. J Biol Chem. 2004;279(40):41903–41910. doi: 10.1074/jbc.M405085200. [DOI] [PubMed] [Google Scholar]
- 47.Lohr HR, Kuntchithapautham K, Sharma AK, Rohrer B. Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp Eye Res. 2006;83(2):380–389. doi: 10.1016/j.exer.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 48.Takano Y, Ohguro H, Dezawa M, et al. Study of drug effects of calcium channel blockers on retinal degeneration of rd mouse. Biochem Biophys Res Commun. 2004;313(4):1015–1022. doi: 10.1016/j.bbrc.2003.12.034. [DOI] [PubMed] [Google Scholar]
- 49.Farber DB, Lolley RN. Cyclic guanosine monophosphate: elevations in degenerating photoreceptor cells of the C3H mouse retina. Science. 1974;186:449–451. doi: 10.1126/science.186.4162.449. [DOI] [PubMed] [Google Scholar]
- 50.Hart AW, McKie L, Morgan JE, et al. Genotype-phenotype correlation of mouse pde6b mutations. Invest Ophthalmol Vis Sci. 2005;46(9):3443–3450. doi: 10.1167/iovs.05-0254. [DOI] [PubMed] [Google Scholar]
- 51.Davis R, Tosi J, Janisch K, et al. Functional rescue of degenerating photoreceptors in mice homozygous for a hypomorphic cGMP phosphodiesterase 6 b allele (Pde6bH620Q) Invest Ophthalmol Vis Sci. 2008;49(11):5067–5076. doi: 10.1167/iovs.07-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Azadi S, Johnson LE, Paquet-Durand F, et al. CNTF+BDNF treatment and neuroprotective pathways in the rd1 mouse retina. Brain Res. 2007;1129(1):116–129. doi: 10.1016/j.brainres.2006.10.031. [DOI] [PubMed] [Google Scholar]
- 53.Jomary C, Cullen J, Jones SE. Inactivation of the Akt survival pathway during photoreceptor apoptosis in the retinal degeneration mouse. Invest Ophthalmol Vis Sci. 2006;47(4):1620–1629. doi: 10.1167/iovs.05-1176. [DOI] [PubMed] [Google Scholar]
- 54.Johnson LE, van Veen T, Ekstrom PA. Differential Akt activation in the photoreceptors of normal and rd1 mice. Cell Tissue Res. 2005;320(2):213–222. doi: 10.1007/s00441-004-1046-8. [DOI] [PubMed] [Google Scholar]
- 55.Hauck SM, Ekstrom PA, Ahuja-Jensen P, et al. Differential modification of phosducin protein in degenerating rd1 retina is associated with constitutively active Ca2+/calmodulin kinase II in rod outer segments. Mol Cell Proteomics. 2006;5(2):324–336. doi: 10.1074/mcp.M500217-MCP200. [DOI] [PubMed] [Google Scholar]
- 56.Roux PP, Topisirovic I. Regulation of mRNA translation by signaling pathways. Cold Spring Harb Perspect Biol. 2012;4(11) doi: 10.1101/cshperspect.a012252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Baal JW, Diks SH, Wanders RJ, et al. Comparison of kinome profiles of Barrett’s esophagus with normal squamous esophagus and normal gastric cardia. Cancer Res. 2006;66(24):11605–11612. doi: 10.1158/0008-5472.CAN-06-1370. [DOI] [PubMed] [Google Scholar]
- 58.Diks SH, Kok K, O’Toole T, et al. Kinome profiling for studying lipopolysaccharide signal transduction in human peripheral blood mononuclear cells. J Biol Chem. 2004;279(47):49206–49213. doi: 10.1074/jbc.M405028200. [DOI] [PubMed] [Google Scholar]
- 59.Davis RJ, Hsu CW, Tsai YT, et al. Therapeutic margins in a novel preclinical model of retinitis pigmentosa. J Neurosci. 2013;33(33):13475–13483. doi: 10.1523/JNEUROSCI.0419-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tosi J, Janisch KM, Wang NK, et al. Cellular and molecular origin of circumpapillary dysgenesis of the pigment epithelium. Ophthalmology. 2009;116(5):971–980. doi: 10.1016/j.ophtha.2008.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tosi J, Davis RJ, Wang NK, Naumann M, Lin CS, Tsang SH. shRNA knockdown of guanylate cyclase 2e or cyclic nucleotide gated channel alpha 1 increases photoreceptor survival in a cGMP phosphodiesterase mouse model of retinitis pigmentosa. J Cell Mol Med. 2011;15(8):1778–1887. doi: 10.1111/j.1582-4934.2010.01201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsang SH, Woodruff ML, Chen CK, et al. GAP-independent termination of photoreceptor light response by excess gamma subunit of the cGMP-phosphodiesterase. J Neurosci. 2006;26(17):4472–4480. doi: 10.1523/JNEUROSCI.4775-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsang SH, Woodruff ML, Hsu CW, et al. Function of the asparagine 74 residue of the inhibitory gamma-subunit of retinal rod cGMP-phophodiesterase (PDE) in vivo. Cell Signal. 2011;23(10):1584–1589. doi: 10.1016/j.cellsig.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsang SH, Woodruff ML, Jun L, et al. Transgenic mice carrying the H258N mutation in the gene encoding the beta-subunit of phosphodiesterase-6 (PDE6B) provide a model for human congenital stationary night blindness. Hum Mutat. 2007;28(3):243–254. doi: 10.1002/humu.20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsang SH, Woodruff ML, Lin CS, et al. Effect of the ILE86TER mutation in the gamma subunit of cGMP phosphodiesterase (PDE6) on rod photoreceptor signaling. Cell Signal. 2012;24(1):181–188. doi: 10.1016/j.cellsig.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wert KJ, Sancho-Pelluz J, Tsang SH. Mid-stage intervention achieves similar efficacy as conventional early-stage treatment using gene therapy in a pre-clinical model of retinitis pigmentosa. Hum Mol Genet. 2014;23(2):514–523. doi: 10.1093/hmg/ddt452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lolley R, Farber D, Rayborn M, Hollyfield J. Cyclic GMP accumulation causes degeneration of photoreceptor cells: simulation of an inherited disease. Science. 1977;196:664–666. doi: 10.1126/science.193183. [DOI] [PubMed] [Google Scholar]
- 68.Wert KJ, Skeie JM, Davis RJ, Tsang SH, Mahajan VB. Subretinal injection of gene therapy vectors and stem cells in the perinatal mouse eye. J Vis Exp. 2012 Nov 25;(69) doi: 10.3791/4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tsang S, Woodruff M, Lin C, et al. Effect of the ILE86TER mutation in the γ subunit of cGMP phosphodiesterase (PDE6) on rod photoreceptor signaling. Cell Signal. 2012;24(1):181–188. doi: 10.1016/j.cellsig.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Janisch KM, Kasanuki JM, Naumann MC, et al. Light-dependent phosphorylation of the gamma subunit of cGMP-phosphodiesterase (PDE6gamma) at residue threonine 22 in intact photoreceptor neurons. Biochem Biophys Res Commun. 2009;390(4):1149–1153. doi: 10.1016/j.bbrc.2009.10.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Woodruff ML, Janisch KM, Peshenko IV, Dizhoor AM, Tsang SH, Fain GL. Modulation of phosphodiesterase6 turnoff during background illumination in mouse rod photoreceptors. J Neurosci. 2008;28(9):2064–2074. doi: 10.1523/JNEUROSCI.2973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tosi J, Wang NK, Zhao J, et al. Rapid and noninvasive imaging of retinal ganglion cells in live mouse models of glaucoma. Mol Imaging Biol. 2010;12(4):386–393. doi: 10.1007/s11307-009-0292-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang NK, Tosi J, Kasanuki JM, et al. Transplantation of reprogrammed embryonic stem cells improves visual function in a mouse model for retinitis pigmentosa. Transplantation. 2010;89(8):911–919. doi: 10.1097/TP.0b013e3181d45a61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mahajan VB, Skeie JM, Assefnia AH, Mahajan M, Tsang SH. Mouse eye enucleation for remote high-throughput phenotyping. J Vis Exp. 2011 Nov 19;(57) doi: 10.3791/3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Freilinger A, Rosner M, Hengstschlager M. Tuberin negatively affects BCL-2’s cell survival function. Amino Acids. 2006;30(4):391–396. doi: 10.1007/s00726-006-0359-1. [DOI] [PubMed] [Google Scholar]
- 76.Rosner M, Freilinger A, Hengstschlager M. Akt regulates nuclear/cytoplasmic localization of tuberin. Oncogene. 2007;26(4):521–531. doi: 10.1038/sj.onc.1209812. [DOI] [PubMed] [Google Scholar]
- 77.Kolb TM, Duan L, Davis MA. Tsc2 expression increases the susceptibility of renal tumor cells to apoptosis. Toxicol Sci. 2005;88(2):331–339. doi: 10.1093/toxsci/kfi310. [DOI] [PubMed] [Google Scholar]
- 78.Punzo C, Kornacker K, Cepko CL. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci. 2009;12(1):44–52. doi: 10.1038/nn.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ruvinsky I, Sharon N, Lerer T, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19(18):2199–2211. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fain GL, Lisman JE. Light, Ca2+, and photoreceptor death: new evidence for the equivalent-light hypothesis from arrestin knockout mice. Invest Ophthalmol Vis Sci. 1999;40(12):2770–2772. [PubMed] [Google Scholar]
- 81.Tsang SH, Chen J, Yamashita CK, et al. Apoptotic photoreceptor death in gene targeted mutant mice and its deceleration by gene therapy. Am J Hum Genet. 1996 Oct;59:A54. [Google Scholar]
- 82.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opinion Cell Biol. 2005;17(6):596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 84.Yu Y, Yoon SO, Poulogiannis G, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332(6035):1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ekim B, Magnuson B, Acosta-Jaquez HA, Keller JA, Feener EP, Fingar DC. mTOR kinase domain phosphorylation promotes mTORC1 signaling, cell growth, and cell cycle progression. Mol Cell Biol. 2011;31(14):2787–2801. doi: 10.1128/MCB.05437-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG., Jr TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003;4(2):147–158. doi: 10.1016/s1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]